Abstract
η6-Coordination catalysis has emerged as an effective strategy for activating electron-rich (hetero)arenes toward nucleophilic substitution. Recent experimental studies on Ru(II)-catalyzed amination of aminopyridines revealed a striking ortho–para reactivity difference, with ortho-substituted substrates undergoing efficient amination while para analogs are unreactive under identical conditions. Herein, we present a density functional theory investigation to elucidate the origin of this divergence. Computed free-energy profiles show that both substitution patterns follow a similar stepwise mechanism involving Ru-bound Meisenheimer intermediates and a proton-transfer relay, with C–N bond cleavage/rearomatization as the rate-determining step. However, the para pathway suffers from a substantially higher overall barrier, originating from the intrinsically less stable Meisenheimer intermediates. Energy decomposition analysis indicates that the decisive factor is weaker orbital interaction between the CpRu(II) fragment and the para-substituted Meisenheimer intermediate, whereas electrostatics and dispersion play negligible roles. These findings highlight the key role of metal–substrate orbital interactions in stabilizing dearomatized intermediates, offering mechanistic insights for rational design of η6-coordination catalysis with enhanced reactivity and selectivity.
1. Introduction
π-Coordination has become a powerful and widely applicable strategy for the activation of inert (hetero)aromatic compounds [1]. In this mode of activation, aromatic substrates form reversible η6-complexes with transition-metal centers through π-electron donation, resulting in perturbation of the electronic structure and reactivity of the arene ring [2]. While the concept of arene π-complexation was first explored decades ago [3], its catalytic potential has seen rapid expansion in recent years [4,5,6,7,8,9,10,11,12,13,14,15,16], enabling a range of transformations that are inaccessible to the corresponding uncoordinated arenes. This activation approach is particularly attractive for electron-rich systems, where conventional strategies like nucleophilic aromatic substitution are typically challenging to provide efficient reactivity.
In recent years, a series of powerful catalytic substitution reactions have been developed based on this π-coordination strategy by Shi and co-workers [17]. Particularly, recent developments have demonstrated that arenophilic metal complexes—most notably low-valent Ru(II) and Rh(III) species—can promote distinctive redox-neutral η6-coordination catalysis to achieve efficient activation and functionalization of otherwise robust C–F, C–OH, and C–NH2 bonds on electron-rich (hetero)arenes [17]. Central to these advances is the use of η6-binding to transiently reverse the polarity of the arene, creating electrophilic character at positions that are otherwise nucleophilic in nature. These reactions proceed under mild conditions, often without additives or oxidants, and tolerate a broad scope of nucleophiles and substitution patterns, establishing a versatile platform for aromatic substitution chemistry.
This new activation paradigm also raises important mechanistic questions. The reactivity patterns observed under η6-coordination catalysis often diverge from those expected in classical aromatic substitution reactions. A striking example appears in a recent report on Ru-catalyzed amination of aminopyridines from the Shi group [18], in which pyridines bearing an amino group at the ortho position undergo efficient C–N bond formation with primary amines, whereas their para-substituted counterparts exhibit no reactivity under otherwise identical conditions (Scheme 1). This observation is particularly noteworthy, as both ortho and para substitution patterns typically exhibit comparable reactivity in conventional aromatic substitution reactions. The unexpected reactivity difference suggests that η6-coordination catalysis may introduce additional factors influencing substrate activation, beyond the classical substituent effects of aromatic substitution described in textbooks. However, Shi’s previous synthetic study [18] did not fully provide an explanation for this reactivity divergence, underscoring the need for further mechanistic investigation.
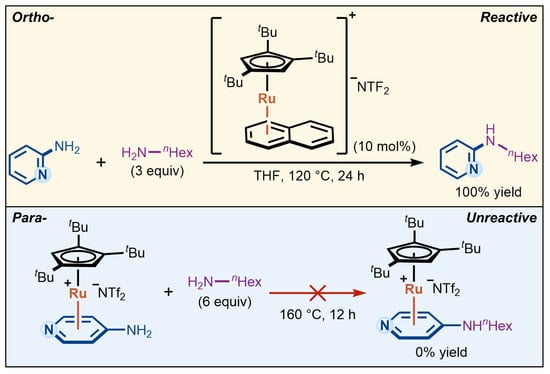
Scheme 1.
Ortho–para reactivity difference in Ru-catalyzed amination of aminopyridines via η6-coordination [18].
To understand the origins of this ortho–para reactivity difference, we conducted a mechanistic investigation using density functional theory (DFT) calculations. Based on the free energy profile analysis of the reaction pathways and electronic structure characterization of key intermediates, we herein report that the observed reactivity pattern is linked to the distinct coordination ability of the Meisenheimer-type intermediates to the Ru(II) center. The enhanced metal binding in the ortho-substituted substrate stabilizes key transient intermediates in the catalytic cycle, whereas the weak coordination in the para-substituted case leads to unfavorable overall barrier. These findings elucidate a fundamental connection between intermediate coordination and catalytic efficiency, providing new design principles for η6-coordination catalysis.
2. Results and Discussion
2.1. Benchmarking Computational Methods for Ru(II)–Aminopyridine π-Complexes
To identify a reliable level of theory for calculating the Ru(II) π-activation complexes, we first performed a comprehensive benchmarking study across a series of commonly used DFT functionals and solvation models. Three crystallographically characterized Ru(II)–6-methylpyridin-2-amine complexes from Shi’s study [18] were selected as reference structures (Figure 1): CPX-A corresponds to the π-complex INT-A featuring a Cp* ligand; CPX-B and CPX-C correspond to two distinct conformers of the π-complex INT-B, which bears a tBu-substituted Cp ligand coordinated to the same aminopyridine substrate. These complexes provide representative models for evaluating the structural accuracy of optimized π-coordinated Ru(II) intermediates.
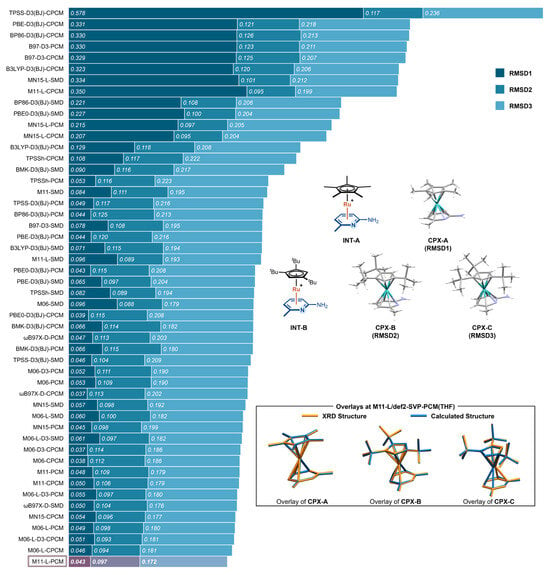
Figure 1.
RMSDs between optimized and crystallographic geometries for CPX-A, CPX-B, and CPX-C using 51 different DFT functional–solvation model combinations. Bars are sorted in descending order of the total RMSDs across the three complexes.
A total of 17 DFT functionals were evaluated: BP86 [19]-D3 [20] (BJ) [21], B3LYP [22]-D3(BJ), PBE [23]-D3(BJ), PBE0 [24]-D3(BJ), B97 [25]-D3, M06 [26], M06-D3, M06-L [27], M06-L-D3, MN15 [28], MN15-L [29], M11 [30], M11-L [31], BMK [32]-D3(BJ), TPSS [33]-D3(BJ), TPSSh [34], and ωB97X-D [35]. Each was tested in combination with three implicit solvation models—PCM, CPCM, and SMD—yielding 51 method combinations. All geometry optimizations were performed using the def2-SVP basis set with THF as the solvent, and basis set variation was not considered in this benchmark. For each structure, the root-mean-square deviation (RMSD) of non-hydrogen atoms was computed relative to the corresponding crystal structure. The three values—RMSD1 (for CPX-A), RMSD2 (for CPX-B), and RMSD3 (for CPX-C)—were used to quantify the agreement between computed and experimental geometries.
The benchmarking results are summarized in Figure 1. The computed RMSD values span a broad range from 0.037 Å to 0.578 Å, reflecting significant differences in structural accuracy across the method combinations. Among the three complexes, CPX-A exhibits the greatest sensitivity to the computational method, with RMSD values ranging from 0.037 Å (M06-D3–CPCM) to 0.578 Å (TPSS-D3(BJ)–CPCM). In contrast, CPX-B is the least sensitive, with all methods yielding RMSD values between 0.093 Å (M06-L-D3-CPCM) and 0.126 Å (BP86-D3(BJ)–CPCM). CPX-C shows moderate sensitivity, with RMSD values from 0.172 Å (M11-L–PCM) to 0.236 Å (TPSS-D3(BJ)–CPCM). These results underscore the varying degrees of geometric responsiveness to methodological choices and highlight the importance of method selection in modeling π-coordinated Ru(II) complexes.
The data also reveal notable synergistic effects between the DFT functional and solvation model. In some cases, the combination of a suboptimal functional and an unfavorable solvent model leads to drastic over-relaxation. A striking example is TPSS-D3(BJ) combined with CPCM, which gives the highest deviation observed—an RMSD of 0.578 Å for CPX-A. By contrast, the same functional with PCM or SMD yields much lower RMSD values (0.049 Å and 0.046 Å, respectively), illustrating the pronounced impact of solvation treatment when coupled with sensitive functionals.
Despite such cases, the choice of functional has a more dominant influence than the solvation model. Once a reliable functional is selected—such as members of the Minnesota family (M06, M06-L, MN15, MN15-L) or ωB97X-D—the differences among PCM, CPCM, and SMD become relatively minor. These functionals deliver consistently low RMSD values across all three complexes, making them broadly applicable regardless of the solvent model. Still, a few extreme functional–solvent combinations, such as MN15–L–SMD and M11–L–PCM, yield significantly higher RMSD values for CPX-A (0.334 Å and 0.350 Å, respectively).
Among all tested combinations, M11–L/def2-SVP-PCM(THF) delivers the best overall performance, with RMSD values of 0.043 Å for CPX-A, 0.097 Å for CPX-B, and 0.172 Å for CPX-C. The method reliably reproduces key structural parameters and overall coordination geometries, as illustrated by the structural overlays in Figure 1. Based on its consistently high accuracy across different ligand environments, M11–L/def2-SVP-PCM(THF) was selected as the level of theory for all subsequent geometry optimizations and mechanistic studies.
2.2. DFT-Computed Mechanistic Profile for the Ortho-Substitution Pathway
We first examined the reaction mechanism for ortho-aminopyridine; the complete free-energy profile is shown in Figure 2. The catalytic cycle is initiated by η6-coordination of the substrate to Ru(II), furnishing the π-complex INT1-ortho. Approach of methylamine gives the more stable, hydrogen-bonded adduct INT2-ortho, from which nucleophilic addition occurs via TS3-ortho to form the Meisenheimer intermediate INT4-ortho. The barrier from INT2-ortho to TS3-ortho is 25.7 kcal/mol, representing the first significant ascent along the pathway.
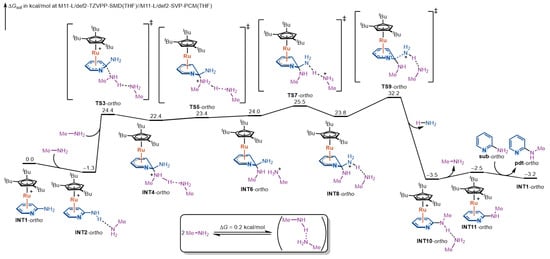
Figure 2.
DFT-computed free-energy profile for the Ru(II)-catalyzed amination of ortho-aminopyridine via η6-coordination.
From INT4-ortho, a stepwise proton-transfer relay enhances the leaving-group ability. The proton on the ammonium moiety (purple-labeled in Figure 2) is first transferred to the hydrogen-bonded methylamine to give INT6-ortho; the newly formed methanaminium then delivers a proton to the departing amino group (blue-labeled in Figure 2), affording INT8-ortho. Because the basicities of the relevant amines are similar, the three Meisenheimer intermediates (INT4-ortho, INT6-ortho, INT8-ortho) are comparable in stability, and each protonation step proceeds with a very small intrinsic barrier within 2 kcal/mol. Therefore, the kinetics of these proton transfer steps do not contribute to rate determination.
After proton transfer, INT8-ortho undergoes C–N bond cleavage at the leaving-group site with rearomatization of the pyridine ring via TS9-ortho. The intrinsic barrier from INT8-ortho to TS9-ortho is 8.4 kcal/mol. Although modest intrinsically, the overall rate-determining step arises here because the dearomatized Meisenheimer manifold is itself high in free energy: measured from the on-cycle resting state INT2-ortho, the overall free-energy barrier is 33.5 kcal/mol via TS9-ortho. The height of this barrier reflects the balance between stabilization provided by Ru-bound Meisenheimer intermediate and the energetic cost of C–N bond cleavage. This C–N cleavage step yields a highly stabilized product-coordinated complex INT10-ortho. Subsequent facile arene exchange releases the product and regenerates the active intermediate INT1-ortho for the next catalytic cycle.
2.3. DFT-Computed Mechanistic Profile for the Para-Substitution Pathway
The computed free-energy profile for the para-aminopyridine amination pathway is shown in Figure 3. As in the ortho case, the reaction begins with η6-coordination of the substrate to the Ru(II) catalyst, forming the π-complex INT1-para. Coordination of methylamine affords the hydrogen-bonded adduct INT2-para, from which nucleophilic attack proceeds via TS3-para to give the Meisenheimer intermediate INT4-para. The barrier from INT2-para to TS3-para is calculated to be 26.6 kcal/mol, comparable to that of the ortho pathway.
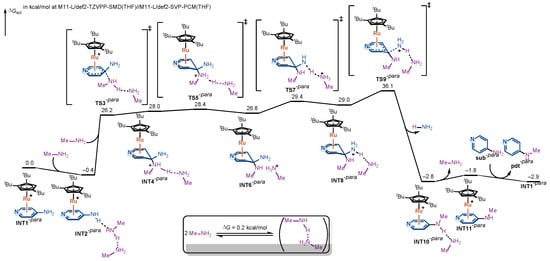
Figure 3.
DFT-computed free-energy profile for the Ru(II)-catalyzed amination of para-aminopyridine via η6-coordination.
From INT4-para, the same stepwise proton-transfer sequence is observed. The proton from the ammonium moiety (purple-labeled in Figure 3) is first delivered to the hydrogen-bonded methylamine, yielding INT6-para; the resulting methanaminium then transfers its proton to the departing amino group (blue-labeled in Figure 3), affording INT8-para. As in the ortho case, these Meisenheimer intermediates (INT4-para, INT6-para, INT8-para) are similar in relative stability, and each proton-transfer step has a small intrinsic barrier, making no significant contribution to the overall rate.
After protonation of the leaving group, INT8-para undergoes C–N bond cleavage with rearomatization through TS9-para. The intrinsic barrier from INT8-para to TS9-para is 7.1 kcal/mol, even lower than in the ortho pathway. However, due to the intrinsically reduced stability of the Meisenheimer intermediates in the para pathway, INT4-para, INT6-para, and INT8-para all lie substantially higher in energy than their ortho counterparts. In particular, INT8-para is calculated to be 29.0 kcal/mol above the on-cycle resting state INT2-para. Consequently, the overall free-energy barrier, measured from INT2-para to TS9-para, reaches 36.5 kcal/mol, which is 3.0 kcal/mol higher than in the ortho pathway. Based on the Eyring equation, this 3.0 kcal/mol free-energy difference would translate into a remarkable difference in the reaction rate constants, further confirming the consistency between our computational results and the experimental observations.
The above comparison reveals that, in the para pathway, the rate-determining step is again the C–N bond cleavage/rearomatization from a high-energy Meisenheimer intermediate. It is the elevated energy of the Meisenheimer intermediate directly translates into the higher overall barrier, accounting for the experimentally observed lack of reactivity of para-substituted aminopyridines.
2.4. Energy Decomposition Analysis of Meisenheimer Intermediate Stability
To further elucidate the origins of the stability difference between the Meisenheimer intermediates in the ortho and para pathways, we performed a quantitative energy decomposition analysis (EDA) using the sobEDA approach developed by Lu and co-workers [36]. In this analysis, the interaction between the CpRu(II) fragment and the substrate fragment was decomposed into electrostatic, exchange-repulsion, Coulomb correlation, and orbital interaction terms. The calculations were carried out for the substrate–Ru π-complex (INT1) and the corresponding protonated Meisenheimer intermediate (INT8) in each pathway, and the results are summarized in Figure 4.
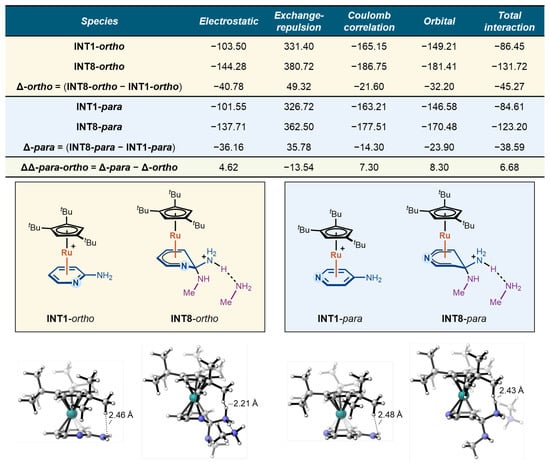
Figure 4.
Energy decomposition analysis of CpRu(II)–substrate interactions for the Meisenheimer intermediates INT1 and INT8.
For each pathway, the change in the interaction energy terms from INT1 to INT8 (Δ-ortho and Δ-para) reflects the energetic contributions of the CpRu(II)–substrate interaction to the stabilization/destabilization of the Meisenheimer intermediate relative to the resting π-complex. Comparing these Δ values between the ortho and para cases yields the ΔΔpara–ortho values, which capture the subtle influence of the substitution pattern on each interaction component and, ultimately, on the stability of the Meisenheimer intermediate.
As shown in Figure 4, the differences in electrostatic, exchange-repulsion, and Coulomb correlation contributions between the two pathways largely cancel each other out (4.62, −13.54, and 7.30 kcal/mol, respectively), indicating that nuclear–nuclear and electrostatic effects are not the decisive factors in the stability difference. Instead, the key distinction lies in the orbital interaction term: the para pathway exhibits orbital interactions that are 8.30 kcal/mol less favorable than in the ortho pathway. This reduced orbital stabilization directly accounts for the less favorable total interaction energy in the para case, with a 6.68 kcal/mol energy difference. Therefore, the weaker orbital stabilization of the para Meisenheimer intermediate—rather than electrostatics or dispersion—accounts for its reduced stability and the higher overall barrier.
To clarify the influence of steric effects on reactivity, we also analyzed the structure of the Meisenheimer intermediate INT8. For both the ortho and para cases, steric congestion between the tert-butyl group of the ligand and the departing amino group is limited, with only one H–H contact of 2.21 Å observed in INT8-ortho. Therefore, the impact of steric effects on the reaction reactivity is limited.
3. Materials and Methods
All density functional theory (DFT) calculation results are obtained with Gaussian 16 program [37]. Default G16 SCF convergence criteria (scf = tight), optimization convergence criteria and integral grid parameters (int = (ultrafine, acc2e = 12)) for Gaussian 16 are applied unless otherwise stated. Geometry optimizations are conducted with M11-L functional, and def2-SVP basis set was used for all atoms, with solvent effects at THF evaluated. Single-point energies and solvent effects at THF are also evaluated with the same functional and def2-TZVPP basis set for all atoms. The solvation energies are calculated with a self-consistent reaction field (SCRF) using the PCM implicit solvent model for geometry optimizations and SMD implicit solvent model for single-point energies. Frequency analysis is also performed at the same level of theory as geometry optimization using harmonic oscillator model to confirm whether optimized stationary points are either local minimum or transition state, as well as to evaluate zero-point vibrational energies and thermal corrections for enthalpies and free energies at 298.15 K.
Energy decomposition analysis (EDA) was conducted with sobEDA [36] and Multiwfn [38] software. The 3D diagrams of optimized structures shown in this Supplementary Information for computations are generated with CYLview software [39].
4. Conclusions
In summary, our DFT study reveals that the pronounced ortho–para reactivity difference in Ru(II)-catalyzed amination of aminopyridines arises from distinct stabilities of the Ru-bound Meisenheimer intermediates. While both substitution patterns proceed through comparable stepwise pathways, the ortho intermediates benefit from stronger CpRu–substrate orbital interactions, leading to lower free-energy barriers for the C–N bond cleavage/rearomatization step. In contrast, the para intermediates exhibit significantly reduced orbital stabilization, rendering them energetically higher and raising the overall barrier beyond the experimentally accessible range. Energy decomposition analysis pinpoints the reduced orbital interaction in the para case as the primary contributor to its lower stability, with electrostatic, exchange-repulsion, and correlation terms largely canceling out. These mechanistic insights underscore the importance of metal–substrate orbital overlap in η6-coordination catalysis and suggest that strategic incorporation of proximal donor groups to enhance such interactions could be a general approach for improving catalytic performance in arene substitution reactions. Building on this mechanistic study, we will further investigate the reactivity differences in various substituted 2-aminopyridine derivatives and employ physical organic parameter-based linear regression [40,41] to establish more broadly applicable theoretical models that can guide the design and development of such reactions.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/inorganics13100316/s1, Computational details, calculated energies, and Cartesian coordinates of all computed species are provided in Supporting Information.
Author Contributions
Conceptualization, X.H. and S.-Q.Z.; calculation, C.W. and S.-Q.Z.; analysis, S.-Q.Z. and X.H.; writing, X.H. and S.-Q.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (22122109, 22271253 and W2512004 X.H.), National Key R&D Program of China (2022YFA1504301, X.H.), New Generation Artificial Intelligence-National Science and Technology Major Project (2025ZD0121905, X.H.), the Starry Night Science Fund of Zhejiang University Shanghai Institute for Advanced Study (SN-ZJU-SIAS-006, X.H.), Fundamental Research Funds for the Central Universities (226-2022-00140, 226-2022-00224, 226-2023-00115 and 226-2024-00003, X.H.), the State Key Laboratory of Physical Chemistry of Solid Surfaces (202210, X.H.), the Leading Innovation Team grant from Department of Science and Technology of Zhejiang Province (2022R01005, X.H.) and Open Research Fund of School of Chemistry and Chemical Engineering of Henan Normal University (2024Z01, X.H.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article/Supplementary Materials. Further inquiries can be directed to the corresponding author.
Acknowledgments
We appreciate the financial support of the National Natural Science Foundation of China (22122109, 22271253 and W2512004 X.H.), National Key R&D Program of China (2022YFA1504301, X.H.), New Generation Artificial Intelligence-National Science and Technology Major Project (2025ZD0121905, X.H.), the Starry Night Science Fund of Zhejiang University Shanghai Institute for Advanced Study (SN-ZJU-SIAS-006, X.H.), Fundamental Research Funds for the Central Universities (226-2022-00140, 226-2022-00224, 226-2023-00115 and 226-2024-00003, X.H.), the State Key Laboratory of Physical Chemistry of Solid Surfaces (202210, X.H.), the Leading Innovation Team grant from Department of Science and Technology of Zhejiang Province (2022R01005, X.H.) and Open Research Fund of School of Chemistry and Chemical Engineering of Henan Normal University (2024Z01, X.H.).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhu, H.; Wu, Y.; Mao, J.; Xu, J.; Walsh, P.J.; Shi, H. C–H functionalization through benzylic deprotonation with π-coordination or cation–π-interactions. Chem. Soc. Rev. 2025, 54, 2520–2542. [Google Scholar]
- Williams, L.J.; Bhonoah, Y.; Wilkinson, L.A.; Walton, J.W. As Nice as π: Aromatic Reactions Activated by π-Coordination to Transition Metals. Chem. Eur. J. 2021, 27, 3650–3660. [Google Scholar] [CrossRef]
- Litvak, V.V.; Kun, P.P.; Shteingarts, V.D. Arene transition metal complexes in reactions with nucleophilic reagents: XIX. Transannular substituent effects in arene-chromium and iron complexes. J. Organomet. Chem. 1988, 348, 219–233. [Google Scholar] [CrossRef]
- Utsunomiya, M.; Hartwig, J.F. Ruthenium-Catalyzed Anti-Markovnikov Hydroamination of Vinylarenes. J. Am. Chem. Soc. 2004, 126, 2702–2703. [Google Scholar] [CrossRef] [PubMed]
- Takaya, J.; Hartwig, J.F. Mechanistic Studies of Ruthenium-Catalyzed Anti-Markovnikov Hydroamination of Vinylarenes: Intermediates and Evidence for Catalysis through π-Arene Complexes. J. Am. Chem. Soc. 2005, 127, 5756–5757. [Google Scholar] [CrossRef]
- Otsuka, M.; Endo, K.; Shibata, T. Catalytic SNAr reaction of non-activated fluoroarenes with amines via Ru η6-arene complexes. Chem. Commun. 2010, 46, 336–338. [Google Scholar] [CrossRef]
- Walton, J.W.; Williams, J.M.J. Catalytic SNAr of unactivated aryl chlorides. Chem. Commun. 2015, 51, 2786–2789. [Google Scholar] [CrossRef]
- Konovalov, A.I.; Gorbacheva, E.O.; Miloserdov, F.M.; Grushin, V.V. Ruthenium-catalyzed nucleophilic fluorination of halobenzenes. Chem. Commun. 2015, 51, 13527–13530. [Google Scholar] [CrossRef]
- Takemoto, S.; Shibata, E.; Nakajima, M.; Yumoto, Y.; Shimamoto, M.; Matsuzaka, H. Ruthenium-Sulfonamide-Catalyzed Direct Dehydrative Condensation of Benzylic C–H Bonds with Aromatic Aldehydes. J. Am. Chem. Soc. 2016, 138, 14836–14839. [Google Scholar] [CrossRef] [PubMed]
- Kang, Q.-K.; Lin, Y.; Li, Y.; Shi, H. Ru(II)-Catalyzed Amination of Aryl Fluorides via η6-Coordination. J. Am. Chem. Soc. 2020, 142, 3706–3711. [Google Scholar] [CrossRef] [PubMed]
- Kang, Q.-K.; Lin, Y.; Li, Y.; Xu, L.; Li, K.; Shi, H. Catalytic SNAr Hydroxylation and Alkoxylation of Aryl Fluorides. Angew. Chem. Int. Ed. 2021, 60, 20391–20399. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Kang, Q.-K.; Li, Y.; Wu, W.-Q.; Zhu, H.; Shi, H. Catalytic Amination of Phenols with Amines. J. Am. Chem. Soc. 2022, 144, 1144–1151. [Google Scholar] [CrossRef]
- Kang, Q.-K.; Li, Y.; Chen, K.; Zhu, H.; Wu, W.-Q.; Lin, Y.; Shi, H. Rhodium-Catalyzed Stereoselective Deuteration of Benzylic C–H Bonds via Reversible η6-Coordination. Angew. Chem. Int. Ed. 2022, 61, e202117381. [Google Scholar] [CrossRef]
- Wu, W.-Q.; Lin, Y.; Li, Y.; Shi, H. Catalytic Dehydrogenative (3 + 2) Cycloaddition of Alkylbenzenes via π-Coordination. J. Am. Chem. Soc. 2023, 145, 9464–9470. [Google Scholar] [CrossRef]
- Schulte, T.; Wang, Z.; Li, C.-C.; Hamad, A.; Waldbach, F.; Pampel, J.; Petzold, R.; Leutzsch, M.; Bahns, F.; Ritter, T. Ruthenium Phenoxo Complexes: An Isolobal Ligand to Cp with Improved Properties. J. Am. Chem. Soc. 2024, 146, 15825–15832. [Google Scholar] [CrossRef]
- Li, Y.; Shi, H. Rhodium-catalyzed addition reactions of benzylic C–H bonds to cyclic N-sulfonyl ketimines via π-coordination. Chin. Chem. Lett. 2024, 35, 108650. [Google Scholar] [CrossRef]
- Chen, K.; Shi, H. Nucleophilic Aromatic Substitution of Halobenzenes and Phenols with Catalysis by Arenophilic π Acids. Acc. Chem. Res. 2024, 57, 2194–2206. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lin, Y.; Wu, W.Q.; Hu, W.Q.; Xu, J.; Shi, H. Amination of Aminopyridines via η6-Coordination Catalysis. J. Am. Chem. Soc. 2024, 146, 22906–22912. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comp. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6169. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. V. Systematic optimization of exchange-correlation functionals. J. Chem. Phys. 1997, 107, 8554–8560. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101. [Google Scholar] [CrossRef]
- Yu, H.S.; He, X.; Li, S.L.; Truhlar, D.G. MN15: A Kohn–Sham global-hybrid exchange–correlation density functional with broad accuracy for multi-reference and single-reference systems and noncovalent interactions. Chem. Sci. 2016, 7, 5032–5051. [Google Scholar] [CrossRef]
- Yu, H.S.; He, X.; Truhlar, D.G. MN15-L: A New Local Exchange-Correlation Functional for Kohn–Sham Density Functional Theory with Broad Accuracy for Atoms, Molecules, and Solids. J. Chem. Theo. Comput. 2016, 12, 1280–1293. [Google Scholar] [CrossRef]
- Peverati, R.; Truhlar, D.G. Improving the Accuracy of Hybrid Meta-GGA Density Functionals by Range Separation. J. Phys. Chem. Lett. 2011, 2, 2810–2817. [Google Scholar] [CrossRef]
- Peverati, R.; Truhlar, D.G. M11-L: A Local Density Functional That Provides Improved Accuracy for Electronic Structure Calculations in Chemistry and Physics. J. Phys. Chem. Lett. 2012, 3, 117–124. [Google Scholar] [CrossRef]
- Boese, A.D.; Martin, J.M.L. Development of density functionals for thermochemical kinetics. J. Chem. Phys. 2004, 121, 3405–3416. [Google Scholar] [CrossRef]
- Tao, J.M.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the Density Functional Ladder: Nonempirical Meta–Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [PubMed]
- Staroverov, V.N.; Scuseria, G.E.; Tao, J.M.; Perdew, J.P. Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes. J. Chem. Phys. 2003, 119, 12129. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. Simple, Efficient, and Universal Energy Decomposition Analysis Method Based on Dispersion-Corrected Density Functional Theory. J. Phys. Chem. A 2023, 127, 7023. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Legault, C.Y. CYLView, version 1.0b; Universitéde Sherbrooke: Montreal, QC, Canada, 2009. Available online: http://www.cylview.org (accessed on 1 January 2025).
- Kuntz, R.; Yan, Y.; Sigman, M.S.; Sanford, M.S. A Physical Organic Chemistry Approach to Developing Cyclopropenium-Based Energy Storage Materials for Redox Flow Batteries. Acc. Chem. Res. 2023, 56, 1239–1250. [Google Scholar] [CrossRef] [PubMed]
- Qian, G.; Zhu, L.; Zeng, Y.; Li, J.; Liu, J.; Fei, C.; Li, Z.; Zhang, S.; Luo, J.; Deng, L.; et al. Mechanism and Origins of Weak Bonding-Controlled Selectivities in Cinchoninium-Catalyzed Umpolung Michael Addition of Imines. CCS Chem. 2025, 7, 1797–1811. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).