
Understanding Dioxygen Activation in the Fe(III)-Promoted Oxidative Dehydrogenation of Amines: A Computational Study

 , , , , and
, , , , and
Abstract

1. Introduction
2. Results
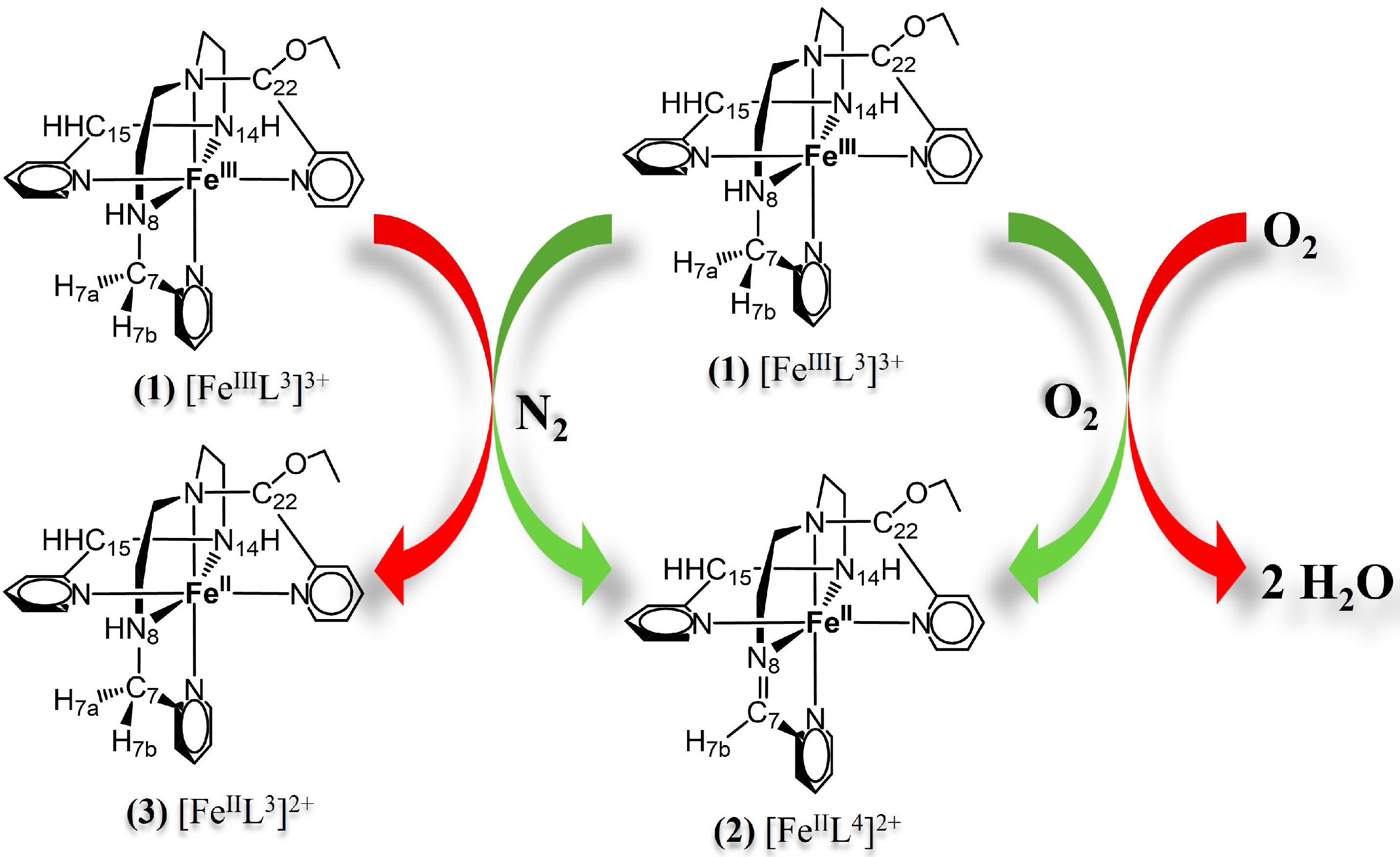
2.1. Ligands and Iron Complexes
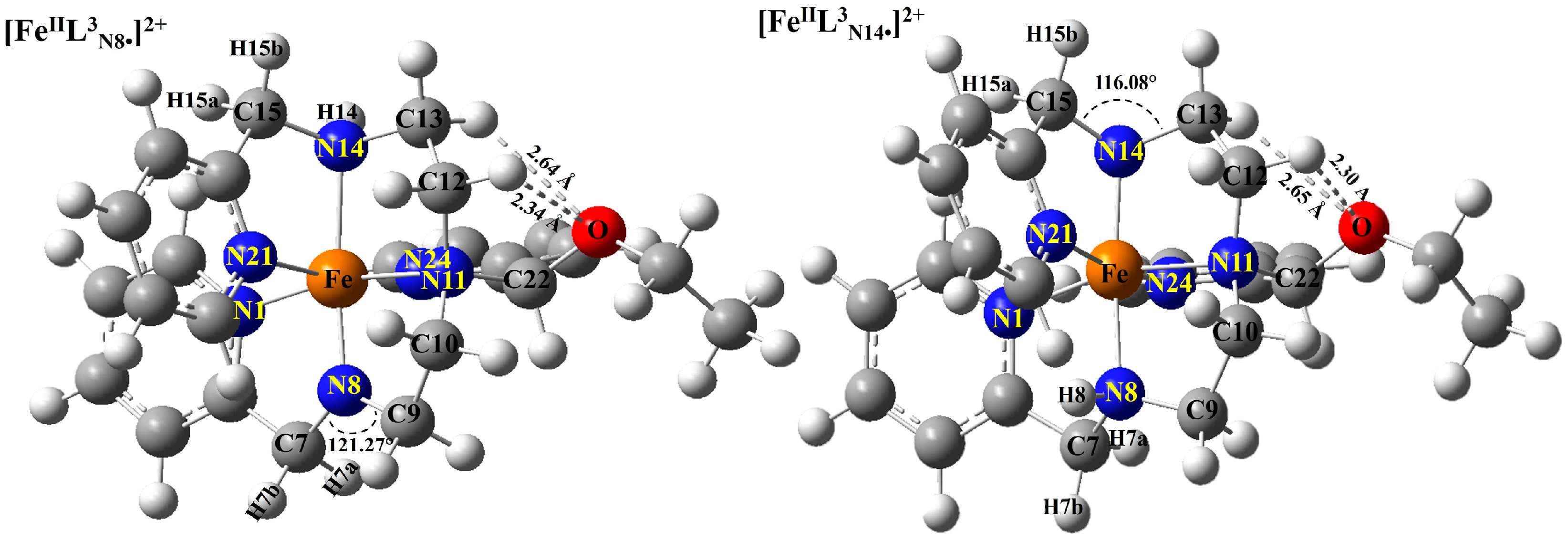
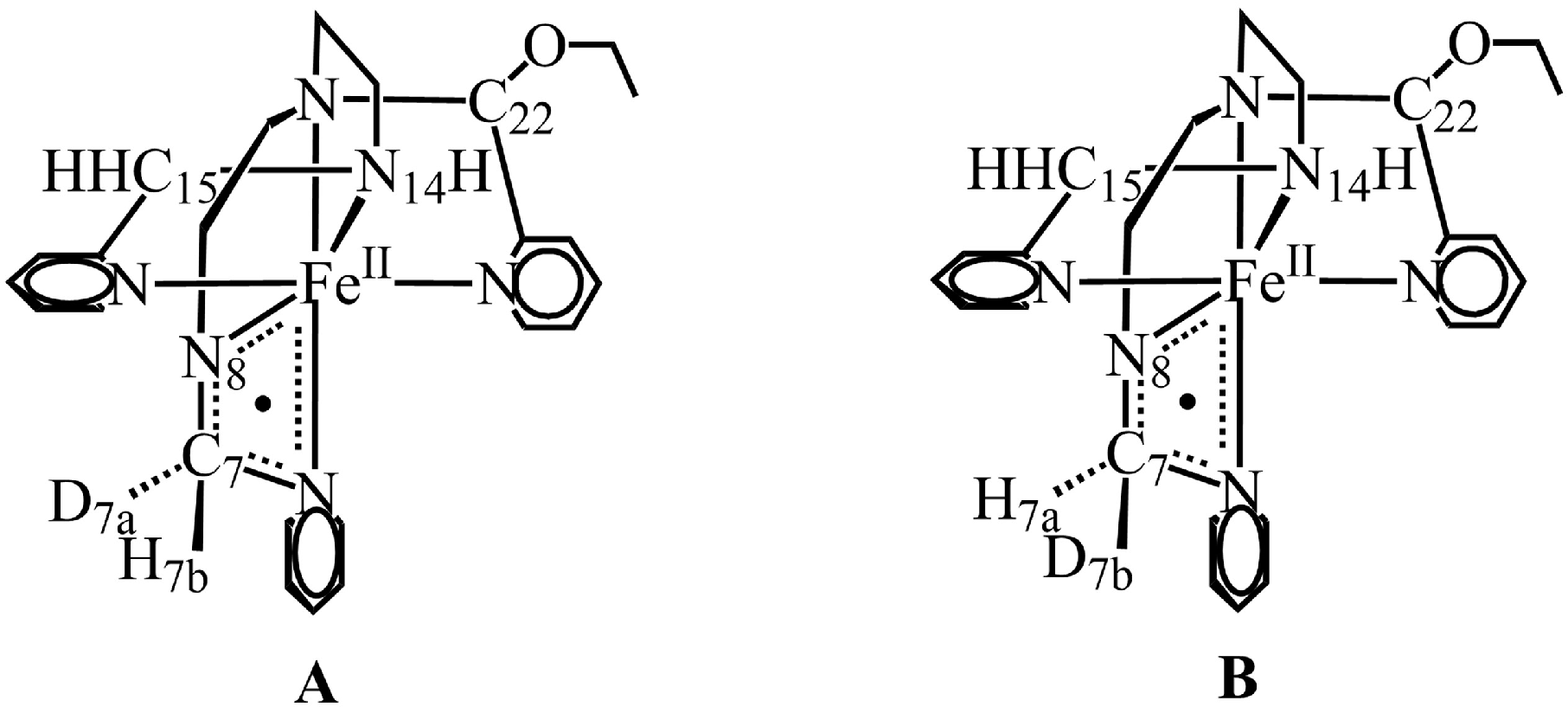
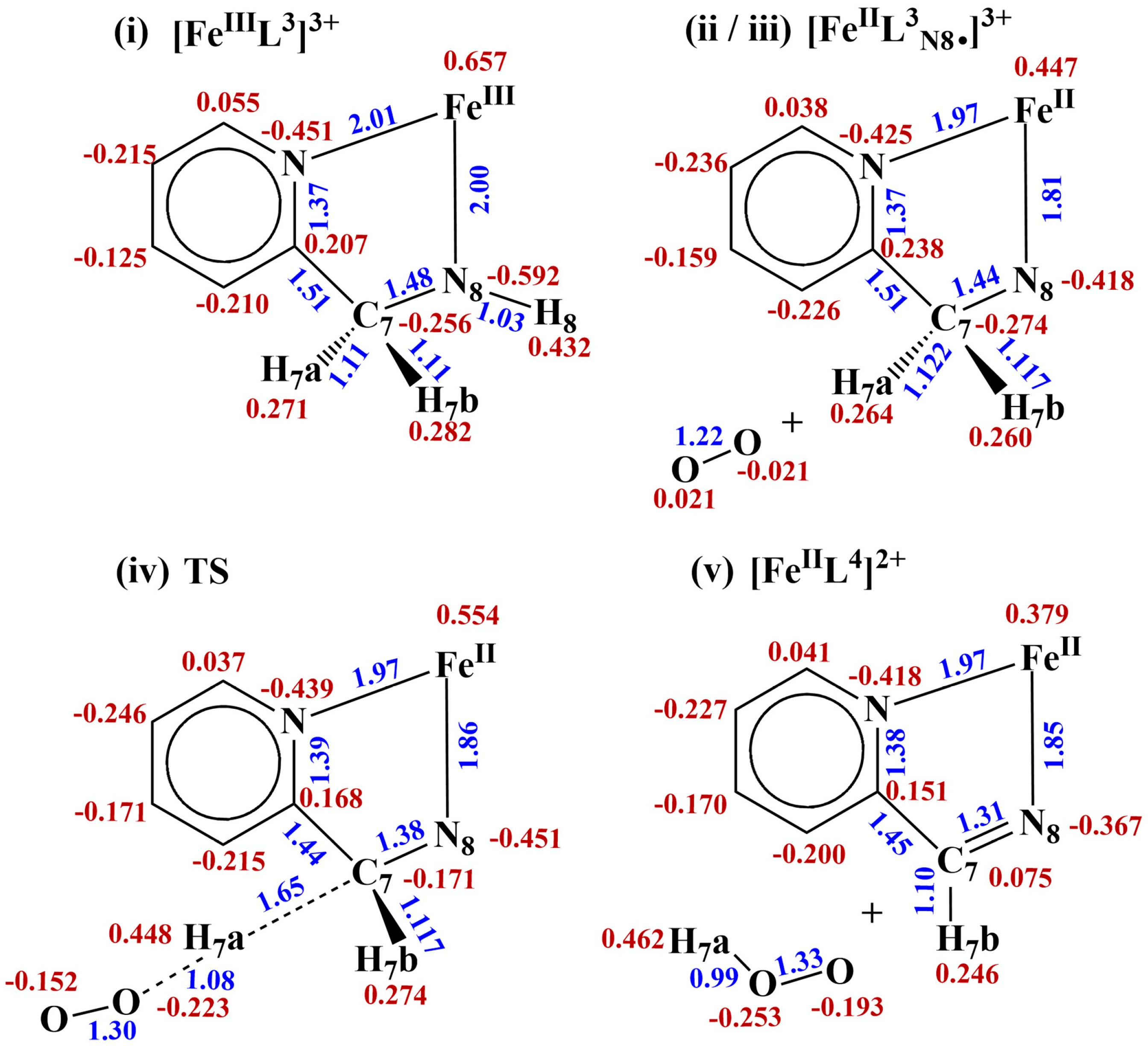
2.2. Nitrogen Radical [FeIIL3N8•]2+ and Site for O2 Attack
2.3. Hydrogen Atom Transfer [FeIIL3N8•]2+ + 3O2 → [FeIIL4]2+ + HO2•
2.3.1. Transition State {[FeIIL3N8• C7•]2+---H7a---3O2}
2.3.2. Spin Density, Charge Density, and Frontier Orbitals
3. Discussion
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| [FeIIL3N8•]2+ | [FeIIL3N14•]2+ | ||
|---|---|---|---|
| Atom | Spin Density | Atom | Spin Density |
| Fe | 0.628 | Fe | 0.638 |
| N8• | 0.359 | N14• | 0.379 |
| C7 | −0.021 | C15 | −0.019 |
| H7a | 0.034 | H15a | 0.021 |
| H7b | 0.015 | H15b | −0.001 |
| N14 | −0.011 | N8 | −0.008 |
| H14 | 0.001 | H8 | 0.000 |
| C15 | 0.002 | C7 | 0.004 |
| H15a | 0.000 | H7a | 0.000 |
| H15b | 0.000 | H7b | 0.003 |
References
- Crichton, R.R. Biological Inorganic Chemistry. A New Introduction to Molecular Structure and Function, 3rd ed.; Academic Press: Cambridge, MA, USA, 2019. [Google Scholar] [CrossRef]
- Solomon, E.I.; Goudarzi, S.; Sutherlin, K.D. O2 Activation by Non-Heme Iron Enzymes. Biochemistry 2016, 55, 6363–6374. [Google Scholar] [CrossRef]
- Maret, W. Zinc Biochemistry: From a Single Zinc Enzyme to a Key Element of Life. Adv. Nutr. 2013, 4, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Koschorreck, K.; Alpdagtas, S.; Urlacher, V.B. Copper-radical oxidases: A diverse group of biocatalysts with distinct properties and a broad range of biotechnological applications. Appl. Eng. Microbiol. 2022, 2, 100037. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Chen, X.-Y.; Stoddart, J.F. Weak bonding strategies for achieving regio- and site-selective transformations. Chem 2022, 8, 414–438. [Google Scholar] [CrossRef]
- Dalton, T.; Faber, T.; Glorius, F. C−H Activation: Toward Sustainability and Applications. ACS Cent. Sci. 2021, 7, 245–261. [Google Scholar] [CrossRef]
- Kumar Sinha, S.; Guin, S.; Maiti, S.; Prasad Biswas, J.; Porey, S.; Maiti, D. Toolbox for Distal C−H Bond Functionalizations in Organic Molecules. Chem. Rev. 2022, 122, 5682–5841. [Google Scholar] [CrossRef]
- Budweg, S.; Wei, Z.; Jiao, H.; Junge, K.; Beller, M. Iron-PNP Pincer-Catalyzed Transfer Dehydrogenation of Secondary Alcohols. ChemSusChem 2019, 12, 2988–2993. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Das, S.; Sikari, R.; Parua, S.; Brandaõ, P.; Demeshko, S.; Meyer, F.; Paul, N.D. Redox Noninnocent Azo-Aromatic Pincers and Their Iron Complexes. Isolation, Characterization, and Catalytic Alcohol Oxidation. Inorg. Chem. 2017, 56, 14084–14100. [Google Scholar] [CrossRef]
- Darcy, J.W.; Koronkiewicz, B.; Parada, G.A.; Mayer, J.M. A Continuum of Proton-Coupled Electron Transfer Reactivity. Acc. Chem. Res. 2018, 51, 2391–2399. [Google Scholar] [CrossRef] [PubMed]
- Ugalde-Saldívar, V.M.; Sosa-Torres, M.E.; Ortiz-Frade, L.; Bernès, S.; Höpfl, H. Novel iron(II) complexes with hexadentate nitrogen ligands obtained via intramolecular redox reactions. Dalton Trans. 2001, 20, 3099–3107. [Google Scholar] [CrossRef]
- Saucedo-Vázquez, J.P.; Ugalde-Saldívar, V.M.; Toscano, A.R.; Kroneck, P.M.H.; Sosa-Torres, M.E. On the Mechanism of Iron(III)-Dependent Oxidative Dehydrogenation of Amines. Inorg. Chem. 2009, 48, 1214–1222. [Google Scholar] [CrossRef] [PubMed]
- Saucedo-Vázquez, J.P.; Kroneck, P.M.H.; Sosa-Torres, M.E. The role of molecular oxygen in the iron(III)-promoted oxidative dehydrogenation of amines. Dalton Trans. 2015, 44, 5510–5519. [Google Scholar] [CrossRef]
- Keene, F.R. Metal-ion promotion of the oxidative dehydrogenation of coordinated amines and alcohols. Coord. Chem. Rev. 1999, 187, 121–149. [Google Scholar] [CrossRef]
- Páez-López, R.D.; Gómez-Soto, M.Á.; Cortés-Hernández, H.F.; Solano-Peralta, A.; Castro, M.; Kroneck, P.M.H.; Sosa-Torres, M.E. Fe(III)-promoted oxidative dehydrogenation of amines by O2—mediated cleavage of C-H bond proceeds via hydrogen atom transfer (HAT). Inorg. Chim. Acta 2025, 578, 122516. [Google Scholar] [CrossRef]
- Victória, H.F.; Ferreira, D.C.; José Filho, B.G.; Martins, D.C.; Pinheiro, M.V.; de AMSáfar, G.; Krambrock, K. Detection of singlet oxygen by EPR: The instability of the nitroxyl radicals. Free Rad. Biol. Med. 2022, 180, 143–152. [Google Scholar] [CrossRef]
- Peng, H.-L.; Chen, X.-Y.; Li, L.-P.; Ye, B.-H. Mechanism study on photo-oxidation dehydrogenation of cyclometalated Ir (III) amino acid complexes. Inorg. Chim. Acta 2020, 513, 119939. [Google Scholar] [CrossRef]
- Zheng, N.; He, X.; Hu, R.; Wang, R.; Zhou, Q.; Lian, Y.; Hu, Z. In-situ production of singlet oxygen by dioxygen activation on iron phosphide for advanced oxidation processes. Appl. Catal. B Environ. 2022, 307, 121157. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009.
- Gao, S.; Li, F. Neutral Stable Nitrogen-Centered Radicals: Structure, Properties, and Recent Functional Application Progress. Adv. Funct. Mater. 2023, 33, 2304291. [Google Scholar] [CrossRef]
- Weinhold, F. Natural bond orbital analysis: A critical overview of relationships to alternative bonding perspectives. J. Comp. Chem. 2012, 33, 2363–2379. [Google Scholar] [CrossRef]
- Glendening, E.D.; Hiatt, D.M.; Weinhold, F. Natural Bond Orbital Analysis of Chemical Structure, Spectroscopy, and Reactivity: How it Works. Compr. Comput. Chem. 2024, 2, 406–421. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Zamora, P.P.; Bieger, K.; Cuchillo, A.; Tello, A.; Muena, J.P. Theoretical determination of a reaction intermediate: Fukui function analysis, dual reactivity descriptor and activation energy. J. Mol. Struct. 2021, 1227, 129369. [Google Scholar] [CrossRef]
- Zaklika, J.; Hładyszowski, J.; Ordon, P.; Komorowski, L. From the Electron Density Gradient to the Quantitative Reactivity Indicators: Local Softness and the Fukui Function. ACS Omega 2022, 7, 7745–7758. [Google Scholar] [CrossRef]
- Pucci, R.; Angilella, G.G.N. Density functional theory, chemical reactivity, and the Fukui functions. Found. Chem. 2022, 24, 59–71. [Google Scholar] [CrossRef]
- Nam, W.; Lee, Y.-M.; Fukuzumi, S. Tuning Reactivity and Mechanism in Oxidation Reactions by Mononuclear Nonheme Iron(IV)-Oxo Complexes. Acc. Chem. Res. 2014, 47, 1146–1154. [Google Scholar] [CrossRef]
- Kal, S.; Que, L. Dioxygen activation by nonheme iron enzymes with the 2-His-1-carboxylate facial triad that generate high-valent oxoiron oxidants. J. Biol. Inorg. Chem. 2017, 22, 339–365. [Google Scholar] [CrossRef]
- De Visser, S.P.; Mukherjee, G.; Ali, H.S.; Sastri, C.V. Local Charge Distributions, Electric Dipole Moments, and Local Electric Fields Influence Reactivity Patterns and Guide Regioselectivities in α-Ketoglutarate-Dependent Non-heme Iron Dioxygenases. Acc. Chem. Res. 2022, 55, 65–74. [Google Scholar] [CrossRef]
- Hou, K.; Börgel, J.; Jiang, H.Z.H.; SantaLucia, D.J.; Kwon, H.; Zhuang, H.; Chakarawet, K.; Rohde, R.C.; Taylor, J.W.; Dun, C.; et al. Reactive high-spin iron (IV)-oxo sites through dioxygen activation in a metal–organic framework. Science 2023, 382, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.S.; Henchman, R.H.; de Visser, S.P. Mechanism of Oxidative Ring-Closure as Part of the Hygromycin BiosynthesisStep by a Nonheme Iron Dioxygenase. ChemCatChem 2021, 13, 3054–3066. [Google Scholar] [CrossRef]
- Goto, M.; Takeshita, M.; Kanda, N.; Sakai, T.; Goedken, V.L. Stoichiometry and Kinetics of Base-Promoted Disproportionation with Concomitant Ligand Oxidation of Tetracyano(l,2-diamine)ferrate(III). Inorg. Chem. 1985, 24, 582–587. [Google Scholar] [CrossRef]
- Barefield, E.K.; Mocella, M.T. Mechanism of Base Promoted Reduction of Nickel(III) Complexes of Macrocyclic Amines. A Coordinated Ligand Radical Intermediate. J. Am. Chem. Soc. 1975, 97, 4238–4246. [Google Scholar] [CrossRef]
- Da Costa Ferreira, A.M.; Toma, H.E. Electron-transfer Kinetics and Mechanism of Di-imine Bond Formation in Tetracyano (et hylenediamine) ferrate (II). J. Chem. Soc. Dalton Trans. 1983, 9, 2051–2055. [Google Scholar] [CrossRef]
- Raleigh, C.J.; Martell, A.E. Oxidative Dehydrogenation of Coordinated l,9-Bis(2-pyridyl)-2,5,8-triazanonane through Formation of a Cobalt Dioxygen Complex intermediate. Inorg. Chem. 1985, 24, 142–148. [Google Scholar] [CrossRef]
- Morgenstern-Badarau, I.; Lambert, F.; Philippe Renault, J.; Cesario, M.; Maréchal, J.-D.; Maseras, F. Amine conformational change and spin conversion induced by metal-assisted ligand oxidation: From the seven-coordinate iron(II)–TPAA complex to the two oxidized iron(II)–(py)3tren isomers. Characterization, crystal structures, and density functional study. Inorg. Chim. Acta 2000, 297, 338–350. [Google Scholar] [CrossRef]
- Christian, G.J.; Arbuse, A.; Fontrodon, X.; Angeles Martinez, M.; Llobet, A.; Maseras, F. Oxidative dehydrogenation of an amine group of a macrocyclic ligand in the coordination sphere of a CuII complex. Dalton Trans. 2009, 30, 6013–6020. [Google Scholar] [CrossRef] [PubMed]
- Stubbe, J.A.; van der Donk, W.A. Protein Radicals in Enzyme Catalysis. Chem. Rev. 1998, 98, 705–762. [Google Scholar] [CrossRef] [PubMed]
- Mandal, A. Radical Chemistry: A Brief History and Overview. Asian J. Chem. 2023, 35, 1539–1562. [Google Scholar] [CrossRef]
- Zard, S.Z. Recent progress in the generation and use of nitrogen-centred radicals. Chem. Soc. Rev. 2008, 37, 1603–1618. [Google Scholar] [CrossRef]
- Pratley, C.; Fenner, S.; Murphy, J.A. Nitrogen-Centered Radicals in Functionalization of sp2 Systems: Generation, Reactivity, and Applications in Synthesis. Chem. Rev. 2022, 122, 8181–8260. [Google Scholar] [CrossRef] [PubMed]
- Büttner, T.; Geier, J.; Frison, G.; Harmer, J.; Calle, C.; Schweiger, A.; Schönberg, H.; Grützmacher, H. A Stable Aminyl Radical Metal Complex. Science 2005, 307, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Olivos Suarez, A.I.; Lyaskovskyy, V.; Reek, J.N.H.; van der Vlugt, J.I.; de Bruin, B. Complexes with Nitrogen-Centered Radical Ligands: Classification, Spectroscopic Features, Reactivity, and Catalytic Applications. Angew. Chem. Int. Ed. 2013, 52, 12510–12529. [Google Scholar] [CrossRef] [PubMed]
- Kaim, W. Manifestations of Noninnocent Ligand Behavior. Inorg. Chem. 2011, 50, 9752–9765. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Raghavachari, K.; Trucks, G.W. Highly correlated systems. Excitation energies of first row transition metals Sc–Cu. J. Chem. Phys. 1989, 91, 1062–1065. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Frisch, M.J.; People, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Liskow, D.H.; Schaefer III, H.F.; Bender, C.F. Geometry and Electronic Structure of the Hydroperoxyl Radical. J. Am. Chem. Soc. 1971, 93, 6734–6737. [Google Scholar] [CrossRef]
- Castro Martínez, F.M.; Páez López, R.D.; Sarmiento Pavía, P.D.; Sosa Torres, M.E.; Kroneck, P.M.H. Cytochrome P450. The Dioxygen-Activating Heme Thiolate. Met. Ions Life Sci. 2020, 20, 165–197. [Google Scholar] [CrossRef]
- Arnold, F.H. Directed Evolution: Bringing New Chemistry to Life. Angew. Chem. Int. Ed. 2018, 57, 4143–4148. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. A perspective on the future of quantum chemical software: The example of the ORCA program package. Faraday Discuss. 2024, 254, 295–314. [Google Scholar] [CrossRef] [PubMed]



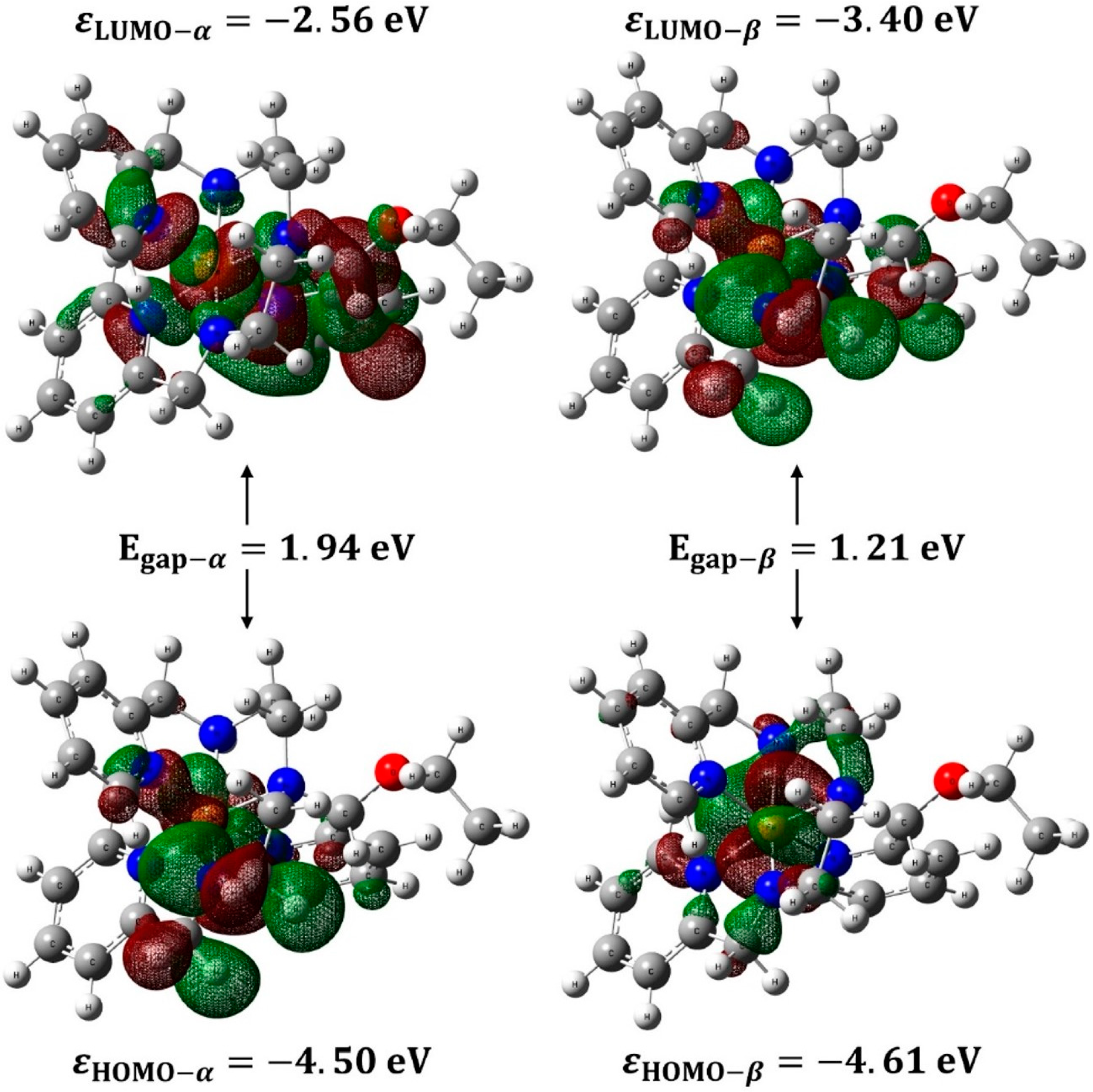
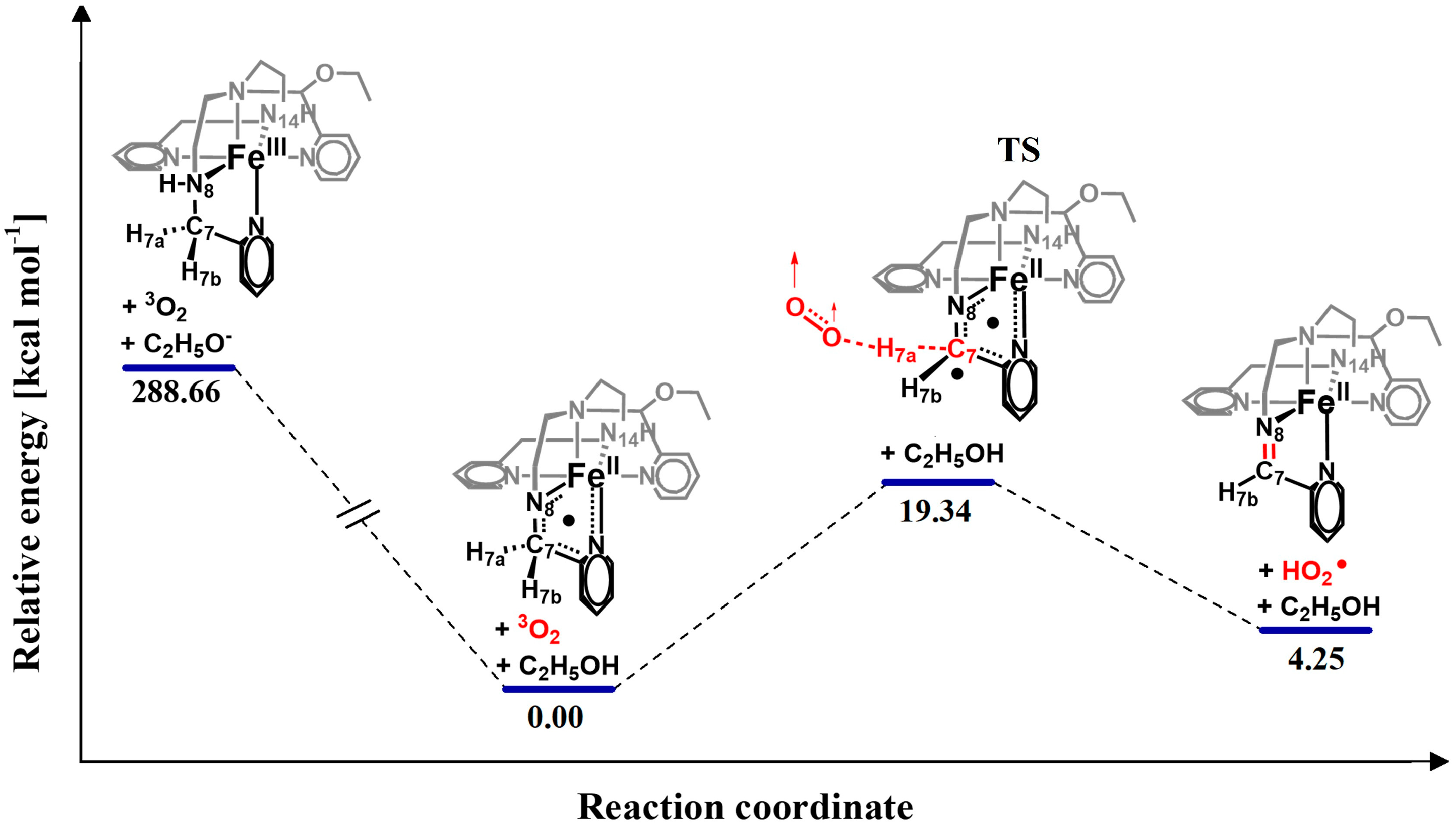
| Species | M | EE + ZPE (kcal mol−1) | EE + TEC (kcal mol−1) |
|---|---|---|---|
| [FeIIIL3]3+, (1) | 2 | −1,629,611.901 | −1,629,593.709 |
| 4 | −1,629,599.929 | −1,629,581.205 | |
| 6 | −1,629,597.183 | −1,629,577.986 | |
| C2H5O− | 1 | −96,657.889 | −96,654.857 |
| [FeIIL3N8•]2+ (2a) | 2 | −1,629,517.246 | −1,629,499.171 |
| 4 | −1,629,500.632 | −1,629,481.836 | |
| 6 | −1,629,495.890 | −1,629,476.821 | |
| C2H5OH | 1 | −97,041.209 | −97,037.904 |
| O2 | 3 | −94,164.405 | −94,162.330 |
| {[FeIIL3N8• C7•]2+---H7a---3O2} (TS) | 4 | −1,723,662.304 | −1,723,642.314 |
| [FeIIL4]2+ (2) | 1 | −1,629,159.347 | −1,629,141.605 |
| 3 | −1,629,138.195 | −1,629,119.575 | |
| 5 | −1,629,138.422 | −1,629,119.446 | |
| HO2• | 2 | −94,518.050 | −94,515.661 |
| Isomer | Bond | Length (Å) | Frequency (cm−1) |
|---|---|---|---|
| [FeIIL3N8•]2+ | N8-C7 | 1.43(8) | |
| C7-H7a | 1.12(2) | 2937.73 (H7a-C7-H7b νas) | |
| C7-H7b | 1.11(7) | 2873.72 (H7a-C7-H7b νs) | |
| N14-C15 | 1.48(2) | ||
| C15-H15a | 1.11(1) | 3023.81 (H15a-C15-H15b νas) | |
| C15-H15b | 1.11(2) | 2976.22 (H15a-C15-H15b νs) | |
| [FeIIL3N14•]2+ | N8-C7 | 1.47(9) | |
| C7-H7a | 1.11(1) | 3031.81 (H7a-C7-H7b νas) | |
| C7-H7b | 1.11(1) | 2978.06 (H7a-C7-H7b νs) | |
| N14-C15 | 1.44(7) | ||
| C15-H15a | 1.11(7) | 2989.45 (H15a-C15-H15b νas) | |
| C15-H15b | 1.11(2) | 2924.56 (H15a-C15-H15b νs) |
| Atom | Fukui Indices | ||
|---|---|---|---|
| [FeIIL3N8•]2+ | f + | f − | Δf |
| N8• | −0.156 | −0.181 | 0.025 |
| C7 | 0.017 | 0.011 | 0.006 |
| H7b | −0.030 | −0.042 | 0.012 |
| H7a | −0.038 | −0.061 | 0.023 |
| C6 | 0.010 | 0.015 | −0.005 |
| [FeIIL3N14•]2+ | f + | f − | Δf |
| N14• | −0.200 | −0.099 | −0.101 |
| C15 | 0.016 | 0.011 | 0.005 |
| H15a | −0.024 | −0.025 | 0.001 |
| H15b | −0.038 | −0.026 | −0.012 |
| C16 | 0.006 | 0.001 | 0.005 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Páez-López, R.D.; Gómez-Soto, M.Á.; Cortés-Hernández, H.F.; Solano-Peralta, A.; Castro, M.; Kroneck, P.M.H.; Sosa-Torres, M.E. Understanding Dioxygen Activation in the Fe(III)-Promoted Oxidative Dehydrogenation of Amines: A Computational Study. Inorganics 2025, 13, 22. https://doi.org/10.3390/inorganics13010022
Páez-López RD, Gómez-Soto MÁ, Cortés-Hernández HF, Solano-Peralta A, Castro M, Kroneck PMH, Sosa-Torres ME. Understanding Dioxygen Activation in the Fe(III)-Promoted Oxidative Dehydrogenation of Amines: A Computational Study. Inorganics. 2025; 13(1):22. https://doi.org/10.3390/inorganics13010022
Chicago/Turabian StylePáez-López, Ricardo D., Miguel Á. Gómez-Soto, Héctor F. Cortés-Hernández, Alejandro Solano-Peralta, Miguel Castro, Peter M. H. Kroneck, and Martha E. Sosa-Torres. 2025. "Understanding Dioxygen Activation in the Fe(III)-Promoted Oxidative Dehydrogenation of Amines: A Computational Study" Inorganics 13, no. 1: 22. https://doi.org/10.3390/inorganics13010022
APA StylePáez-López, R. D., Gómez-Soto, M. Á., Cortés-Hernández, H. F., Solano-Peralta, A., Castro, M., Kroneck, P. M. H., & Sosa-Torres, M. E. (2025). Understanding Dioxygen Activation in the Fe(III)-Promoted Oxidative Dehydrogenation of Amines: A Computational Study. Inorganics, 13(1), 22. https://doi.org/10.3390/inorganics13010022