

Complexation of Boron and Aluminum with a Bidentate Hydroxy-BN-naphthalene Ligand

 , , and
, , and 
Abstract
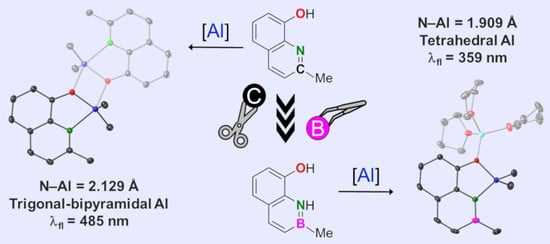
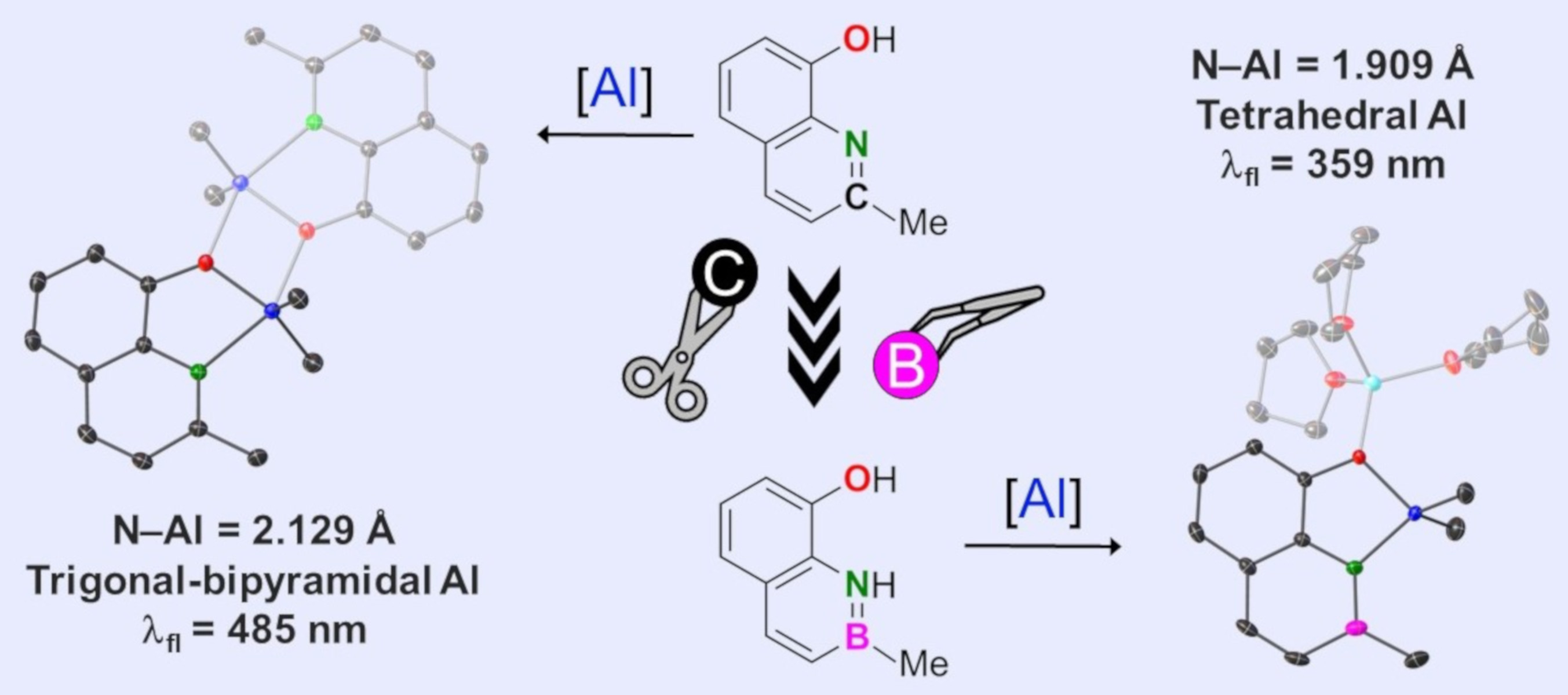
1. Introduction

2. Results and Discussion
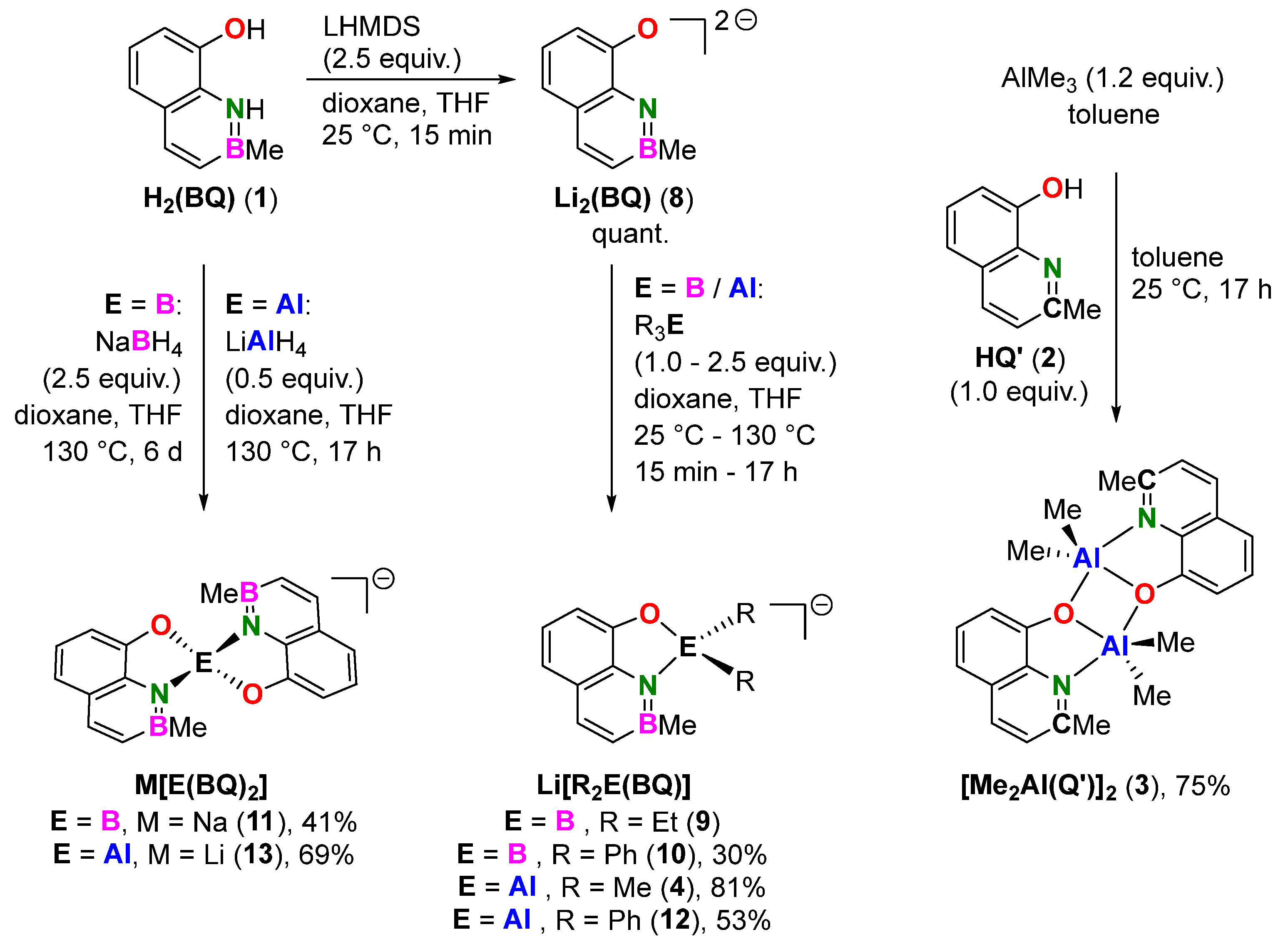
2.1. Syntheses
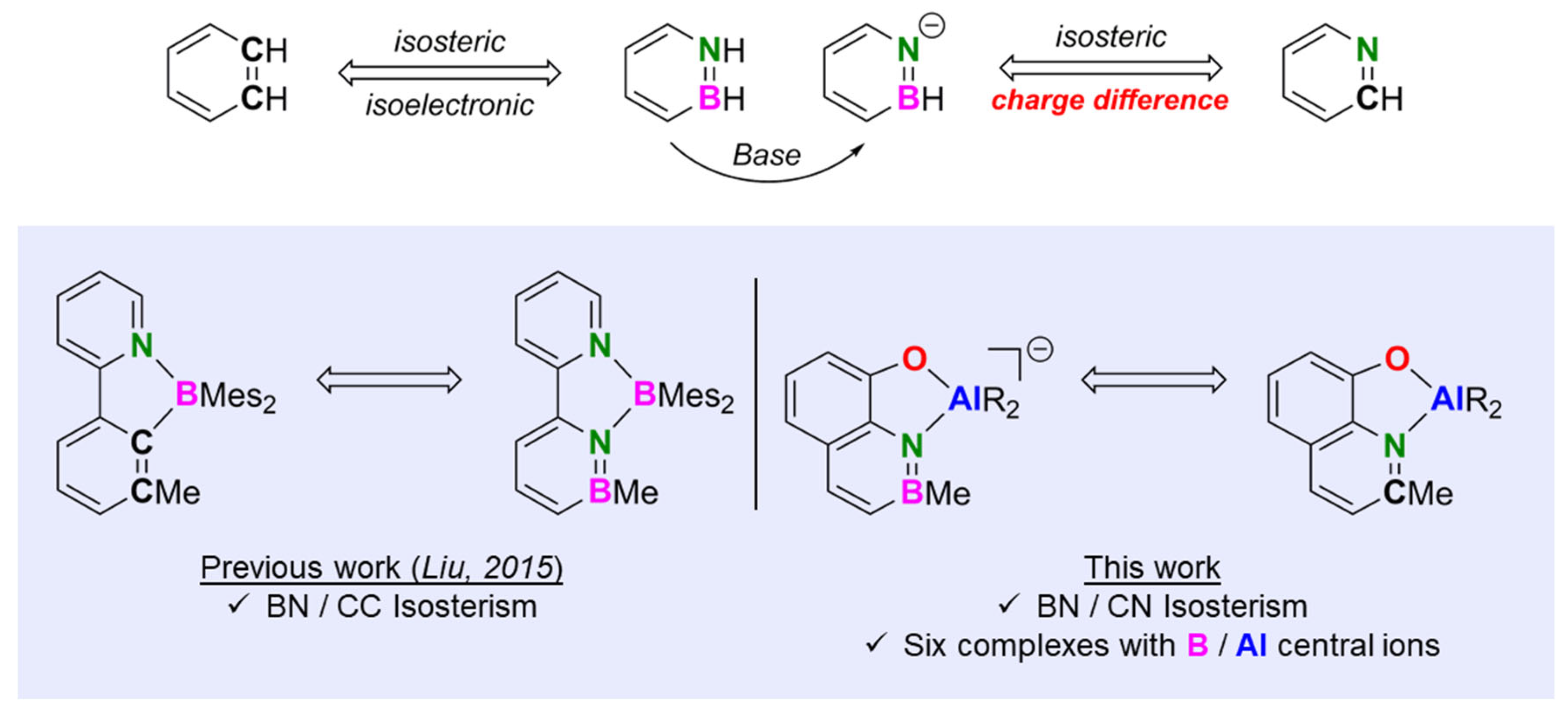
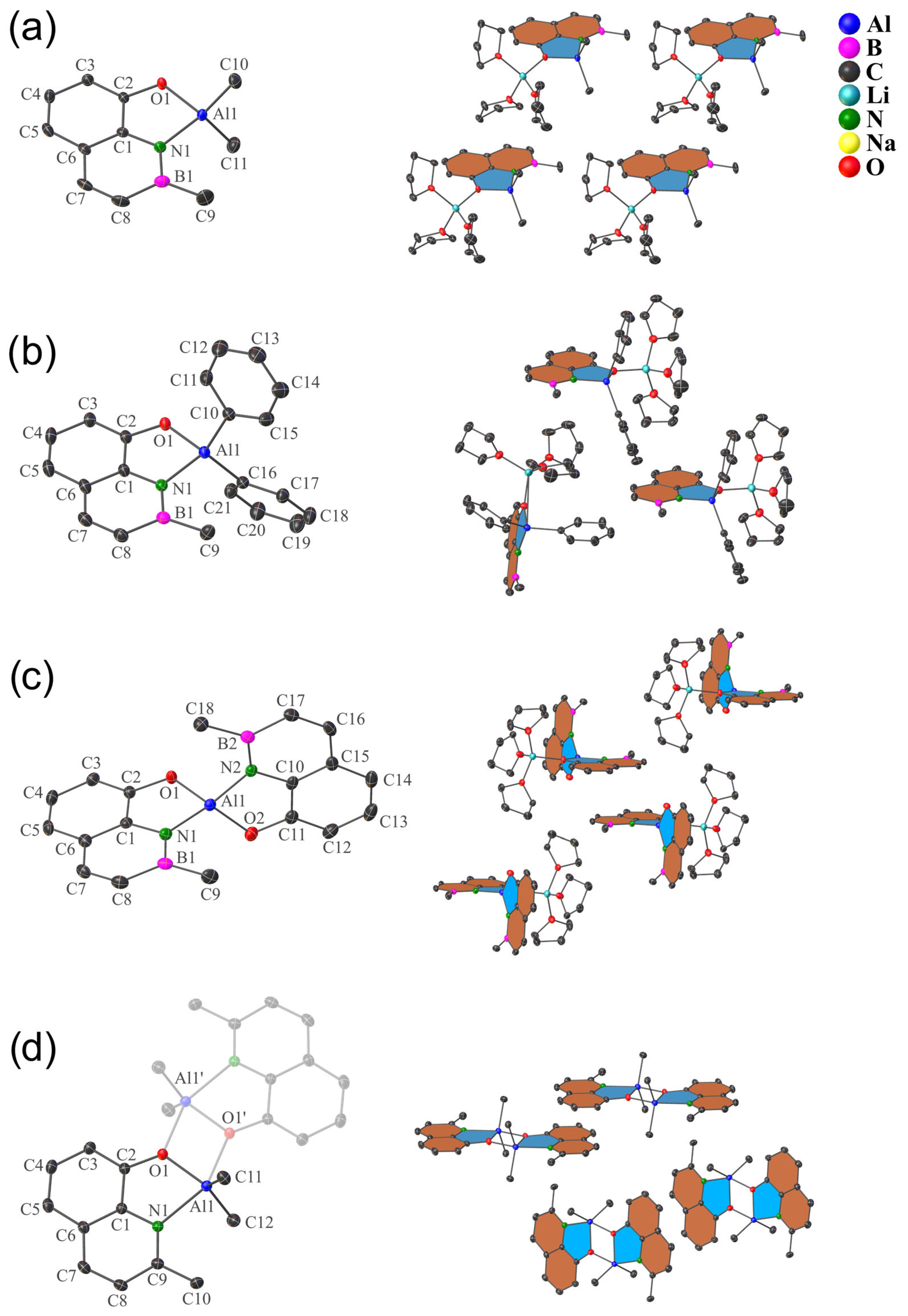
2.2. X-ray Diffraction Analysis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
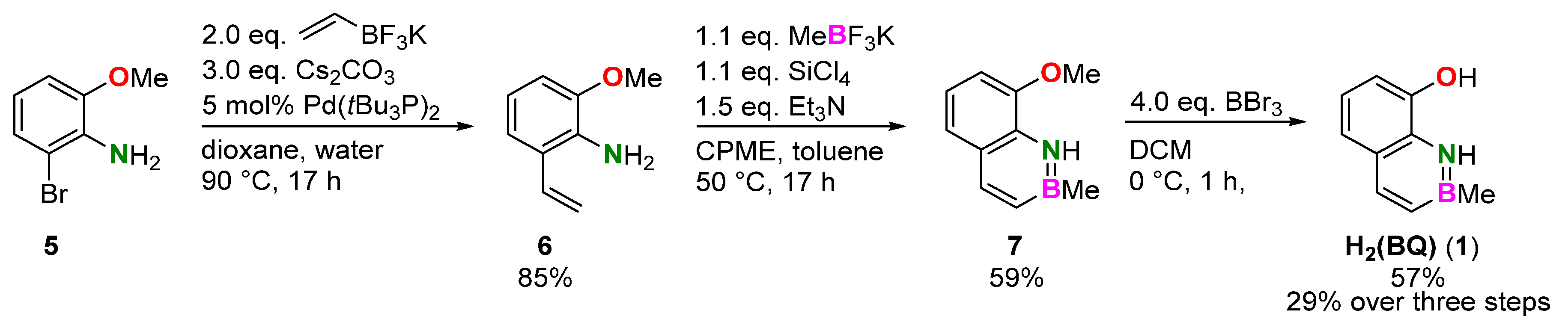{kind=link}
{kind=link}
| Entry | Complex Structure | B–O [Å] | B–N [Å] | B–C [Å] | O–M [Å] [a] | V [Å3] [b] |
|---|---|---|---|---|---|---|
| 1 | ([Li(THF)2(diox)0.5] [Et2B(BQ)])2 (9) [c] | 1.590 (4) | 1.587 (4) | 1.610 (5), 1.616 (4) | 1.885 (5) | 2.077 |
| 2 | [Li(THF)3] [Ph2B(BQ)] (10) [d] | 1.573 (2) | 1.574 (2) | 1.619 (2), 1.621 (2) | 1.935 (3) | 2.059 |
| 3 | ([Na(THF)2] [B(BQ)2])2 (11) | 1.499 (2), 1.518 (2) | 1.593 (2), 1.546 (2) | - | 2.3558 (12) | 1.797 |

| Entry | Complex Structure | Al–O [Å] | Al–N [Å] | Al–C [Å] | O–M [Å] [a] | V [Å3] [b] |
|---|---|---|---|---|---|---|
| 1 | [Li(THF)3] [Me2Al(BQ)] (4) | 1.8368 (14) | 1.9091 (17) | 1.969 (2), 1.974 (2) | 1.892 (3) | 3.471 |
| 2 | [Li(THF)3] [Ph2Al(BQ)] (12) | 1.832 (2) | 1.890 (3) | 1.976 (3), 1.977 (3) | 1.916 (6) | 3.452 |
| 3 | [Li(THF)3] [Al(BQ)2] (13) | 1.7741 (14), 1.7879 (15) | 1.8565 (17), 1.8570 (17) | - | 1.923 (4) | 2.876 |
| 4 | [Me2Al(Q’)]2 (3) | 1.8732 (9) | 2.1293 (11) | 1.9756 (12), 1.9787 (11) | 2.0082 (8) | - |
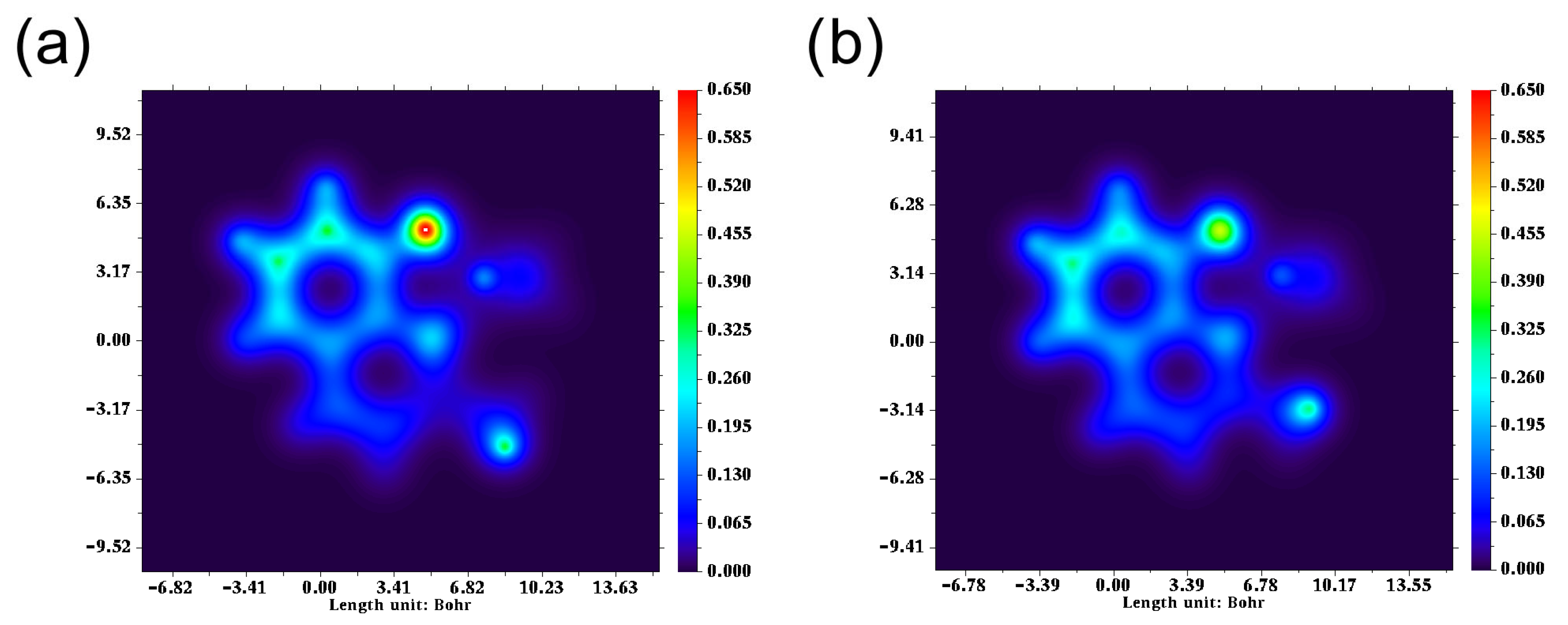
2.3. Theoretical Analysis

| Li[Me2Al(BQ)] (4) | [Me2Al(Q’)]2 (3) | |
|---|---|---|
| Bond Lengths [a]/Wiberg Bond Indices (WBI) | ||
| Al1–O1 | Calcd. 1.900 Å | Calcd. 1.913 Å |
| X-ray: 1.837 Å | X-ray: 1.873 Å | |
| (Calcd. 14: 1.846 Å) | (Calcd. 15: 1.838 Å) | |
| WBI: 0.33 | WBI: 0.25 | |
| Al1–N1 | Calcd. 1.901 Å | Calcd. 2.220 Å |
| X-ray: 1.909 Å | X-ray: 2.129 Å | |
| (14: 1.953 Å) | (15: 2.058 Å) | |
| WBI: 0.38 | WBI: 0.19 | |
| Al1–O1′ | - | Calcd. 2.055 Å |
| X-ray: 2.010 Å | ||
| WBI: 0.18 | ||
| Natural Atomic Charges/Electron Configurations [a][b] | ||
| Al1 | +1.86/3s0.513p0.60 | +1.91/3s0.503p0.57 |
| O1 | –1.00/2s1.682p5.32 | –0.92/2s1.632p5.28 |
| N1 | –1.09/2s1.382p4.70 | –0.58/2s1.312p4.25 |
| B1/C9 | +0.89 (B1) | +0.30 (C9) |
| C8 | –0.56 | –0.27 |
| C9/C10 | –1.01 (C9) | –0.68 (C10) |
| Sum of Donor–Acceptor Orbital Interactions (E(2))[a][b][c] | ||
| LP O→LV Al | 143.6 kcal mol−1 | 201.6 kcal mol−1 |
| LP N→LV Al | 127.7 kcal mol−1 | 73.7 kcal mol−1 |
| LP C→LV Al | 459.8 kcal mol−1 | 442.3 kcal mol−1 |
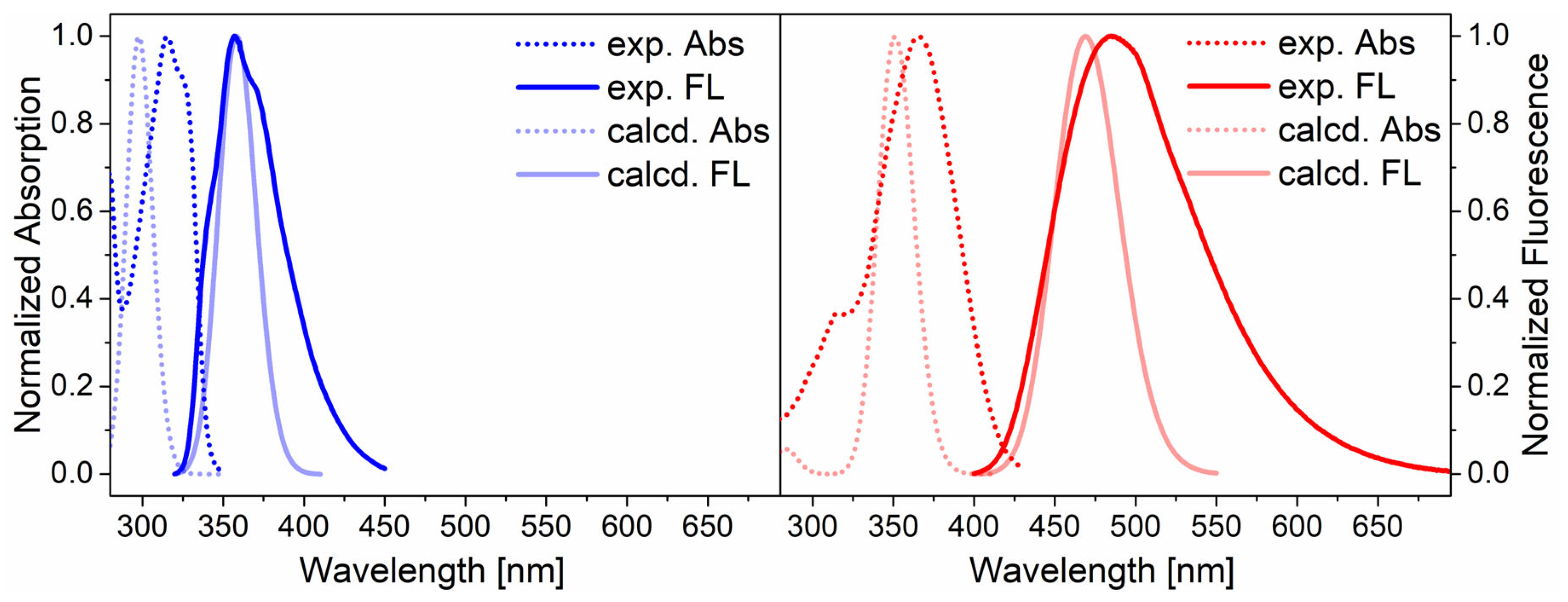
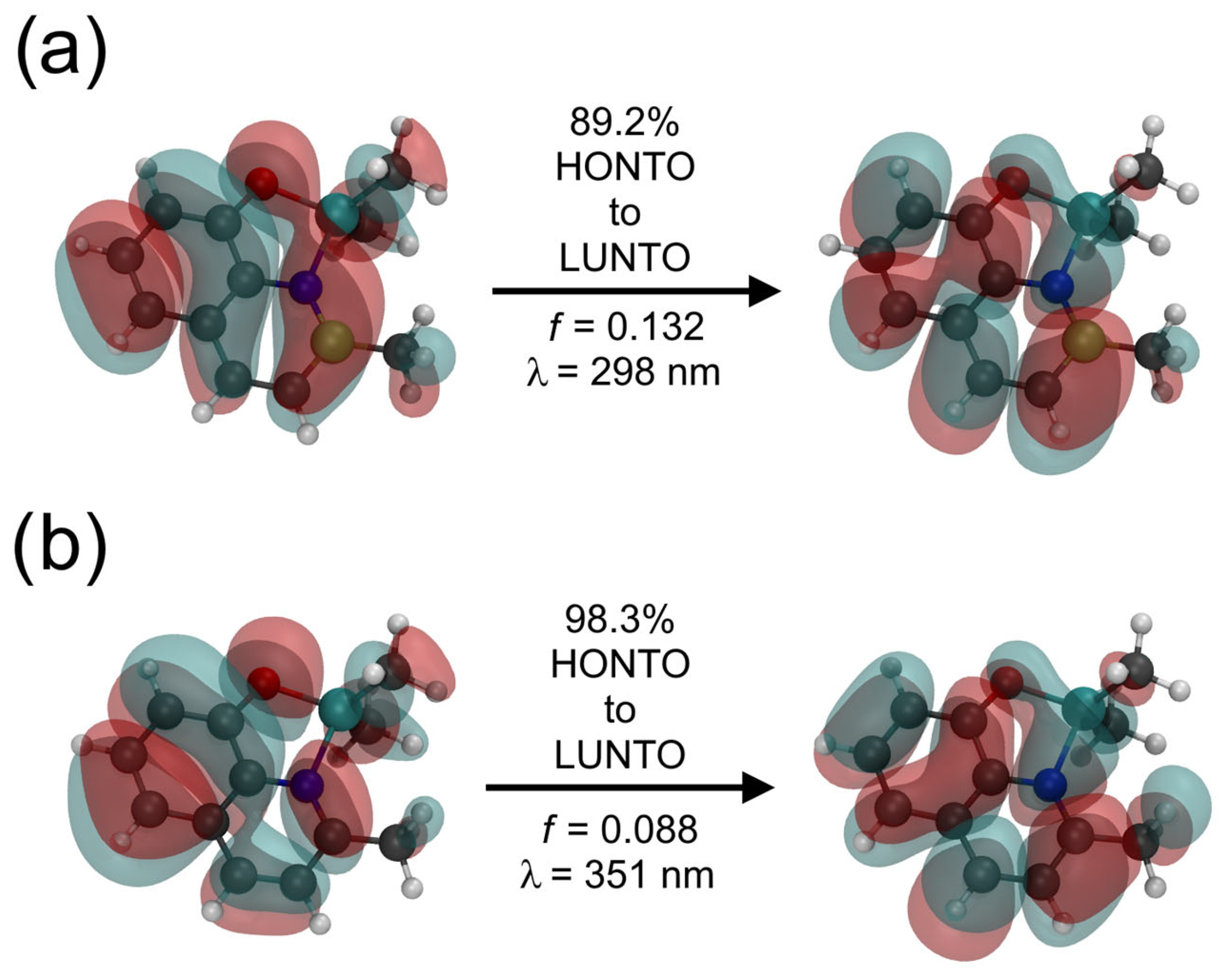
2.4. Optical Properties
3. Materials and Methods
3.1. General Information
3.2. Synthesis of 2-Methoxy-6-vinylaniline (6)

3.3. Synthesis of 8-Methoxy-2-methyl-1,2-dihydro-1-aza-2-boranaphthalene (7)

3.4. Synthesis of 8-Hydroxy-2-methyl-1,2-dihydro-1-aza-2-boranaphthalene (1)

3.5. General Procedure for the Complexation of Group 13 Elements
3.6. 1:1. Complex of H2(BQ) with Triethylborane (Li[Et2B(BQ)])

3.7. 1:1. Complex of H2(BQ) with Triphenylborane (Li[Ph2B(BQ)])

3.8. 2:1. Complex of H2(BQ) with Sodium Borohydride (Na[B(BQ)2])

3.9. 1:1. Complex of H2(BQ) with Trimethylaluminum (Li[Me2Al(BQ)])

3.10. 1:1. Complex of H2(BQ) with Triphenylaluminum (Li[Ph2Al(BQ)])

3.11. 2:1. Complex of H2(BQ) with Lithium Aluminum Hydride (Li[Al(BQ)2])

3.12. 1:1. Complex of HQ’ with Trimethylaluminum ([Me2Al(Q’)]2)

4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Campbell, P.G.; Marwitz, A.J.V.; Liu, S.-Y. Recent Advances in Azaborine Chemistry. Angew. Chem. Int. Ed. 2012, 51, 6074–6092. [Google Scholar] [CrossRef]
- Bélanger-Chabot, G.; Braunschweig, H.; Roy, D.K. Recent Developments in Azaborinine Chemistry. Eur. J. Inorg. Chem. 2017, 38–39, 4353–4368. [Google Scholar] [CrossRef]
- Campbell, P.G.; Abbey, E.R.; Neiner, D.; Grant, D.J.; Dixon, D.A.; Liu, S.-Y. Resonance Stabilization Energy of 1,2-Azaborines: A Quantitative Experimental Study by Reaction Calorimetry. J. Am. Chem. Soc. 2010, 132, 18048–18050. [Google Scholar] [CrossRef] [PubMed]
- Abbey, E.R.; Zakharov, L.N.; Liu, S.-Y. Crystal Clear Structural Evidence for Electron Delocalization in 1,2-Dihydro-1,2-azaborines. J. Am. Chem. Soc. 2008, 130, 7250–7252. [Google Scholar] [CrossRef]
- Appiarius, Y.; Staubitz, A. Azaborinine. Chem. Unserer Zeit 2023, 57, 180–190. [Google Scholar] [CrossRef]
- McConnell, C.R.; Liu, S.-Y. Late-stage functionalization of BN-heterocycles. Chem. Soc. Rev. 2019, 48, 3436–3453. [Google Scholar] [CrossRef] [PubMed]
- Appiarius, Y.; Stauch, T.; Lork, E.; Rusch, P.; Bigall, N.C.; Staubitz, A. From a 1,2-azaborinine to large BN-PAHs via electrophilic cyclization: Synthesis, characterization and promising optical properties. Org. Chem. Front. 2021, 8, 10–17. [Google Scholar] [CrossRef]
- Appiarius, Y.; Gliese, P.J.; Segler, S.A.W.; Rusch, P.; Zhang, J.; Gates, P.J.; Pal, R.; Malaspina, L.A.; Sugimoto, K.; Neudecker, T.; et al. BN-Substitution in Dithienylpyrenes Prevents Excimer Formation in Solution and in the Solid State. J. Phys. Chem. C 2022, 126, 4563–4576. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Dietz, R. New heteroaromatic compounds. Part III. 2,1-Borazaro-naphthalene (1,2-dihydro-1-aza-2-boranaphthalene). J. Chem. Soc. 1959, 546, 2728–2730. [Google Scholar] [CrossRef]
- Paetzold, P.; Stanescu, C.; Stubenrauch, J.R.; Bienmüller, M.; Englert, U. 1-Azonia-2-boratanaphthalenes. Z. Anorg. Allg. Chem. 2004, 630, 2632–2640. [Google Scholar] [CrossRef]
- Wisniewski, S.R.; Guenther, C.L.; Argintaru, O.A.; Molander, G.A. A Convergent, Modular Approach to Functionalized 2,1-Borazaronaphthalenes from 2-Aminostyrenes and Potassium Organotrifluoroborates. J. Org. Chem. 2014, 79, 365–378. [Google Scholar] [CrossRef]
- Liu, Z.; Ishibashi, J.S.A.; Darrigan, C.; Dargelos, A.; Chrostowska, A.; Li, B.; Vasiliu, M.; Dixon, D.A.; Liu, S.-Y. The Least Stable Isomer of BN Naphthalene: Toward Predictive Trends for the Optoelectronic Properties of BN Acenes. J. Am. Chem. Soc. 2017, 139, 6082–6085. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Kampf, J.W.; Ashe, A.J. 1,2-Azaboratabenzene: A Heterocyclic π-Ligand with an Adjustable Basicity at Nitrogen. Organometallics 2004, 23, 5626–5629. [Google Scholar] [CrossRef]
- Pan, J.; Kampf, J.W.; Ashe Iii, A.J. The Preparation and Crystal Structures of η1-Derivatives of 2-Phenyl-1,2-azaboratabenzene. Organometallics 2008, 27, 1345–1347. [Google Scholar] [CrossRef]
- Lindl, F.; Lamprecht, A.; Arrowsmith, M.; Khitro, E.; Rempel, A.; Dietz, M.; Wellnitz, T.; Bélanger-Chabot, G.; Stoy, A.; Paprocki, V.; et al. Aromatic 1,2-Azaborinin-1-yls as Electron-Withdrawing Anionic Nitrogen Ligands for Main Group Elements. Chem. Eur. J. 2023, 29, e202203345. [Google Scholar] [CrossRef]
- Baschieri, A.; Aleotti, F.; Matteucci, E.; Sambri, L.; Mancinelli, M.; Mazzanti, A.; Leoni, E.; Armaroli, N.; Monti, F. A Pyridyl-1,2-azaborine Ligand for Phosphorescent Neutral Iridium(III) Complexes. Inorg. Chem. 2023, 62, 2456–2469. [Google Scholar] [CrossRef]
- Baggett, A.W.; Vasiliu, M.; Li, B.; Dixon, D.A.; Liu, S.-Y. Late-Stage Functionalization of 1,2-Dihydro-1,2-azaborines via Regioselective Iridium-Catalyzed C–H Borylation: The Development of a New N,N-Bidentate Ligand Scaffold. J. Am. Chem. Soc. 2015, 137, 5536–5541. [Google Scholar] [CrossRef] [PubMed]
- Linnell, R. Notes- Dissociation Constants of 2-Substituted Pyridines. J. Org. Chem. 1960, 25, 290. [Google Scholar] [CrossRef]
- Davies, G.H.M.; Jouffroy, M.; Sherafat, F.; Saeednia, B.; Howshall, C.; Molander, G.A. Regioselective Diversification of 2,1-Borazaronaphthalenes: Unlocking Isosteric Space via C–H Activation. J. Org. Chem. 2017, 82, 8072–8084. [Google Scholar] [CrossRef]
- Kwak, S.W.; Hong, J.H.; Lee, S.H.; Kim, M.; Chung, Y.; Lee, K.M.; Kim, Y.; Park, M.H. Synthesis and Photophysical Properties of a Series of Dimeric Indium Quinolinates. Molecules 2021, 26, 34. [Google Scholar] [CrossRef]
- Qi, Y.; Kang, R.; Huang, J.; Zhang, W.; He, G.; Yin, S.; Fang, Y. Reunderstanding the Fluorescent Behavior of Four-Coordinate Monoboron Complexes Containing Monoanionic Bidentate Ligands. J. Phys. Chem. B 2017, 121, 6189–6199. [Google Scholar] [CrossRef]
- Bakewell, C.; White, A.J.P.; Long, N.J.; Williams, C.K. 8-Quinolinolato Gallium Complexes: Iso-selective Initiators for rac-Lactide Polymerization. Inorg. Chem. 2013, 52, 12561–12567. [Google Scholar] [CrossRef] [PubMed]
- Hellstrom, S.L.; Ugolotti, J.; Britovsek, G.J.P.; Jones, T.S.; White, A.J.P. The effect of fluorination on the luminescent behaviour of 8-hydroxyquinoline boron compounds. New J. Chem. 2008, 32, 1379–1387. [Google Scholar] [CrossRef]
- Qin, Y.; Pagba, C.; Piotrowiak, P.; Jäkle, F. Luminescent Organoboron Quinolate Polymers. J. Am. Chem. Soc. 2004, 126, 7015–7018. [Google Scholar] [CrossRef] [PubMed]
- Wrackmeyer, B. Organoboranes and tetraorganoborates studied by 11B and 13C NMR spectroscopy and DFT calculations. Z. Naturforsch. B 2015, 70, 421–424. [Google Scholar] [CrossRef]
- Zhang, N.; Wang, C.; Zou, G.; Tang, J. Palladium-catalyzed cross-coupling of aryl chlorides with O, N-chelate stabilized diarylborinates. J. Organomet. Chem. 2017, 842, 54–58. [Google Scholar] [CrossRef]
- Yamaguchi, I.; Iijima, T.; Yamamoto, T. Synthesis and reactivity of Al(Et)q′2 (q′=2-methyl-8-quinolinolato) and crystal structures of [Al(Et)2q]2 and Alq′2q (q=8-quinolinolato). J. Organomet. Chem. 2002, 654, 229–232. [Google Scholar] [CrossRef]
- Pan, J.; Kampf, J.W.; Ashe, A.J. Tricarbonylchromium Complexes of 1,2-Dihydro-1,2-benzazaborines. Organometallics 2009, 28, 506–511. [Google Scholar] [CrossRef]
- Wu, Q.; Esteghamatian, M.; Hu, N.-X.; Popovic, Z.; Enright, G.; Tao, Y.; D’Iorio, M.; Wang, S. Synthesis, Structure, and Electroluminescence of BR2q (R = Et, Ph, 2-Naphthyl and q = 8-Hydroxyquinolato). Chem. Mater. 2000, 12, 79–83. [Google Scholar] [CrossRef]
- Höpfl, H.; Barba, V.; Vargas, G.; Farfan, N.; Santillan, R.; Castillo, D. X-ray crystallographic study of three (N→B)-borinates prepared from 8-hydroxyquinoline and 2-hydroxypyridine. Chem. Heterocycl. Compd. 1999, 35, 912–927. [Google Scholar] [CrossRef]
- Hu, T.-C.; Wu, M.-C.; Wu, S.-S.; Hu, C.-H.; Lin, C.-H.; Datta, A.; Lin, T.-H.; Huang, J.-H. Synthesis, characterization and reactivity study of aluminum compounds incorporating bi- and tri-dentate pyrrole–piperazine ligands. RSC Adv. 2016, 6, 16331–16339. [Google Scholar] [CrossRef]
- Francis, J.A.; Bott, S.G.; Barron, A.R. Aluminium compounds containing bidentate ligands: Chelate ring size and rigid conformation effects. J. Chem. Soc. Dalton Trans. 1998, 3305–3310. [Google Scholar] [CrossRef]
- Bulteau, Y.; Tarrat, N.; Pébère, N.; Lacaze-Dufaure, C. 8-Hydroxyquinoline complexes (Alq3) on Al(111): Atomic scale structure, energetics and charge distribution. New J. Chem. 2020, 44, 15209–15222. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. B 1964, 136, 864–871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. A 1965, 140, 1133–1138. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.; Handy, N. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Karafiloglou, P.; Landis, C.R.; Weinhold, F. NBO 7.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2018. [Google Scholar]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Vogler, A.; Kunkely, H. Luminescent Metal Complexes: Diversity of Excited States. In Transition Metal and Rare Earth Compounds: Excited States, Transitions, Interactions; Yersin, H.I., Ed.; Springer: Berlin/Heidelberg, Germany, 2001; pp. 143–182. [Google Scholar]
- Burrows, H.D.; Fernandes, M.; Seixas de Melo, J.; Monkman, A.P.; Navaratnam, S. Characterization of the Triplet State of Tris(8-hydroxyquinoline)aluminium(III) in Benzene Solution. J. Am. Chem. Soc. 2003, 125, 15310–15311. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.L. Natural transition orbitals. J. Chem. Phys. 2003, 118, 4775–4777. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Pérez-Bolívar, C.; Montes, V.A.; Anzenbacher, P. True Blue: Blue-Emitting Aluminum(III) Quinolinolate Complexes. Inorg. Chem. 2006, 45, 9610–9612. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Mohammadnezhad, G.; Amini, M.M.; Khavasi, H.R.; Plass, W. Structural and spectroscopic characterizations of aluminum phenoxide. Spectrochim. Acta A 2016, 157, 238–243. [Google Scholar] [CrossRef]




| Entry | Reagent (Equiv.) | Ligand | T | t | Product (#) [a] | Yield |
|---|---|---|---|---|---|---|
| 1 | BEt3 (2.50) | 8 | 130 °C | 17 h | Li[Et2B(BQ)] (9) | [b] |
| 2 | BPh3 (2.50) | 8 | 130 °C | 17 h | Li[Ph2B(BQ)] (10) | 30% |
| 3 | NaBH4 (2.50) | 1 | 130 °C | 6 d | Na[B(BQ)2] (11) | 41% |
| 4 | AlMe3 (2.50) | 8 | 25 °C | 15 min | Li[Me2Al(BQ)] (4) | 81% |
| 5 | AlPh3 (1.00) | 8 | 25 °C | 15 min | Li[Ph2Al(BQ)] (12) | 53% |
| 6 | LiAlH4 (0.50) | 1 | 130 °C | 17 h | Li[Al(BQ)2] (13) | 69% |
| 7 | AlMe3 (1.20) | 2 | 25 °C | 17 h | [Me2Al(Q’)]2 (3) | 75% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Appiarius, Y.; Puylaert, P.; Werthschütz, J.; Neudecker, T.; Staubitz, A. Complexation of Boron and Aluminum with a Bidentate Hydroxy-BN-naphthalene Ligand. Inorganics 2023, 11, 295. https://doi.org/10.3390/inorganics11070295
Appiarius Y, Puylaert P, Werthschütz J, Neudecker T, Staubitz A. Complexation of Boron and Aluminum with a Bidentate Hydroxy-BN-naphthalene Ligand. Inorganics. 2023; 11(7):295. https://doi.org/10.3390/inorganics11070295
Chicago/Turabian StyleAppiarius, Yannik, Pim Puylaert, Julius Werthschütz, Tim Neudecker, and Anne Staubitz. 2023. "Complexation of Boron and Aluminum with a Bidentate Hydroxy-BN-naphthalene Ligand" Inorganics 11, no. 7: 295. https://doi.org/10.3390/inorganics11070295
APA StyleAppiarius, Y., Puylaert, P., Werthschütz, J., Neudecker, T., & Staubitz, A. (2023). Complexation of Boron and Aluminum with a Bidentate Hydroxy-BN-naphthalene Ligand. Inorganics, 11(7), 295. https://doi.org/10.3390/inorganics11070295