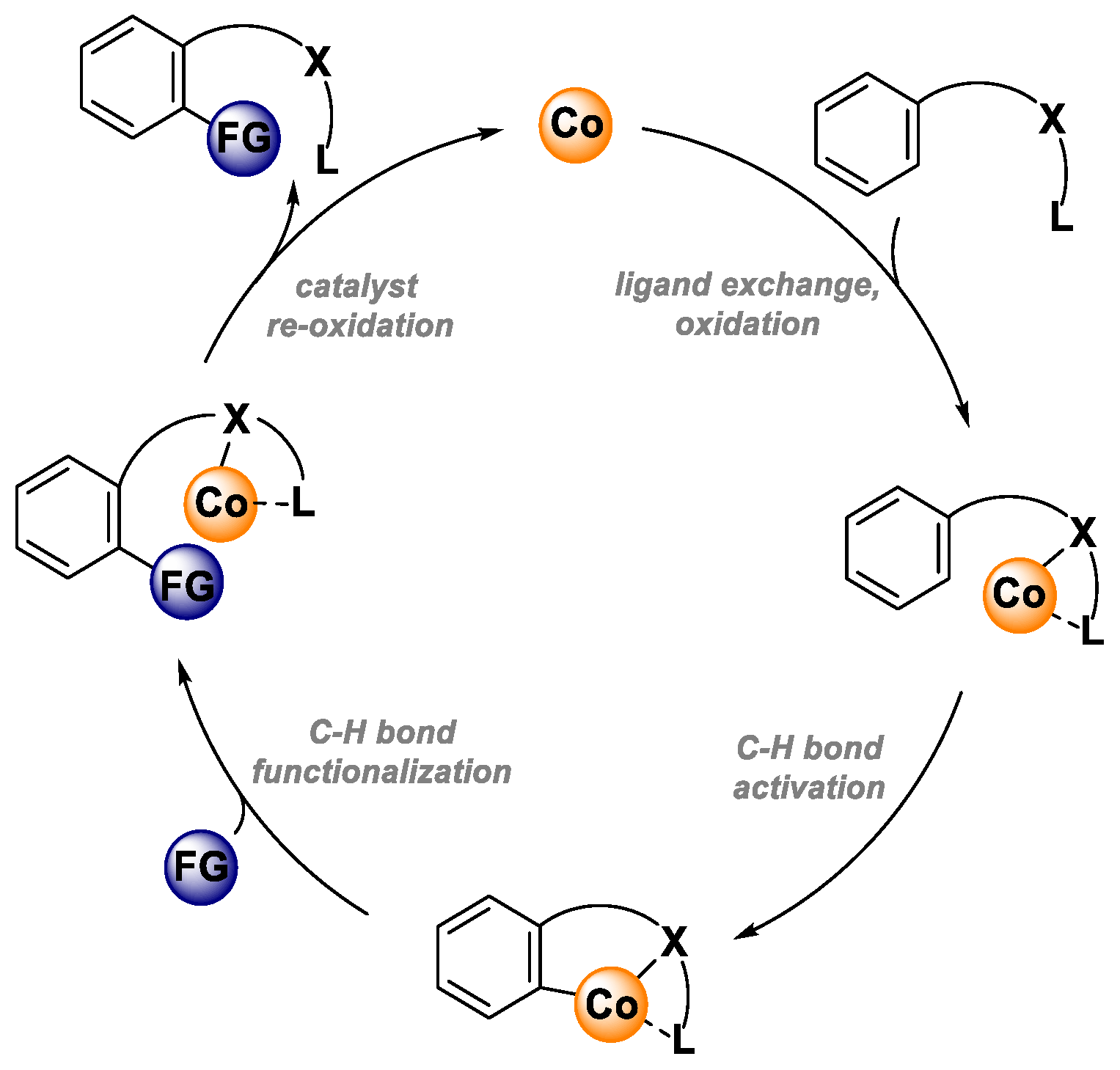
3. Ligand Exchange, Oxidation
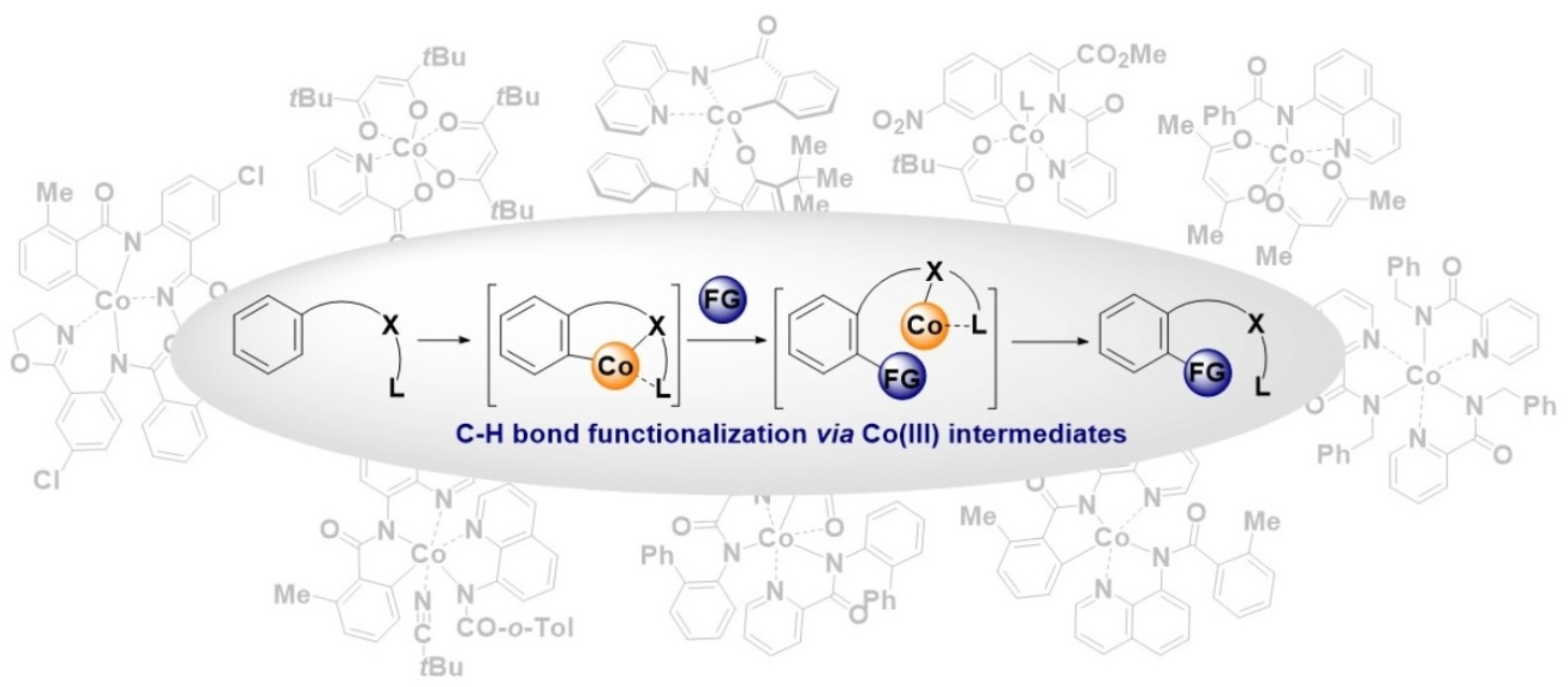
The first step of the catalytic cycle is ligand exchange/substrate coordination and oxidation of Co(II) species to Co(III) species. According to the literature data, two operative pathways are plausible for this step. First, the catalytic cycle could be initiated with substrate coordination to Co(II) salt to form a Co(II)-substrate complex, which is further oxidized to a Co(III) complex that undergoes the C-H activation step. In the second operative pathway, the Co(II) catalyst might be first oxidized to Co(III) salt. Next, substrate coordination takes place. In the literature, there is support for both of these pathways. Most likely, the operative pathway depends on the reaction conditions and/or substrate used for the transformation [
23].
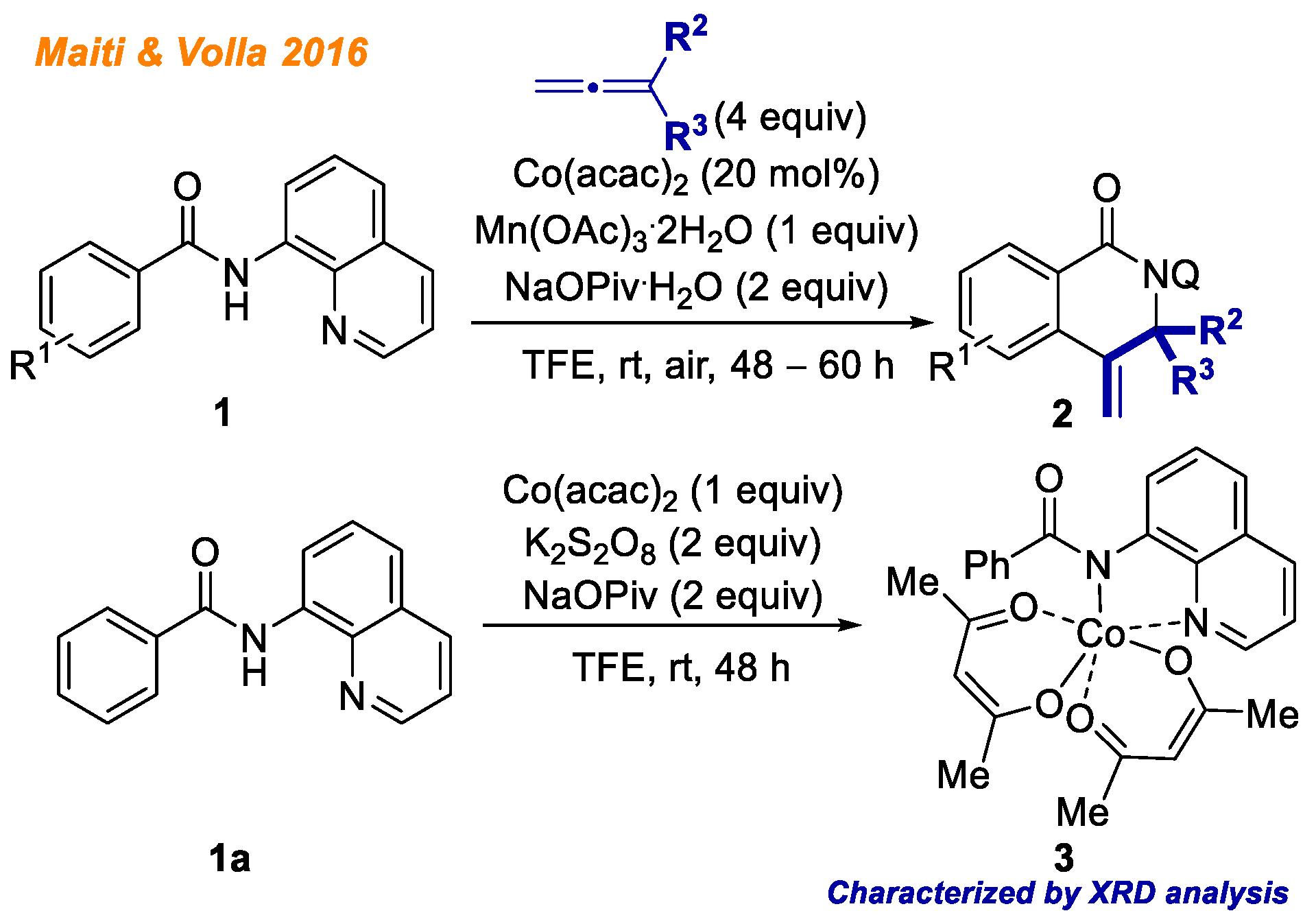
In 2016, Maiti and Volla reported a novel cobalt-catalyzed methodology for the intermolecular heterocyclization of benzamides
1 (
Scheme 2) [
24]. In their work, the following conditions were used: allene as the C-H bond functionalization reagent, Co(acac)
2 as the catalyst, Mn(OAc)
3·2H
2O/air as reaction oxidant, and sodium pivalate in TFE. Using the developed methodology, authors were able to provide a broad substrate scope with respect to both allenes and benzamides, delivering 41 different products with yields up to 90%. The authors conducted series of mechanistic experiments to study the reaction mechanism in detail. The authors concluded that electrophilic cobaltation is unlikely based on competitive experiments between methoxy- and fluoro-substituted benzamides. Experiments with deuterium labeled substrates indicated that C-H bond activation might not be the rate-limiting step. Additionally, authors were able to isolate cobalt(III)-benzamide intermediate
3, whose structure was confirmed with XRD analysis. Along with complex
3, authors detected the formation of C-H activated Co(III)-intermediate by HRMS, although they were not able to isolate corresponding complex. Based on the mechanistic experiments as well as isolated complex
3, the authors proposed the plausible reaction mechanism, which is consistent with the general Co(II)-Co(III)-Co(I) catalytic cycle.
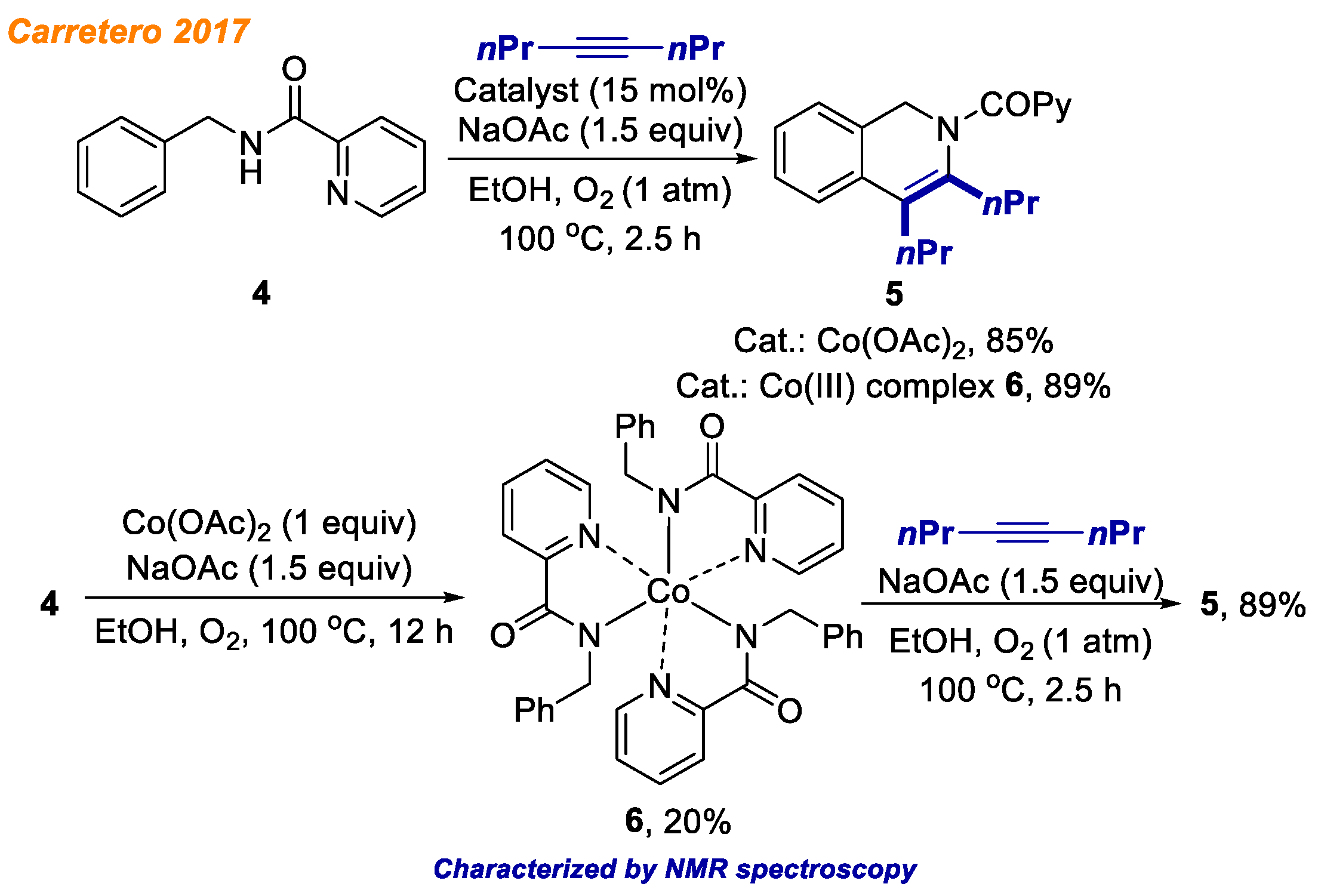
In 2017, the Carretero group reported an efficient protocol for the cobalt-catalyzed, picolinamide-directed C-H bond functionalization/annulation of benzylamine derivatives
4 with various alkynes (
Scheme 3) [
25]. The reaction proceeds in the presence of Co(OAc)
2 catalyst, O
2 oxidant, and NaOAc additive in EtOH at 100 °C temperature. The authors were able to ensure good functional group tolerance under the reaction conditions as well as a variety of terminal and internal alkynes delivered the desired products
5 predominantly with good and excellent yields. According to the mechanistic hypothesis, the authors propose that this reaction proceeds through the octahedral cobalt intermediate
6, which was successfully isolated. The structure of Co(III) complex
6 was proven by ESI-HRMS and NMR analysis, although no crystals for X-ray diffraction analysis were obtained. In the stoichiometric experiment, Co(III)-species
6 reacted with 4-octyne to afford product
5 in 89% yield. Moreover, Co(III) complex
6 was found to be catalytically competent in the reaction of
4 with alkyne, delivering product
5 in 89% yield. In comparison, under the standard reaction conditions using Co(OAc)
2 salt, the same product
5 was obtained in 85% yield. These results indicated that complex
6 could be the active catalyst precursor for the transformation. Additionally, the authors performed ESI-HRMS experiments of the reaction mixture to shed some more light on the reaction mechanism, and deciphered different cobalt complexes being present in the reaction mixture, although none of them was isolated and characterized.
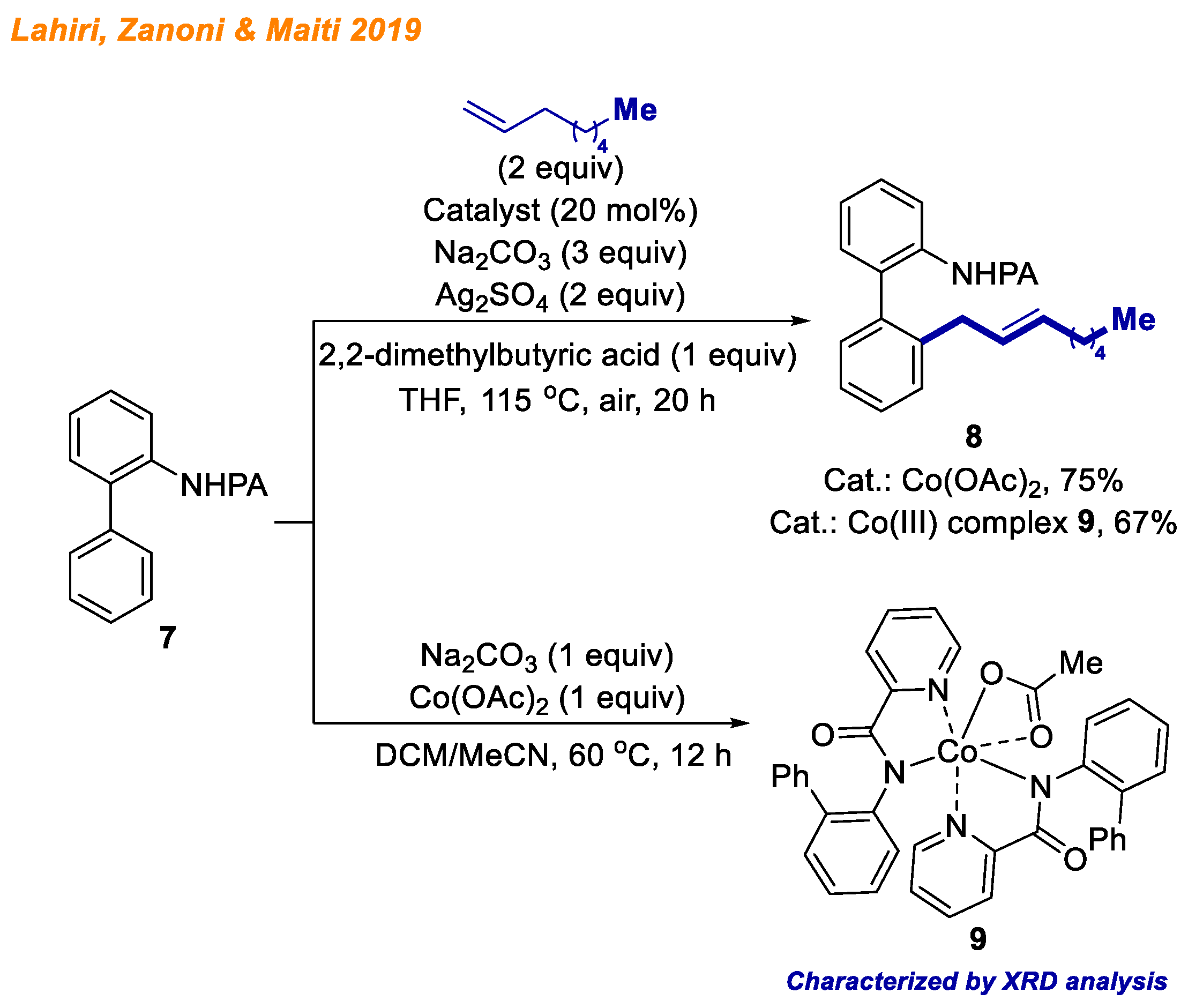
Two years later, in 2019, Lahiri, Zanoni and Maiti reported a cobalt-catalyzed C-H bond allylation reaction using arylanilines
7 as substrates and unbiased terminal olefins (
Scheme 4) [
26]. The most common problem in these reactions is products’ styrenyl/allylic regioselectivity of the double bond, which arises from the metal center’s ability to unselectively perform β-hydride elimination. In this context, authors successfully overcame the challenge and were able to deliver 36 different aryl(allyl)anilines
8 with yields up to 96% in a highly selective fashion. In addition, the authors demonstrated that picolinamide directing group (PA) can be easily removed upon slight heating in basic conditions. To gain insight into the reaction mechanism, among kinetic and labeling studies, authors were able to isolate five-membered Co(III) intermediate
9, whose structure was confirmed using XRD and ESI-MS analyses. The isolated Co(III) complex
9 was found to be catalytically competent, delivering product
8 in 67% yield, suggesting the involvement of a catalytically active high-valent Co(III) species.
In 2020, Wang and colleagues demonstrated a novel cobalt-catalyzed C-H/C-H bond cross-coupling reaction between benzoxazole and aryl aniline
7 (
Scheme 5) [
27]. The use of Co(OAc)
2 catalyst, Ag
2CO
3 oxidant, and Ole-ONa base in fluorobenzene was found to represent the optimal conditions for the successful reaction. The main advantage of the developed transformation was the straightforward access to biphenyls
10 in good yields (up to 73%), which possess antifungal activities and COX-2 inhibition potency. To investigate the reaction mechanism, the authors performed series of control experiments, including H/D exchange and KIE experiments, which led to conclusion that the C-H activation step is irreversible, but not the rate-determining step. Additionally, two Co(III) intermediates
11 and
12 were obtained by the reaction of aniline
7 with a stoichiometric amount of Co(OAc)
2, oxidant and base. Both Co(III) complexes
11 and
12 were characterized using NMR spectroscopy and high-resolution mass spectrometry. According to the suggested reaction mechanism, Co(III) complex
12 could be obtained from Co(III) complex
11 via a C-H activation step, which most likely occurs via a base-promoted concerted metalation-deprotonation mechanism. Notably, cobaltacycles
11 and
12 provided the desired product
8 in 41% and 78% yield, respectively, whereas using Co(OAc)
2 as the catalyst, the product yield was 71%, which confirmed the hypothesis that both isolated Co(III) complexes are most likely intermediates of the developed reaction.
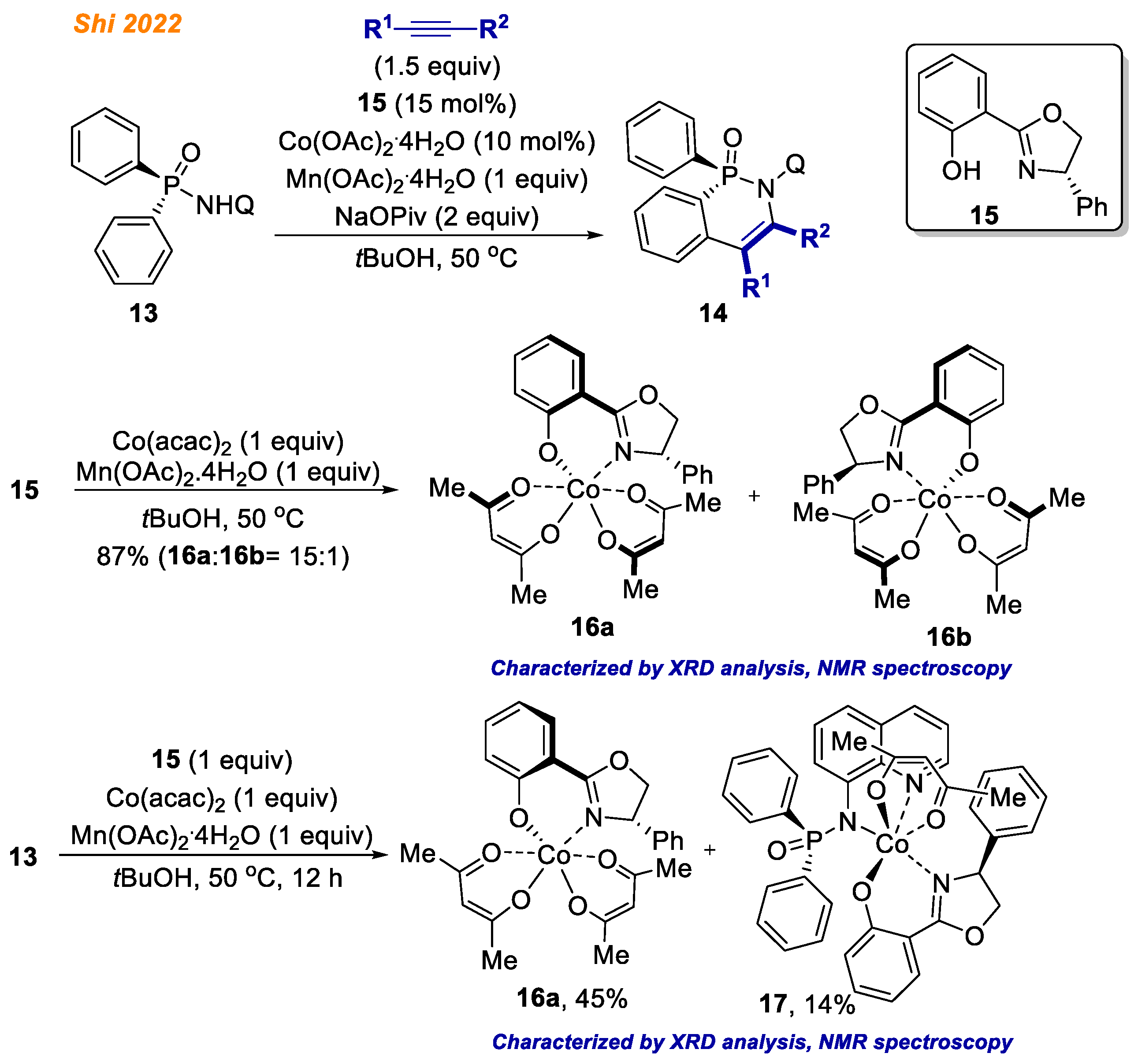
Recently, in 2022, the Shi group reported an elegant enantioselective C-H bond functionalization methodology exploiting diarylphosphinamides
13 (
Scheme 6) [
28]. In their study, employing Co(OAc)
2·4H
2O catalyst and Salox ligand
15, azaphosphinines
14 were obtained with yields up to 99% with fascinating product enantioselectivities (up to >99% ee). The authors demonstrated great substrate/alkyne scope, delivering 45 different enantiopure products with excellent yields. Great emphasis was put on the understanding of the reaction mechanism and isolation of potential intermediates of the catalytic cycle.
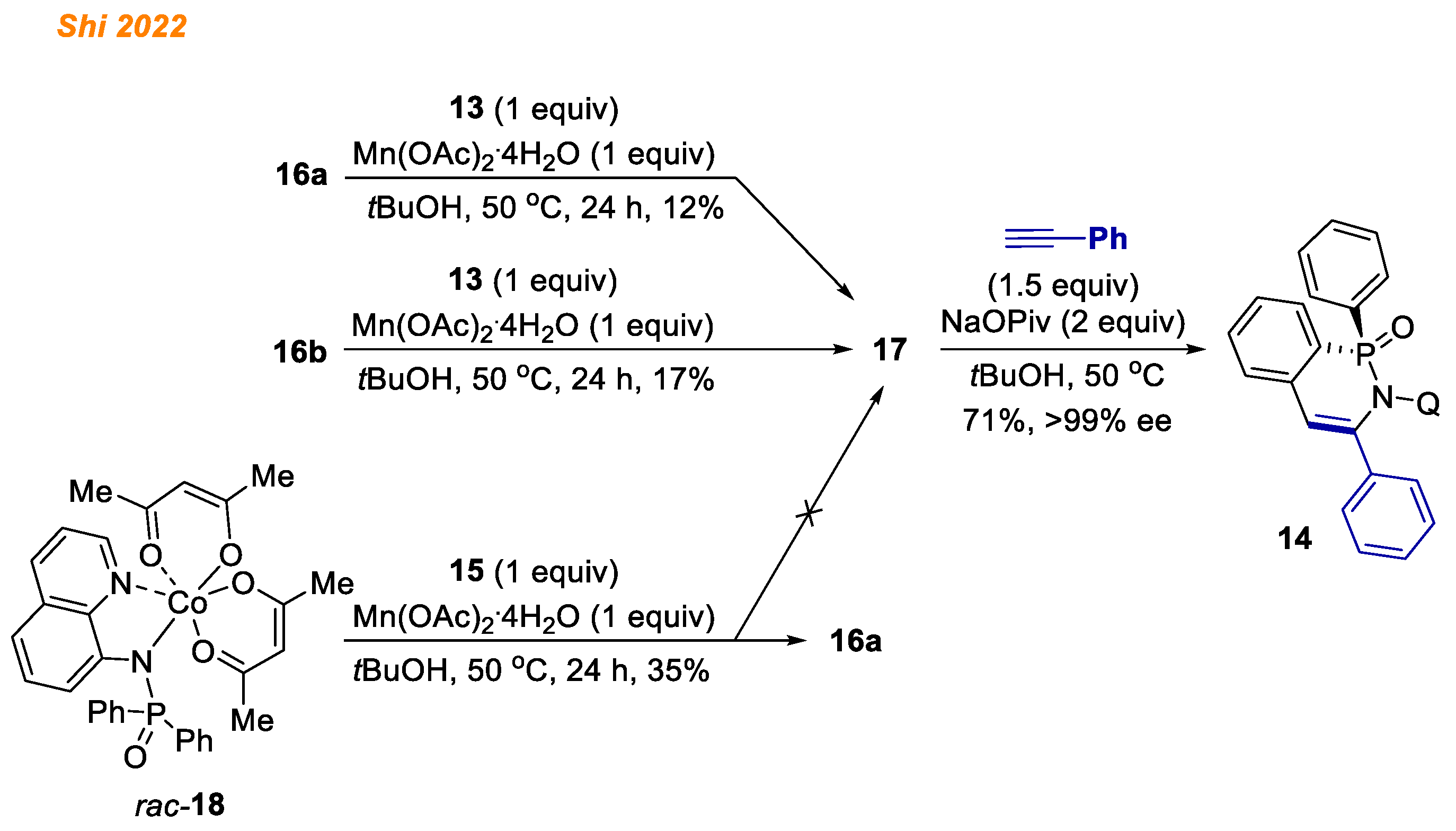
First, to test the proof of concept, authors synthesized chiral octahedral Co(III)-Salox complexes 16, which were hypothesized to act as the reaction catalysts. Accordingly, Co(acac)2 in combination with Mn(OAc)2·4 H2O in the presence of chiral ligand 15 gave Co(III)-Salox complex diastereomers 16a and 16b (16a:16b = 15:1) in high yield.
With both Co(III) complex isomers
16a and
16b in hand, the authors proved their hypothesis and demonstrated that both complexes
16 are suitable chiral catalysts for enantioselective desymmetrizing C-H annulation of diarylphosphinamides
13 with alkynes thereby demonstrating the structure of the active cobalt catalyst which participates in developed reaction. Next, the reaction of diarylphosphinamide
13 with ligand
15 and Co(acac)
2 under oxidative conditions resulted in the simultaneous formation of Co(III) complexes
16a and
17 with 45% and 14% yield, respectively. Additionally, both
16a and its diastereomer
16b in the reaction with diarylphosphinamide
13 in the presence Mn(OAc)
2·4H
2O gave Co(III) complex
17 (
Scheme 7) [
28]. The authors speculated that Mn(OAc)
2·4H
2O likely promotes the formation of
17 by facilitating ligand exchange, as without Mn(OAc)
2·4H
2O, complex
17 was not observed. In contrast, ligand exchange between the pre-formed
rac-18 and ligand
15 provided complex
16a, not
17. Finally, complex
17 in reaction with phenylacetylene gave product
14 in 71% yield, suggesting that all of the obtained cobalt complexes
16–
18 might be the reaction intermediates.
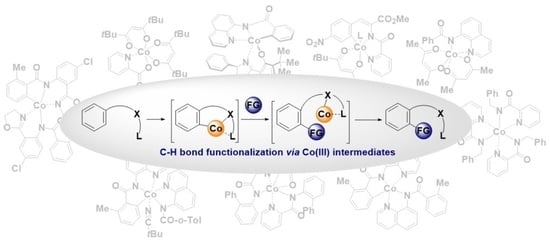
4. C-H Bond Activation
The key elementary step of C-H bond functionalization is the C-H bond activation. For high-valent cobalt catalysis, several C-H bond activation mechanisms leading to the formation of Co(III)-aryl complex are considered to be operative:
electrophilic aromatic substitution;
base-assisted intramolecular electrophilic substitution;
concerted metalation–deprotonation;
single-electron transfer [
23,
29].
In the literature, there are several examples of isolated relatively stable Co(III)-aryl complexes obtained via direct C-H bond activation. Such complexes are proven to be invaluable assets for the mechanistic studies.
In early 2014, the Daugulis group developed benzamide
1a C(sp
2)-H bond alkenylation with alkynes, using 8-aminoquinoline (Q) as a directing group (
Scheme 8) [
22]. The reaction conditions were mild and provided products
19 in good to excellent yields, tolerating a wide range of alkynes and substituents at benzene ring moiety. The authors hypothesized that due to the aminoquinoline stabilization of metals in high oxidation state, cobalt complex
20 could be the reaction intermediate. In addition, they successfully synthesized complex
20, the structure of which was confirmed by NMR analysis, providing strong evidence for C(sp
2)-H bond activation of phenyl moiety and Co(III) species.
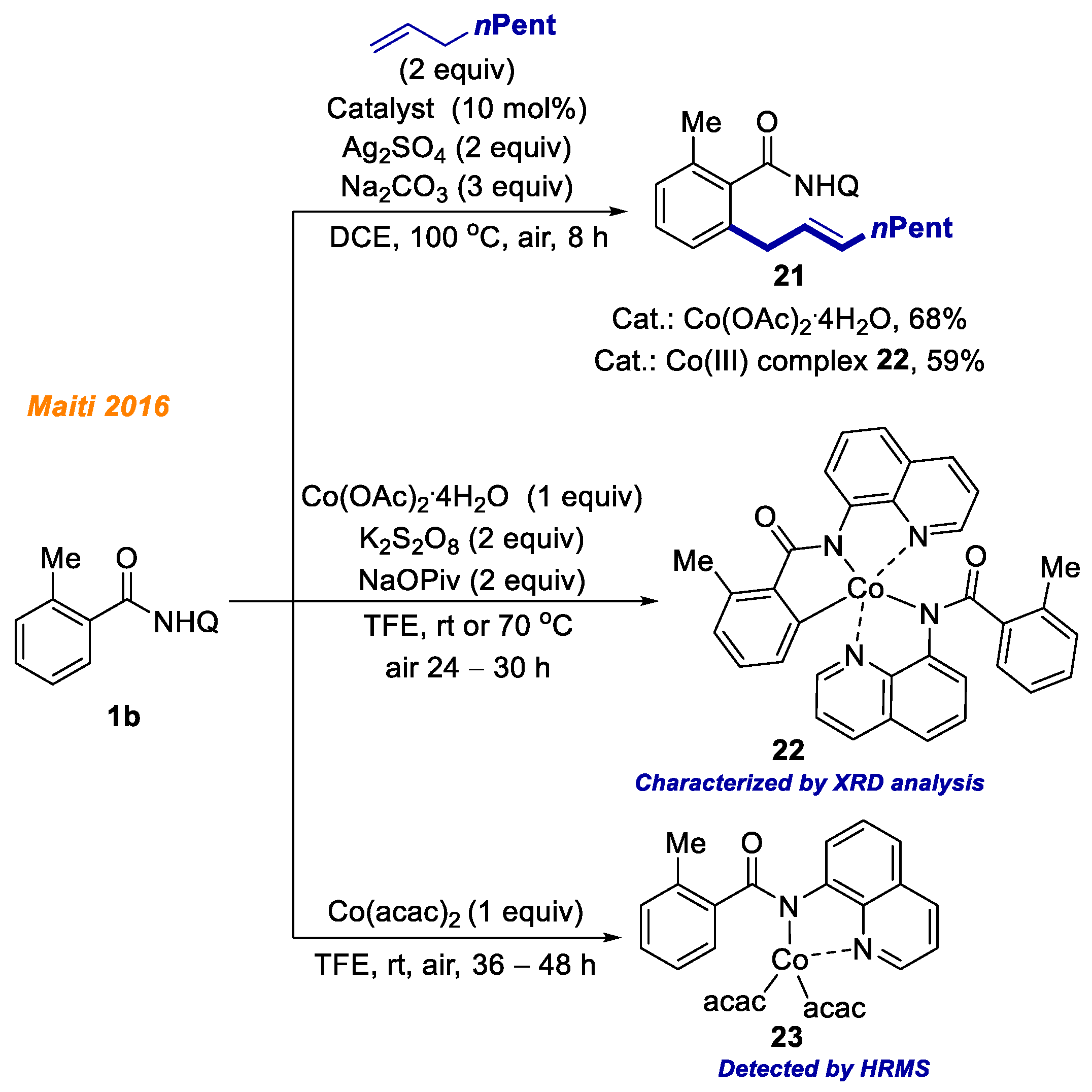
Maiti and co-workers in 2016 disclosed a novel methodology for benzamide
1b C(sp
2)-H bond allylation (
Scheme 9) [
30]. The optimization studies showed that the combination of Co(OAc)
2·4H
2O catalyst, Ag
2SO
4 oxidant, and 8-aminoquinoline directing group was the most suitable for the developed transformation. Both electron-donating and electron-withdrawing amides were successfully applied and yielded allylamides
21 with moderate to very good yields. In order to thoroughly outline all the aspects of this reaction, a series of control experiments were performed, including kinetic and labeling studies along with radical quenching experiments. Moreover, the authors succeeded in isolation of C-H activated Co(III)-aryl intermediate
22, and proved its catalytic competency towards developed reaction. Employment of complex
22 as the catalyst yielded allylamide
21 in 59% yield. Additionally, they identified and characterized cobalt(III) complex
23 by HRMS, which underwent C-H bond activation/ligand exchange steps and formed Co(III)-aryl complex
22 after the addition of a stoichiometric amount of NaOPiv to the reaction mixture.
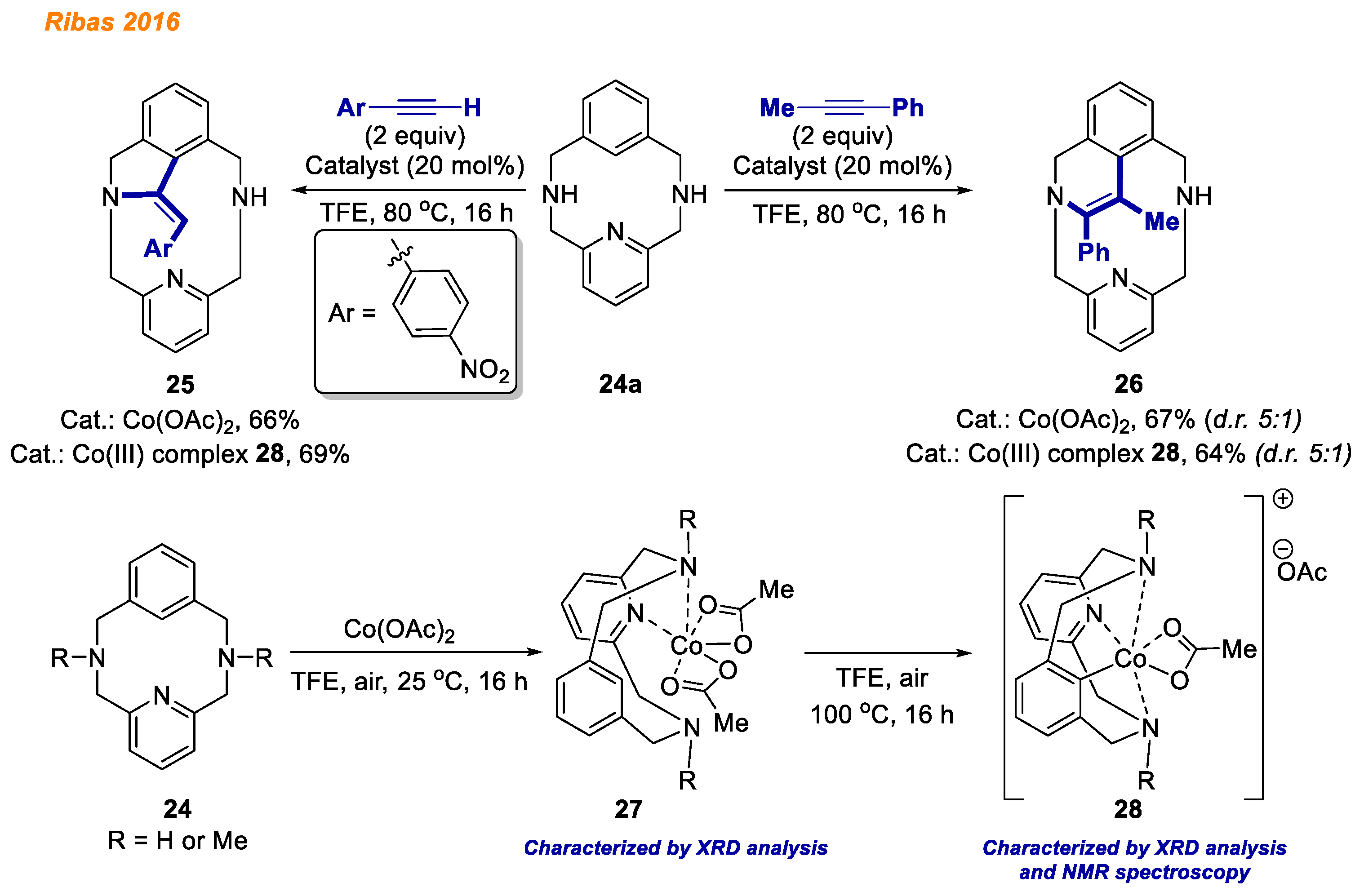
In 2016, Ribas and co-workers described the synthesis and characterization of benchtop-stable organometallic aryl-Co(III) complexes obtained through C-H bond activation, using a 12-membered macrocyclic substrate
24a (
Scheme 10) [
31]. Cobalt (II) coordination compounds
27 were prepared by the reaction of Co(OAc)
2 with macrocycles
24 (R = H or Me) at room temperature in TFE solution, and their structures were initially confirmed by HRMS and XRD analysis. Careful analysis of solid-state molecular structure indicated that Co(II) complexes
27 possess two acetates coordinated in a bidentate fashion. Notably, tridentate macrocycles
24 act as bidentate ligands, coordinated only through the pyridine and one amine. Upon increasing the temperature, the desired C-H activated cobalt(III) complexes
28 were obtained. Due to the stability of obtained Co(III)-aryl complexes
28, they were successfully characterized by NMR and HRMS, providing spectra consistent with a Co(III) low spin diamagnetic metal center. Additionally, the authors explored the reactivity of isolated Co(III)-aryl compounds
28 in stoichiometric reactions with terminal and internal alkynes. Employing internal alkynes, the expected six-membered 1,2-dihydroisoquinolines
26 could be obtained in yields up to 72%, whereas terminal alkynes led to the formation of dihydroisoindoline
25 (5-membered ring) as a thermodynamically more stable product. It was observed that decreasing the reaction temperature or changing the electronic effects in alkynes, e.g., using phenylacetylene instead of 4-nitrophenylacetylene, led to the formation of a kinetic product (six-membered ring). In addition, using isolated Co(III) intermediates
28, annulation reactions were also studied in a catalytic fashion. As a result, the desired products were obtained in very good yields, indicating that Co(III)-aryl complexes
28 are the reaction intermediates.
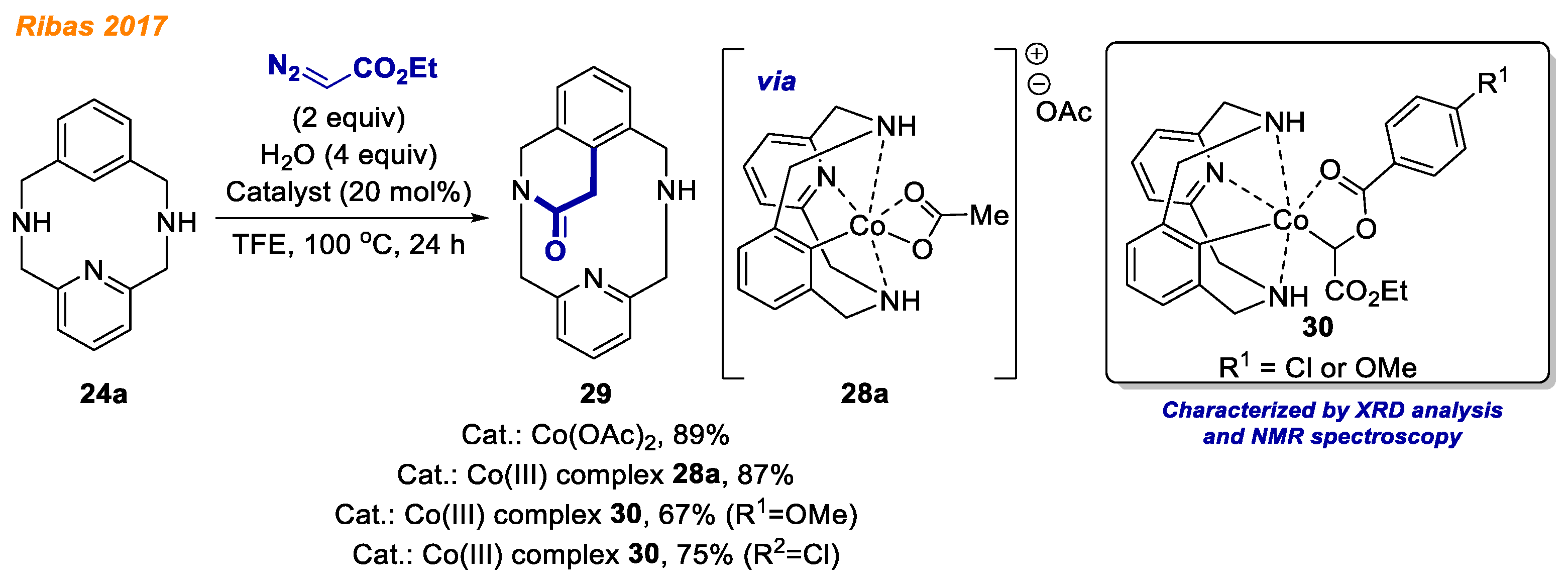
One year later, in 2017, Ribas group explored the formation of Aryl-Co(III) masked carbenes in cobalt-catalyzed C-H bond functionalization with diazo esters as a continuation of their previous work (
Scheme 11) [
32]. Optimization studies revealed that the developed protocol requires Co(OAc)
2 catalyst and H
2O as an additive to afford the desired isoquinoline
29 via annulation reaction with ethyl diazoacetate (EDA). The authors utilized previously isolated cobalt complex
28a as the substrate and performed detailed mechanistic investigation under anhydrous reaction conditions. When
28a reacted with ethyl diazoacetate, a single peak was observed by HRMS analysis. Authors proposed the formation of a putative aryl-Co(III)-carbene intermediate. Although attempts to unravel its nature by crystallographic analysis were unsuccessful, suitable crystals for XRD analysis were obtained by replacement of acetate anion in
28a with
p-substituted benzoates. This anion exchange facilitated a rapid color switch from red to orange in the reaction with EDA, and recrystallization from CHCl
3/pentane afforded orange crystals of Co(III) complex
30. Similar to a previous report [
31], isolated cobalt intermediates
28a and
30 were used as catalysts, providing the desired product
29 with yields 67–87%, indicating that organometallic complexes
28a and
30 are catalytically active species. Ribas and co-workers are continuing the investigation of Co(III)-aryl complex
28 reactivity towards other transformations [
33].
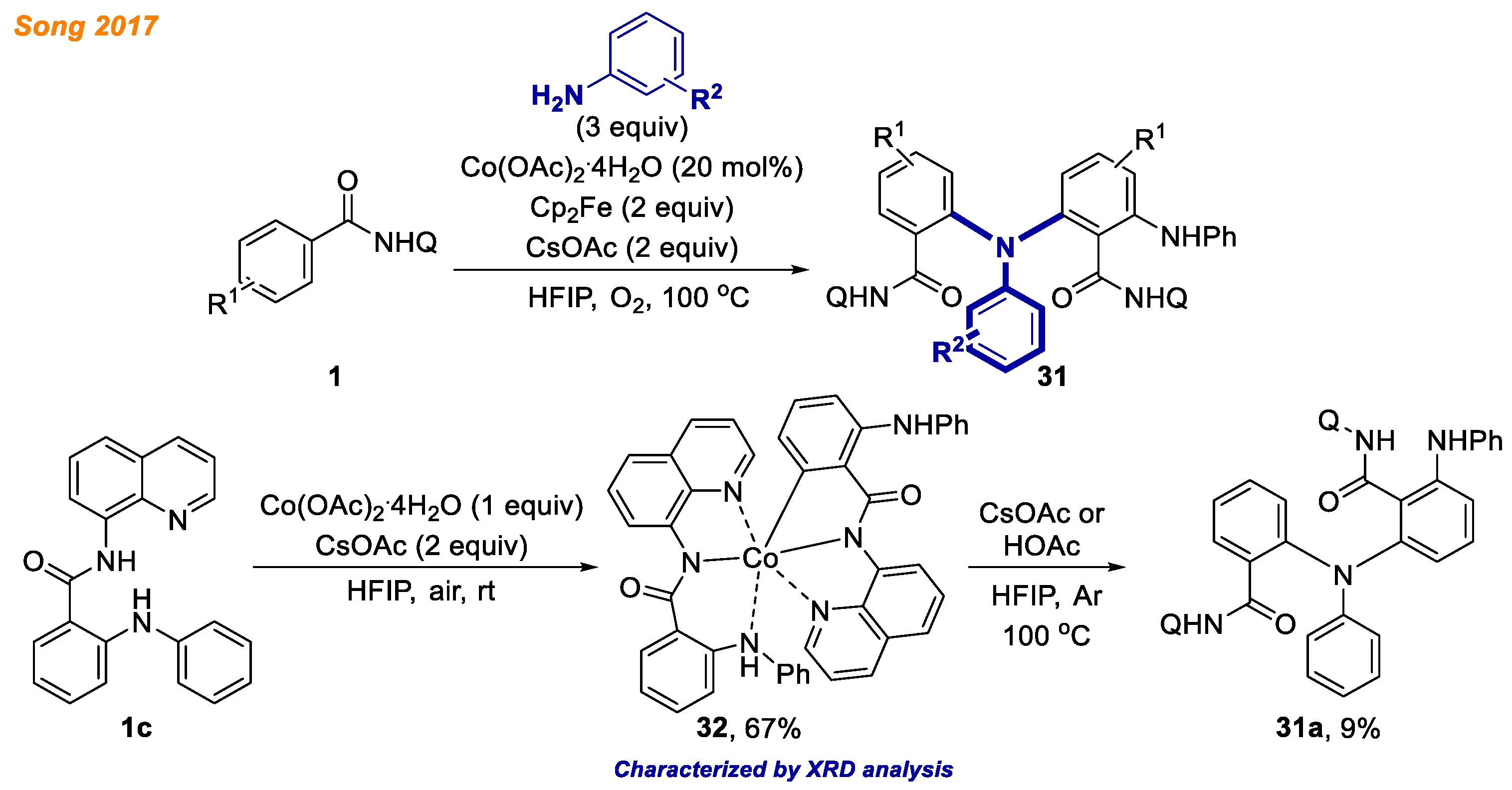
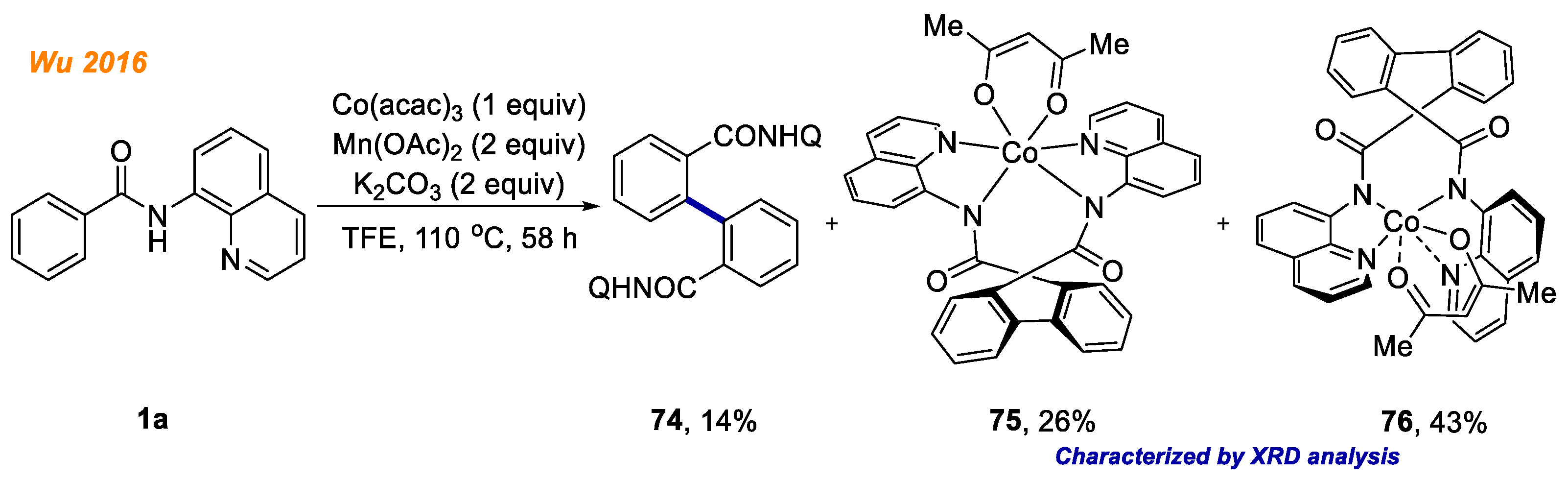
In the same year, Song and co-workers reported a selective and facile access to triarylamines
31 via cobalt-catalyzed oxidative C-H/N-H cross-coupling reaction (
Scheme 12) [
34]. During optimization of reaction conditions, authors were able to push the selectivity towards the desired product
31 over the dimerization side reaction of benzamides
1. The best results were achieved, using optimized catalytic system, which consisted of Co(OAc)
2·4H
2O catalyst, ferrocene cooxidant and CsOAc in HFIP at 100 °C temperature under aerobic conditions. Furthermore, Co(III) complex
32 was obtained from the reaction of benzamide
1c with stoichiometric amount of Co(OAc)
2·4H
2O at room temperature under air. Pleasingly, its structure was confirmed by XRD analysis. The authors demonstrated that cobaltacycle
32 under basic or acidic conditions delivered the triarylamine product
31a only in 9% yield. Although other mechanistic experiments suggest a Co(II)-Co(III)-Co(I) catalytic cycle, based on the low yield of product
31a formation from Co(III) complex
32, the involvement of Co(IV) catalytic species cannot be excluded.
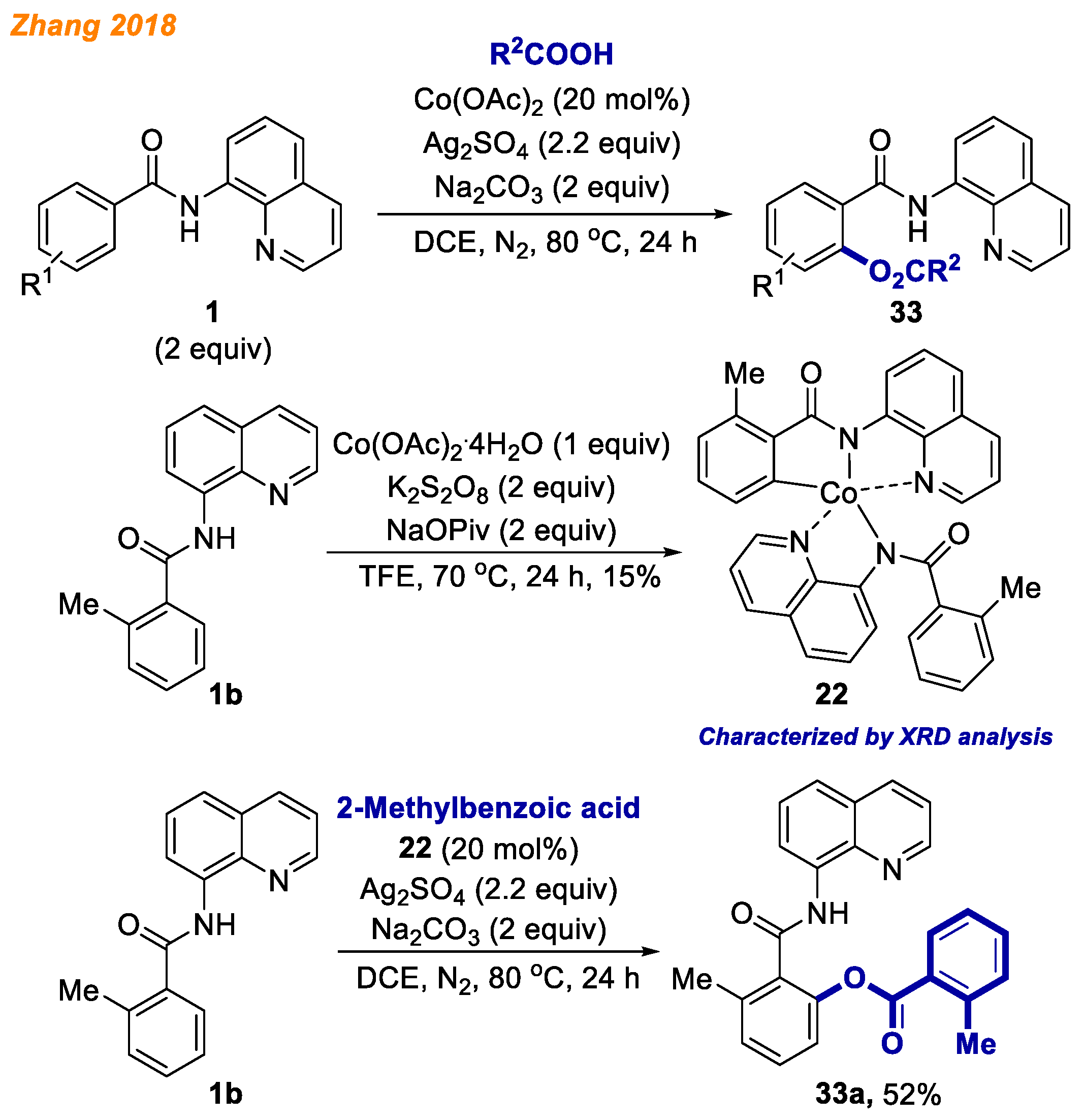
In 2018, Zhang and co-workers reported a facile and powerful protocol for the cobalt-catalyzed C-H bond acyloxylation of benzamides
1 (
Scheme 13) [
35]. In the developed methodology anhydrous Co(OAc)
2 in combination with Ag
2SO
4 and Na
2CO
3 in DCE was found to be the catalytic system of choice. The optimized reaction conditions were compatible with a diverse substrate scope, delivering a broad variety of
o-substituted benzamides
33 (44 products) with yields up to 99%. Additionally, radical-trapping and deuterium-labeling experiments were performed to understand the reaction mechanism in detail. Besides, authors synthesized Co(III)-aryl complex
22 based on a procedure previously described by Maiti and co-workers in 2016 [
30], starting from methylbenzamide
1b, and confirmed its structure by single-crystal X-ray diffraction analysis. Authors suggested that Co(III) complex
22 could be the key intermediate in this reaction as it catalyzed the model reaction, delivering product
33a in 52% yield.
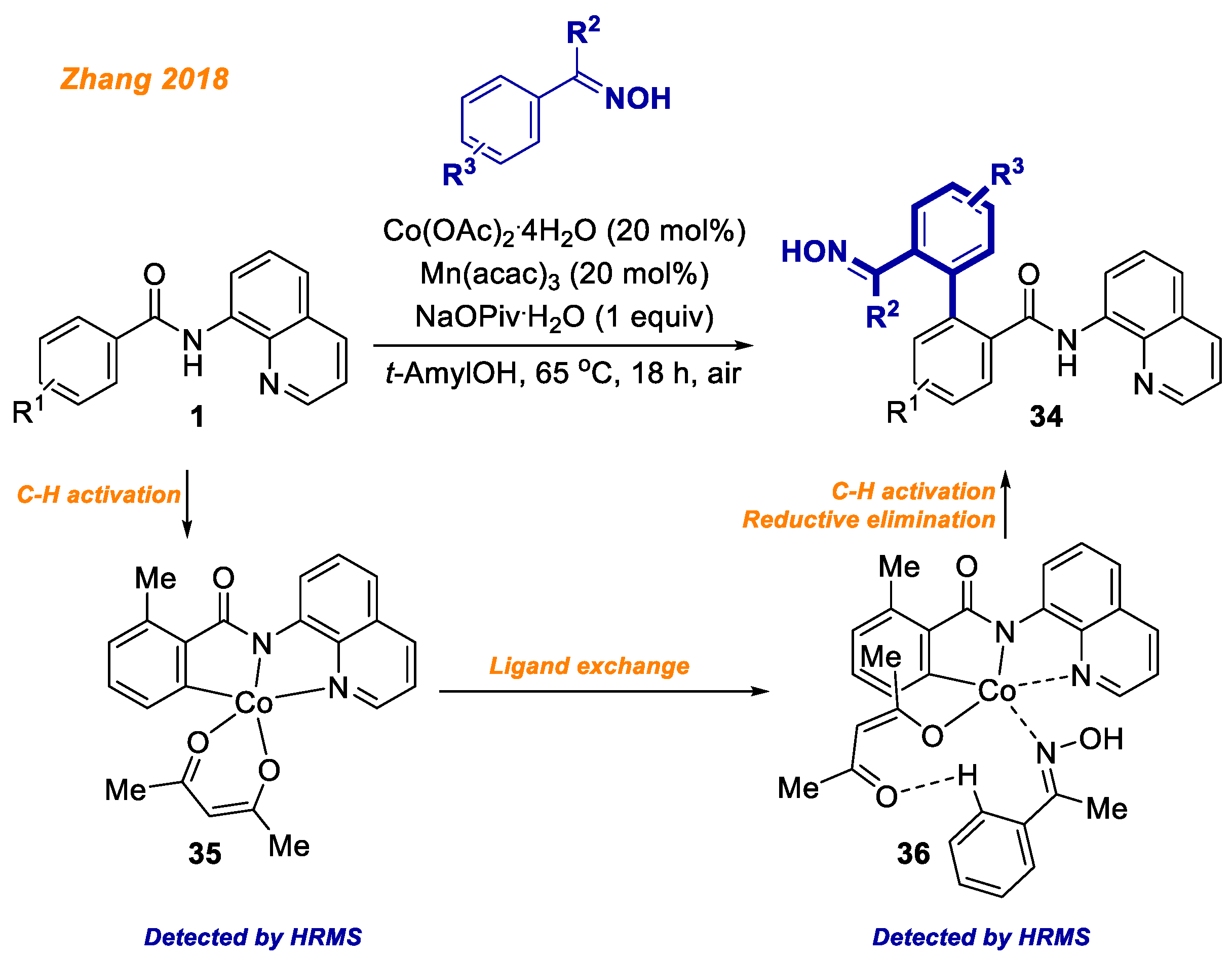
In the same year, the Zhang group reported a novel strategy for the synthesis of difunctional biaryls
34 from readily available benzamides
1 and oximes (
Scheme 14) [
36]. In contrast to the majority of cobalt-catalyzed benzamide
1 C-H bond functionalization reactions, the reported protocol is very mild as this transformation takes place at 65 °C temperature under air. The reported methodology demonstrates broad substrate scope as well as remarkable chemoselectivity towards cross-coupling. To gain insight into the reaction mechanism, the authors performed an intermolecular competition experiment of electronically differentiated benzamides as well as KIE experiments. In addition, both, C-H activated Co(III)-aryl complex
35 and oxime-coordinated Co(III)-aryl complex
36 were successfully detected by HRMS. Mechanistic studies along with detected Co(III) intermediates helped authors to propose the reaction mechanism, which is in accordance with the general Co(II)-Co(III)-Co(I) mechanism.
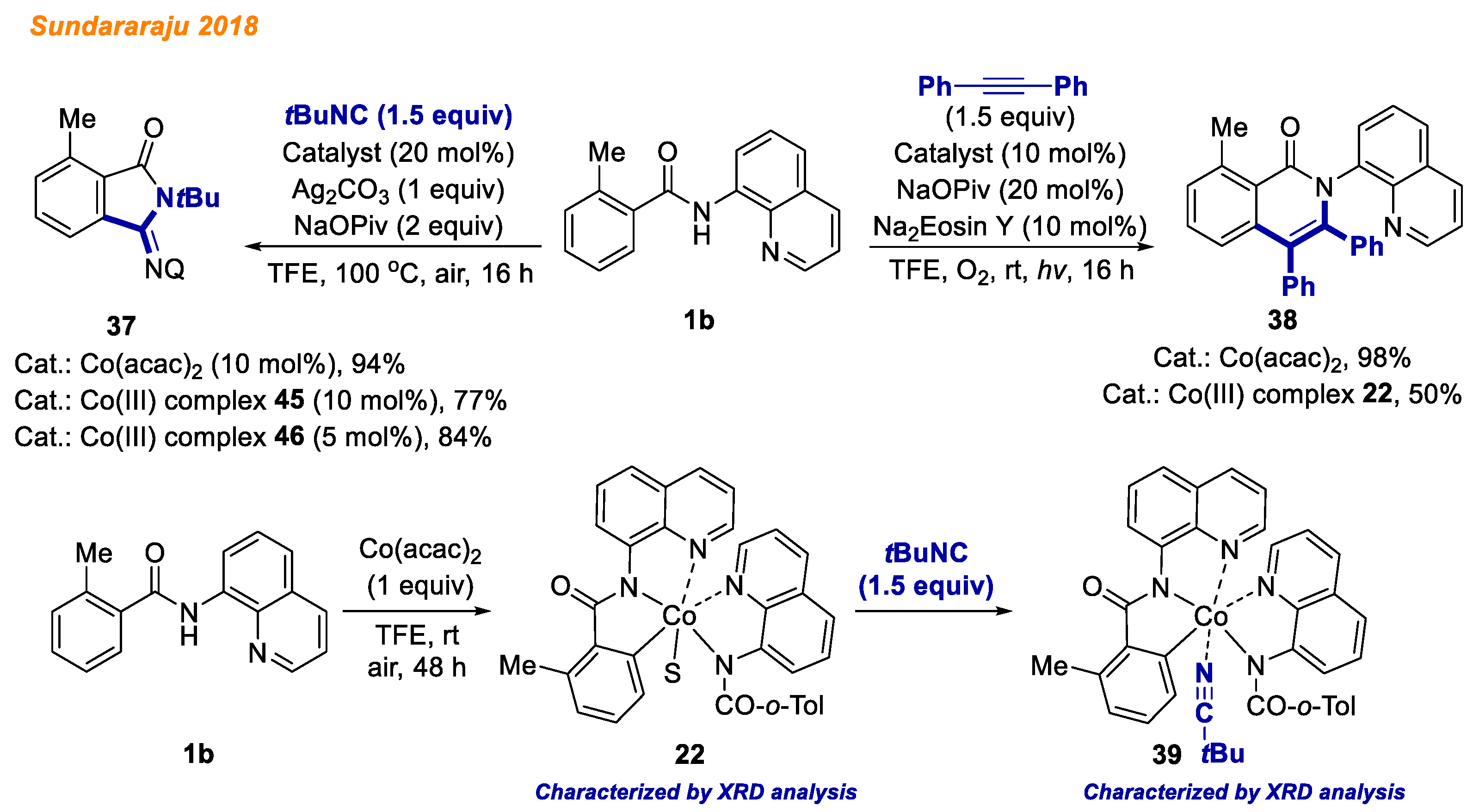
In 2018, the Sundararaju group developed a novel pathway for isonitrile insertion/acyl group migration between N-H and C-H bonds of benzamides
1b through intramolecular
trans-amidation catalyzed by Co(acac)
2 (
Scheme 15) [
37]. The authors demonstrated broad substrate scope yielding a wide variety of iminoisoindolinone derivatives
37 (42 products, yields up to 94%). Besides, significant mechanistic studies were performed, including KIE, H/D exchange, and radical trapping experiments. Moreover, the stoichiometric reaction of benzamide
1b with Co(acac)
2 yielded C-H activated Co(III)-aryl complex
22, which was previously described by Maiti [
30], as well as later by the Zhang group [
35]. The reaction of Co(III)-aryl complex
22 with
tert-butyl isocyanide at room temperature in 10 min yielded Co(III) intermediate
39, which was isolated and its structure was confirmed by HRMS and XRD analysis. Both intermediates
22 and
39 were used as catalysts for the transformation of benzamide
1b to iminoisoindolinone derivative
37 under standard reaction conditions and were found to be active in catalysis.
Later the same year, Sundararaju and colleagues reported a strategy for benzamide
1b C-H and N-H bond annulation with alkynes by merging cobalt-mediated catalysis with photocatalysis (
Scheme 15) [
38]. Employing relatively similar reaction conditions to the group’s previous report [
35] and Na
2Eosin Y as the photoredox catalyst, various electron-donating and electron-withdrawing benzamides delivered isoquinolones with yields up to 99%. In addition, Co(III) complex
22 was found to be catalytically active in reaction with benzamide
1b, delivering isoquinolone
38 with 50% yield in contrast to Co(acac)
2, which furnished the desired product with 98% yield.
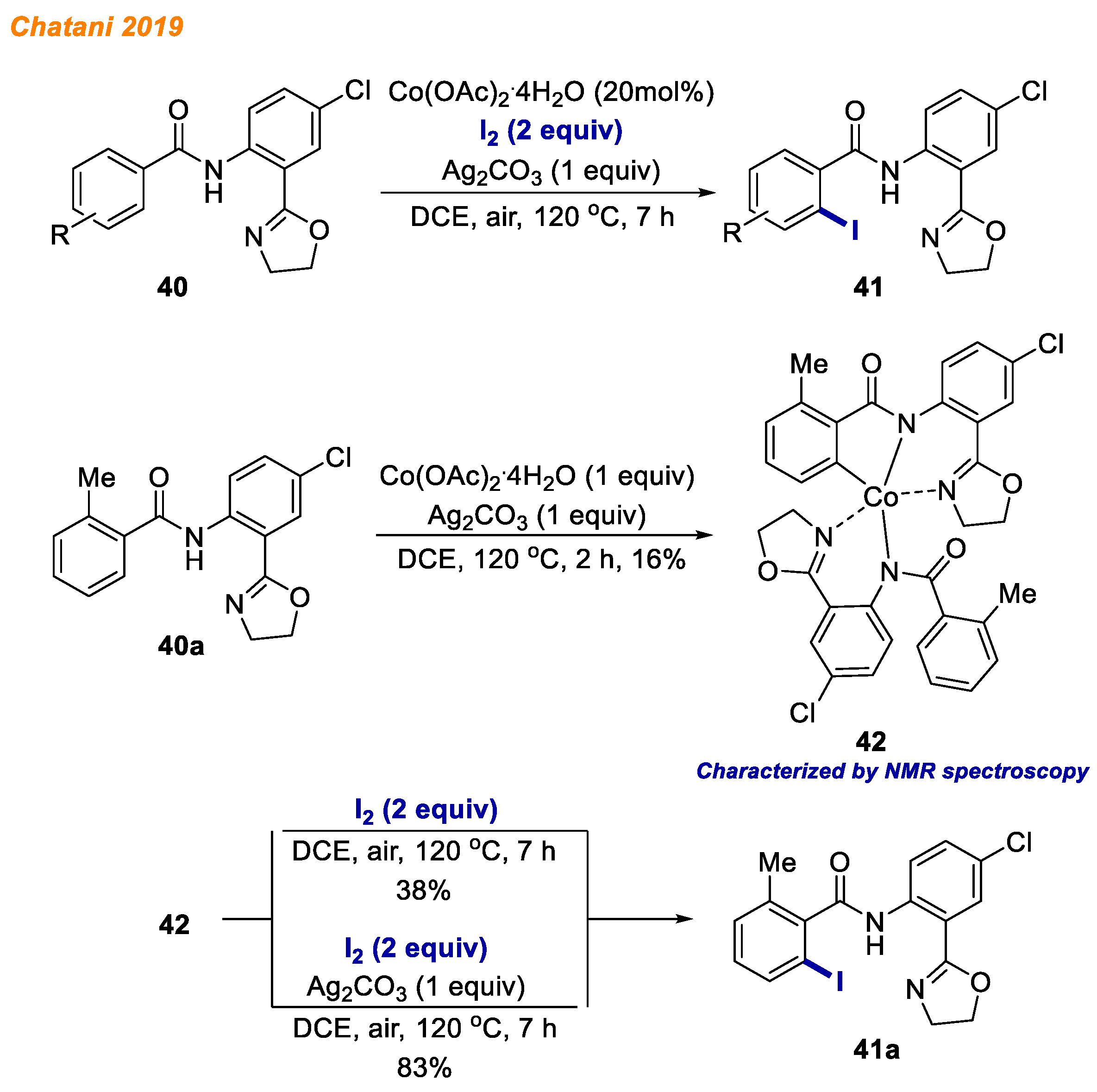
A year later, in 2019, Chatani reported an efficient protocol for cobalt-catalyzed C-H bond iodination of benzamides
40 with elemental iodine (
Scheme 16) [
39]. Thus, 2-Aminophenyloxazoline-based bidentate chelation as a directing group in combination with Co(OAc)
2·4H
2O catalyst and Ag
2CO
3 oxidant were found to represent the optimal catalytic system for the synthesis of 2-iodobenzamides
41 in moderate to great yields. When methylbenzamide
40a was treated with equimolar amount of Co(OAc)
2·4H
2O and Ag
2CO
3 in DCE at 120 °C for 2 h, the Co(III) complex
42 was successfully isolated and characterized by HRMS and NMR spectroscopy. Interestingly, without the oxidant Co(III), complex
42 furnished the iodination product only in 38% yield along with a significant amount of unidentified byproducts, whereas in the presence of Ag
2CO
3 the yield increased to 83%, and no byproducts were detected. Based on these experiments, the authors concluded that despite the catalytic activity of Co(III) complex
42, silver oxidant is an essential component of the reaction, promoting the iodination reaction and eliminating the formation of byproducts. The authors therefore speculated that Co(III) complex
42 is not the key intermediate of the reaction, but exists as a resting state.
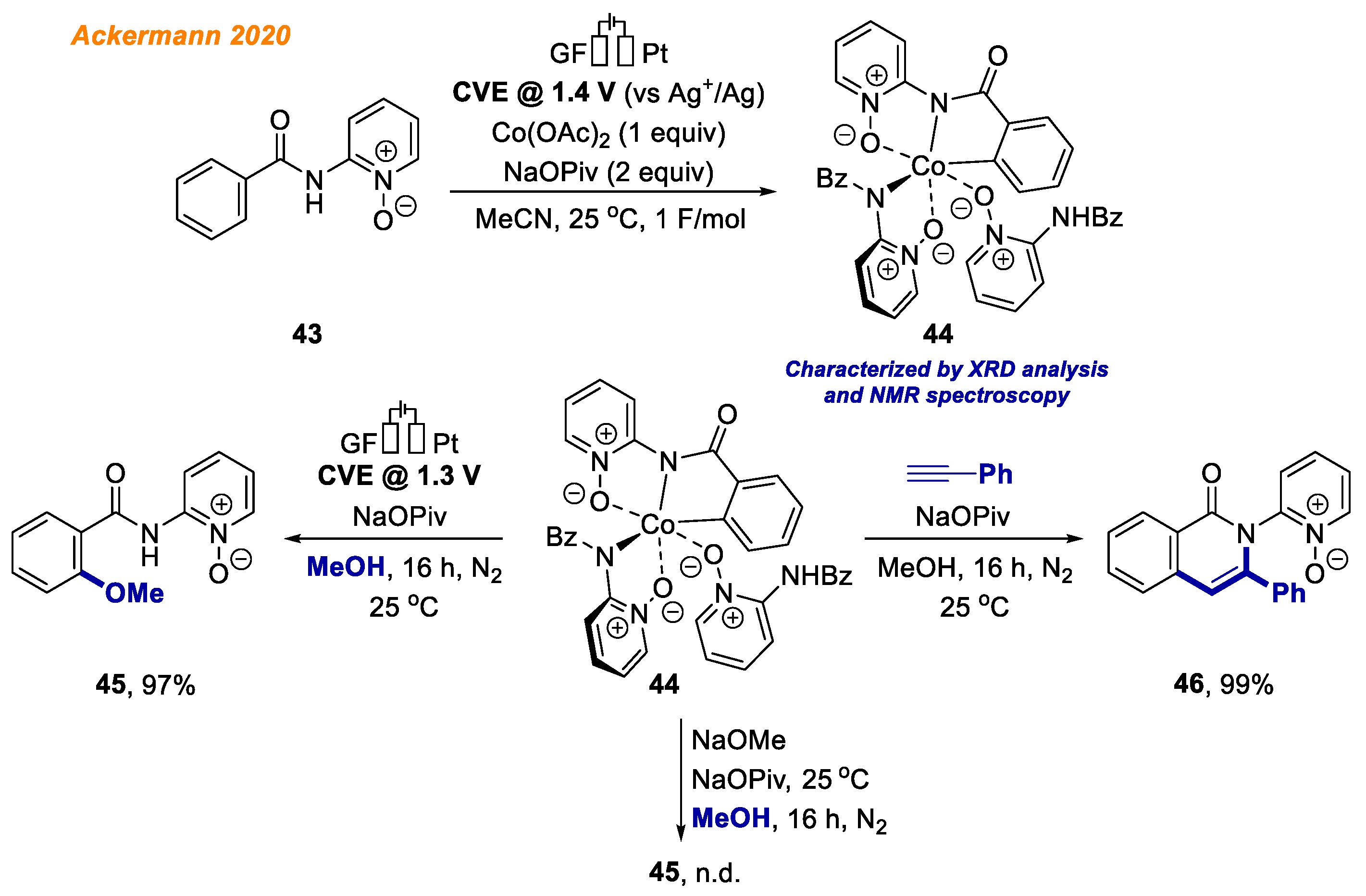
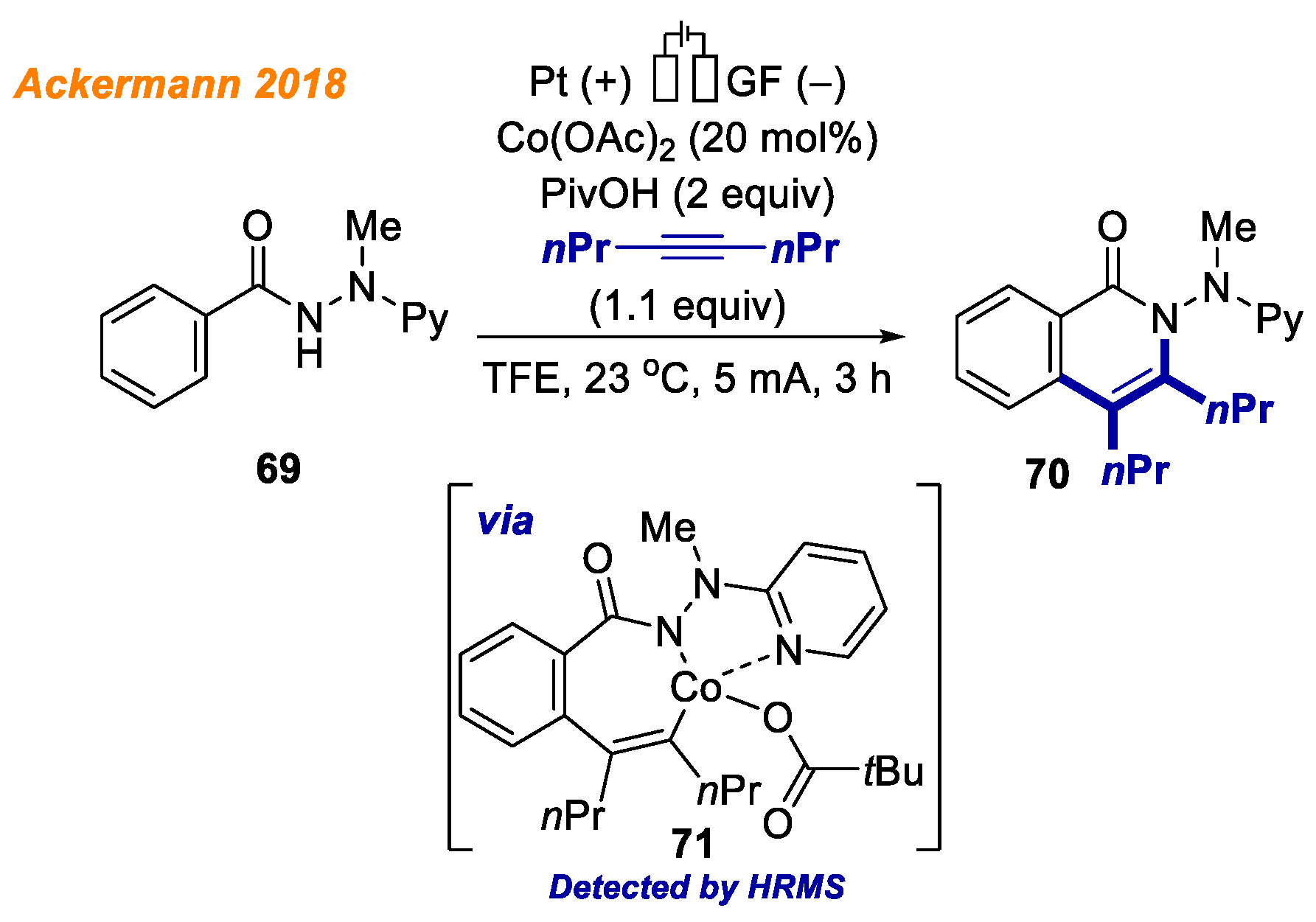
In 2020, the Ackermann group identified and characterized electrochemically generated high valent cobalt (III/IV) complexes as crucial intermediates in electrochemical cobalt-catalyzed C-H bond functionalization reactions (
Scheme 17) [
40].
The envisioned 18e− cobaltacycle 44 was electrochemically synthesized starting from benzamide 43 and an equimolar amount of Co(OAc)2, and its structure was unambiguously confirmed by NMR spectroscopy, ESI-MS, and XRD analysis. Investigating the red-ox potential by means of voltammetry, the authors were able to affirm the anodic generation of Co(IV) complexes. Interestingly, the authors observed the formation of alkoxylated product 45 from Co(III)-aryl complex 44 only when voltage was applied. Such a result supports the oxidation-induced reductive elimination pathway involving Co(IV) species. At the same time, Co(III) complex 44 reaction with phenylacetylene proceeded smoothly, allowing to obtain product 46 in 99% yield. These findings are indicative of different mechanisms being operative for the C-O versus C-H formations.
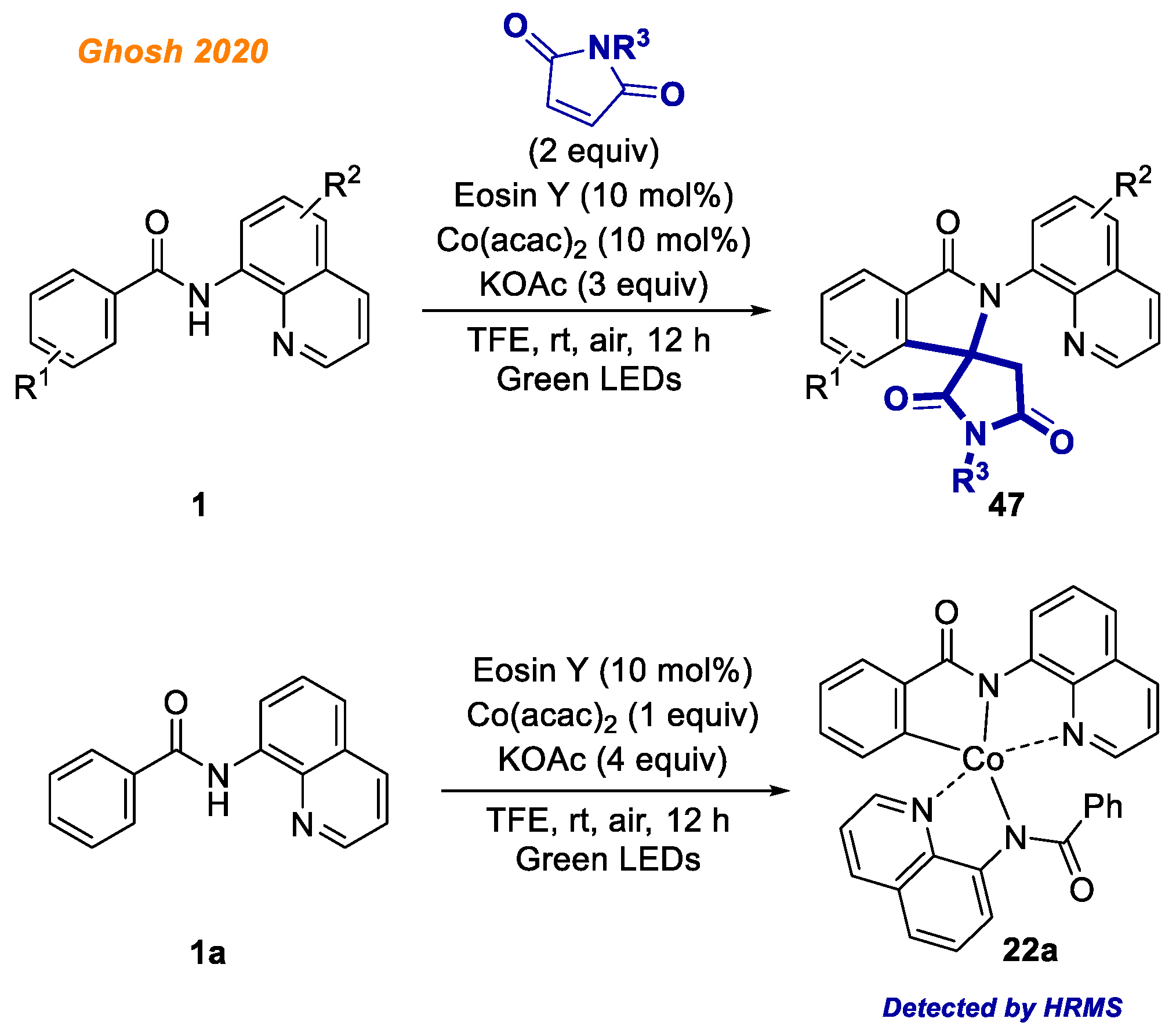
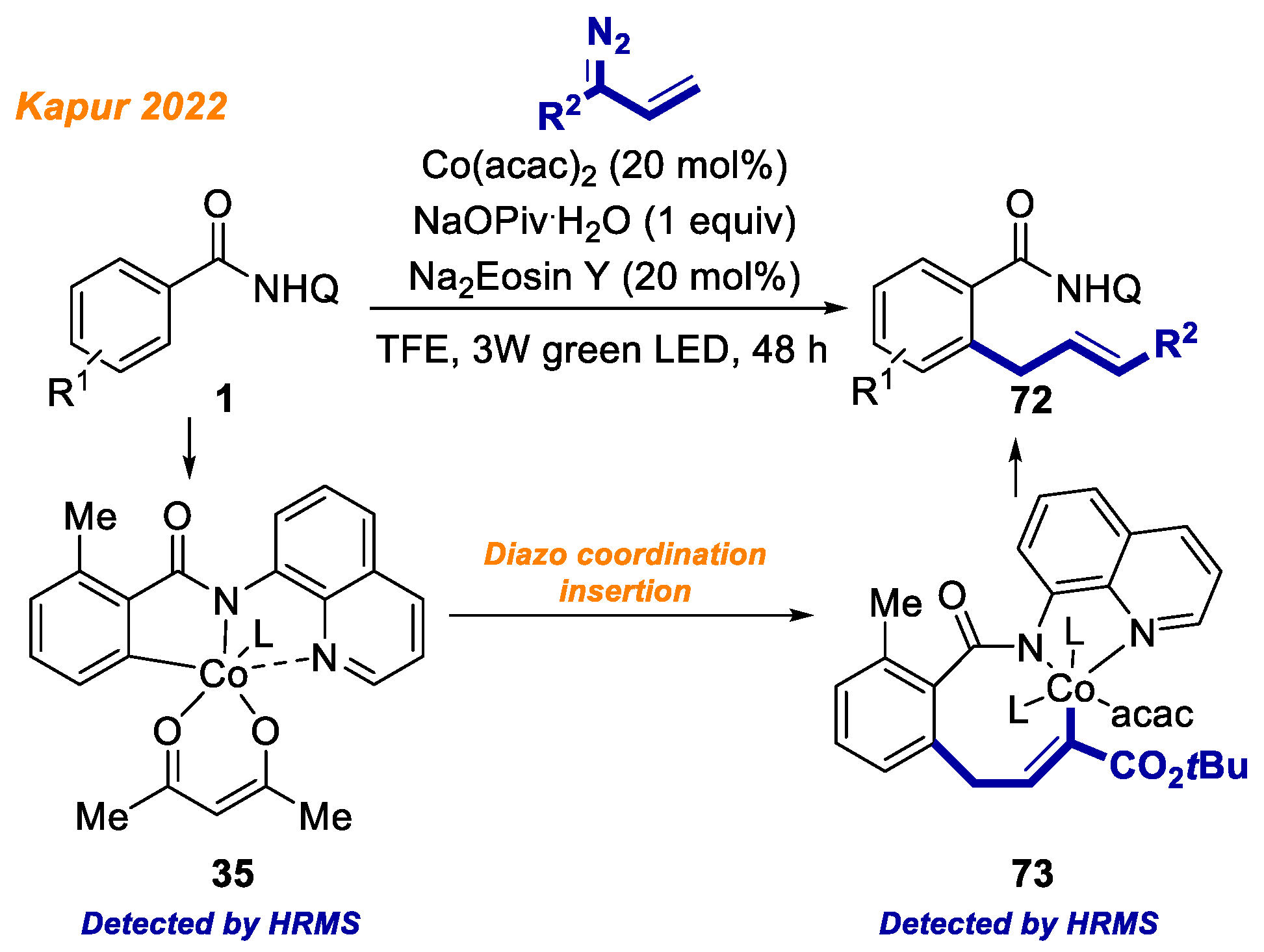
A novel methodology merging visible-light photocatalysis and cobalt catalysis was reported by the Ghosh group in 2020 (
Scheme 18) [
41].
The developed protocol gives an efficient access to isoindolone spirosuccinimides 47 by the oxidative cyclization of benzamides 1 with maleimides. The main advantage of the developed transformation was the use of photocatalyst Eosin Y, a commonly available organic dye, instead of sacrificial metal oxidant to reoxidize the in-situ formed low-valent Co(I) to active high valent Co(II)/Co(III) species to continue the catalytic cycle. Mild reaction conditions tolerated well a broad variety of benzamides 1 containing substituents both in phenyl moiety and in the aminoquinoline directing group moiety, delivering isoindolones 47 with yields up to 92%. It should be noted that authors were able to identify a five-membered Co(III)intermediate 22a by HRMS, which validated the involvement of Co(III) complex 22a in the reaction mechanism, although no further identification of reaction intermediates was performed.
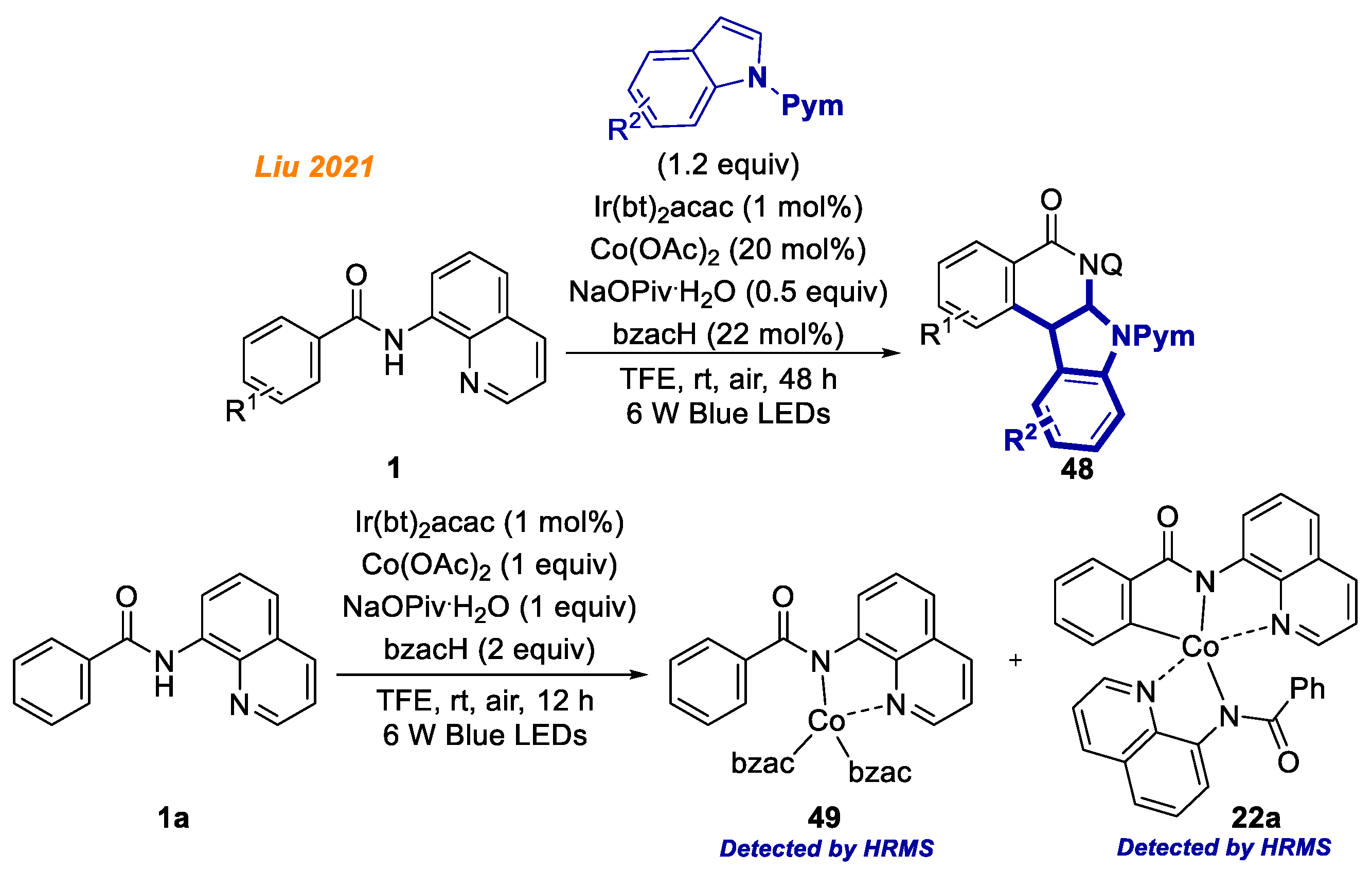
One year later, in 2021, Liu and co-workers depicted metallaphotoredox dearomatization of indoles for the facile generation of indoloisoquinolinones
48 via [4 + 2] annulation reaction with benzamides
1 (
Scheme 19) [
42]. The developed catalytic system employed Co(OAc)
2 catalyst and Ir(bt)
2acac photocatalyst, sodium pivalate additive, and benzoylacetone ligand in TFE at room temperature. Similar to Ghosh’s report [
41], no external oxidant was required due to photocatalytic reoxidation of the catalyst. The substrate scope studies showed that both electron-rich and electron-poor substrates displayed similar reactivity and gave the products mostly in good to excellent yield (up to 92%). Control experiments revealed the involvement of single electron transfer processes, as with the addition of radical scavengers, the generation of product was entirely suppressed. To gain insight into the mechanism, the authors performed deuterium-labeling experiments, as well as studied KIE of the reaction. In addition, the authors synthesized Co(bzac)
3, which was found to be catalytically competent. Moreover, two Co(III) complexes
49 and
22a were detected by ESI-HRMS, establishing the intermediacy of the related Co(III) system in the present reaction.
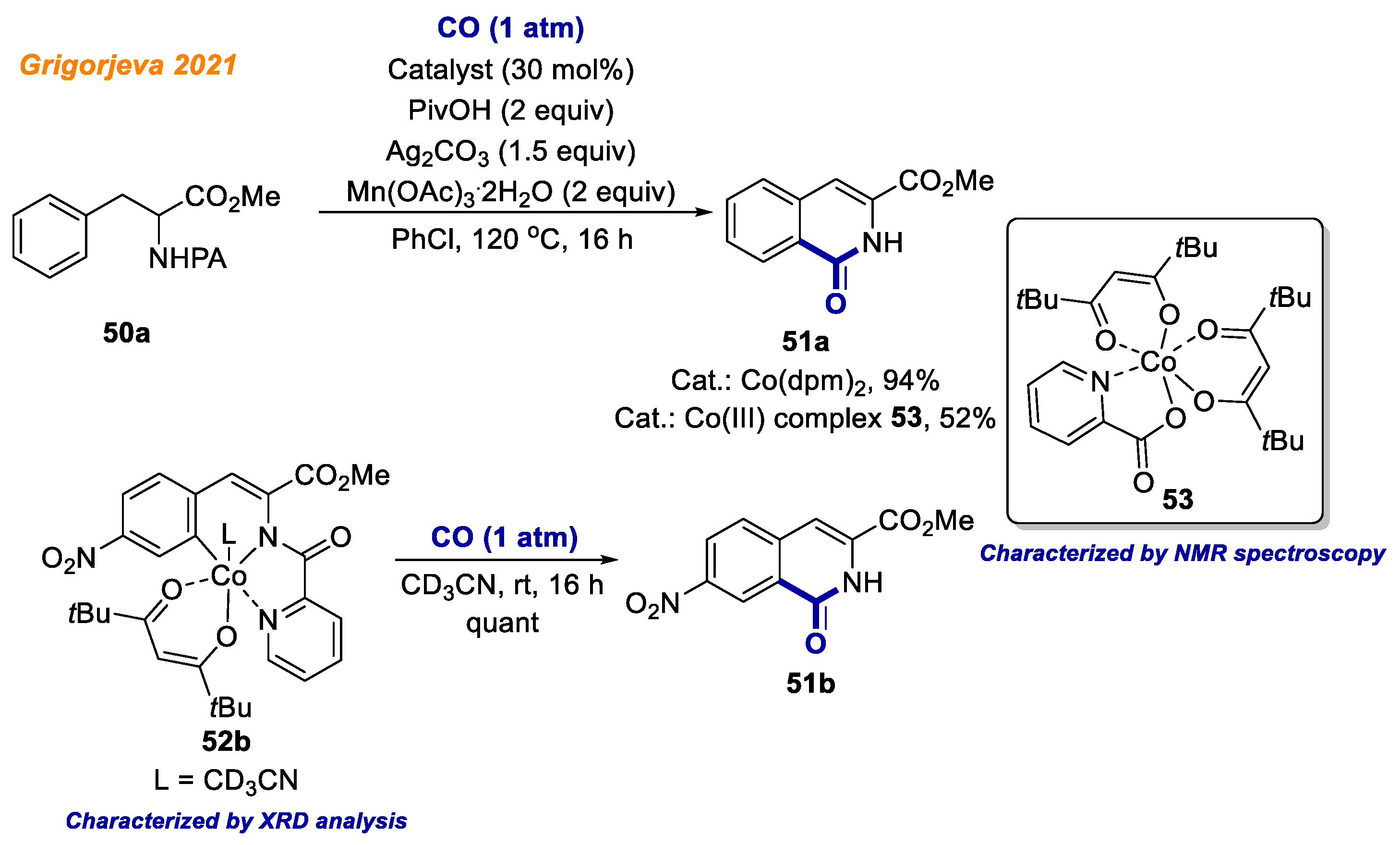
In the same year, Grigorjeva and co-workers reported an efficient methodology for the synthesis of dihydroisoquinolinolones
51 via cobalt catalyzed C-H bond carbonylation of phenylalanine derivatives
50 (
Scheme 20) [
43]. Interestingly, in this transformation, the picolinamide directing group revealed its traceless nature as it was cleaved in situ under the reaction conditions. The authors were able to demonstrate a diverse substrate scope, delivering the desired
N-unsubstituted cyclization products
51 with moderate to excellent yields (up to 95%). Furthermore, it was shown that the developed methodology may be applicable to a late-stage functionalization of short peptides, although partial racemization of some products was observed. Besides, under the standard reaction conditions, authors were able to isolate the key intermediates, including Co(III) complex
53. The employment of Co(III) intermediate
53 as the reaction catalyst gave product
51a in 52% yield. In addition, C-H activated Co(III)-aryl intermediate
52b was successfully isolated, and its structure was unambiguously confirmed by XRD analysis. Pleasingly, under CO (1 atm) at room temperature, cobaltacycle
52b quantitatively formed C-H carbonylation product
51b, which supported the author’s conclusion this complex most likely was the key intermediate of the reaction.
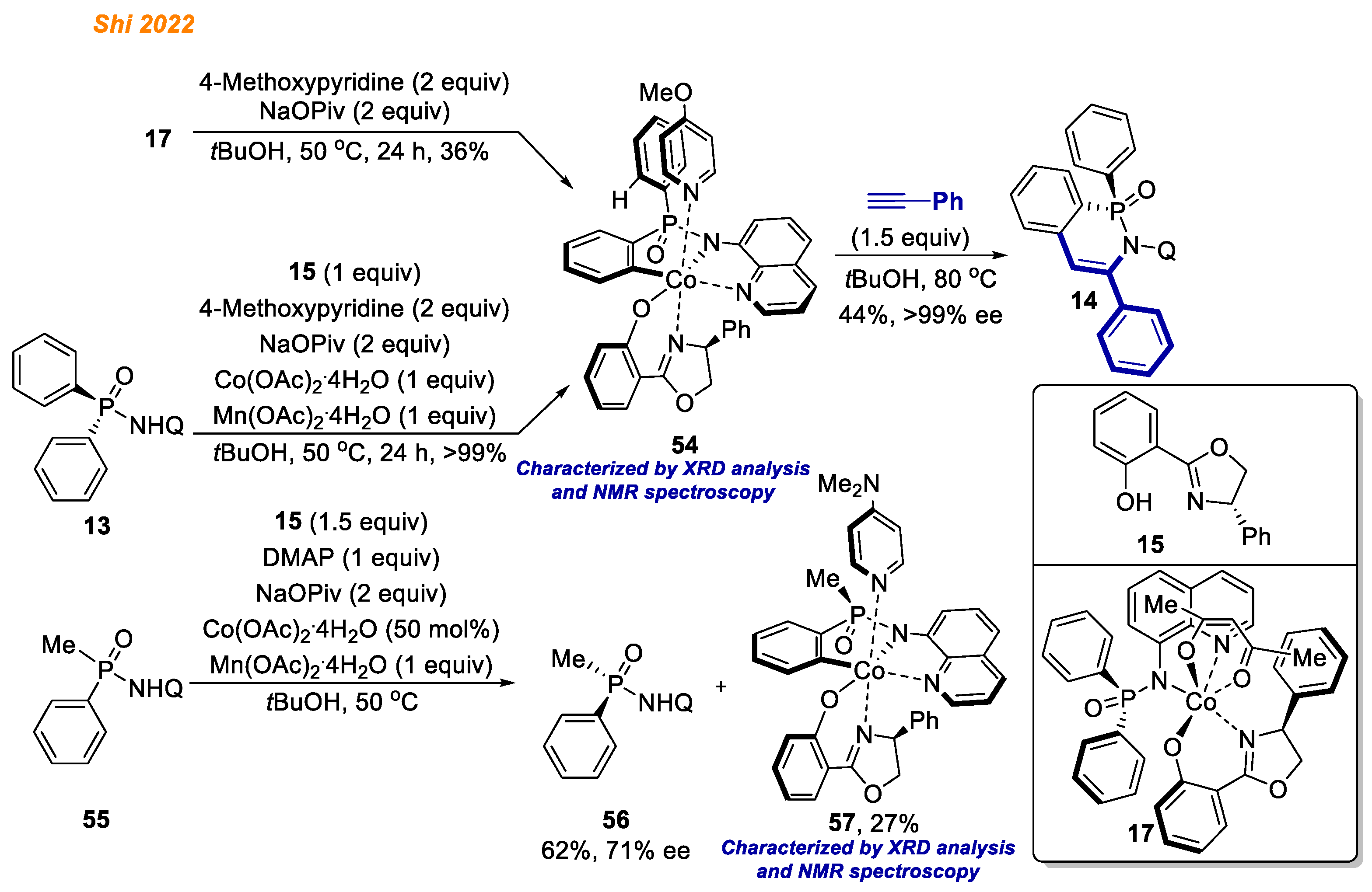
In 2022, along with cobalt complexes formed in ligand exchange/oxidation step, the Shi group were able to synthesize and isolate C-H activated Co(III)-aryl intermediates (
Scheme 21) [
28]. For example, the reaction of previously isolated Co(III) complex
17 was conducted in the presence of NaOPiv and 4-methoxypyridine as a neutral ligand to stabilize the resulting cobalt complex. As a result, Co(III)-aryl complex
54 was obtained in 36% yield as a single octahedral diastereomer via asymmetric C-H activation step.
Alternatively, cobalt(III) intermediate 54 was also obtained in quantitative yield directly from phosphinic amide 13 by the reaction with equimolar amount of ligand 15, Co(II) acetate, oxidant, NaOPiv, and 4-methoxypyridine as a stabilizing ligand. Notably, the configuration of azaphosphinine oxide 14 matches the stereochemistry of phosphorus center in 17, which led authors to consider enantio-determining C-H bond cleavage. Regarding this, authors performed stoichiometric kinetic resolution of racemic 55. It was revealed that only one enantiomer gave corresponding Co(III) intermediate 57, which further suggested that the chirality of the phosphorus center was established through enantio-determining C-H bond cleavage.
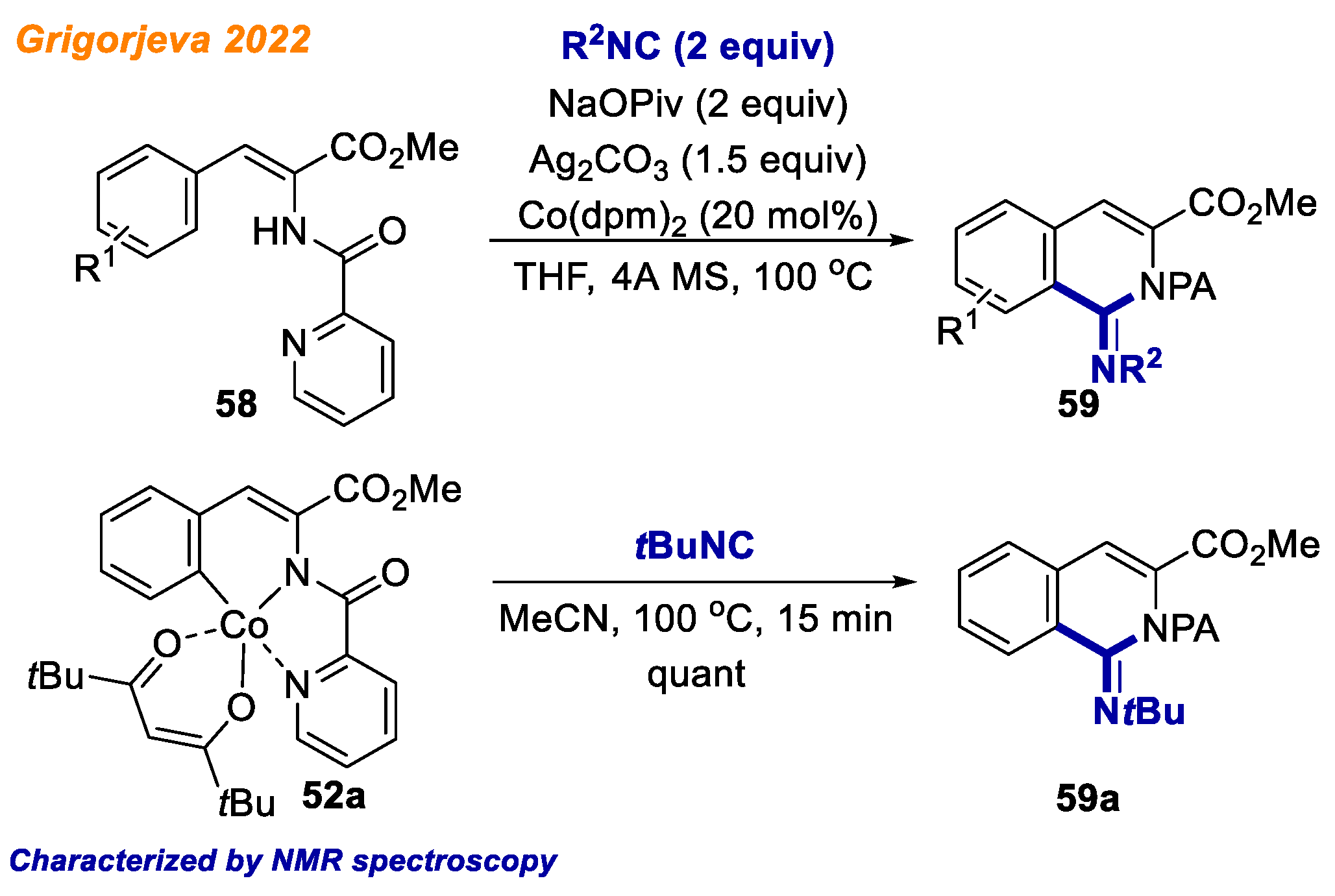
As a continuation of their previous work on amino acid C-H functionalization, in 2022, Grigorjeva and co-workers reported novel protocol for the cobalt-catalyzed C-H bond imination of α,β-unsaturated phenylalanine derivatives
58 using isocyanides (
Scheme 22) [
44]. The substrate scope was explored using several α,β-unsaturated phenylalanines
58 and different isocyanides, delivering 26 different iminoisoquinolines
59 in good to excellent yields (up to 96%). Although picolinamide did not act as a traceless directing group, as shown in their previous report [
43], it can be easily cleaved under the reductive conditions using LiAlH
4 or Zn/AcOH. In addition, the developed methodology was applied for the synthesis of PDE5 inhibitor. In order to gain insight into the reaction mechanism, the authors performed series of control experiments, including ligand exchange and competition experiments, H/D scrambling and KIE studies. Besides, a stoichiometric experiment with Co(III)-aryl complex
52a was performed. Interestingly,
52a in the reaction with
tert-butyl isocyanide gave product
59a in quantitative NMR yield in the absence of external oxidant, indicating that
59a is very likely the intermediate of proposed catalytic cycle.
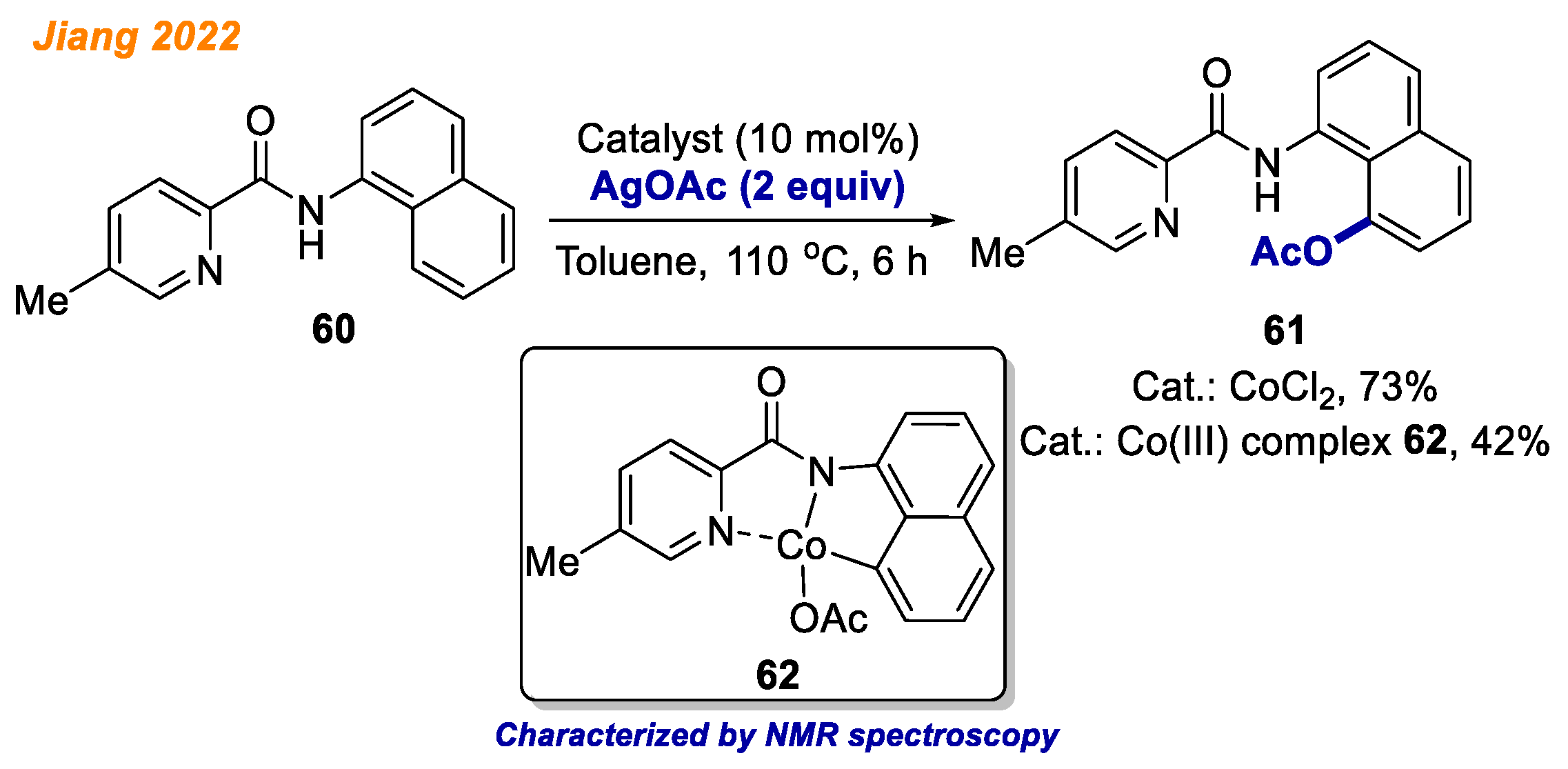
In the same year, Jiang and colleagues demonstrated an interesting approach towards C-H bond acyloxylation of picolinamides
60 with silver carboxylates under cobalt catalysis (
Scheme 23) [
45]. Generally, substituted silver carboxylates and substituted picolinamides
60 were reactive, delivering a very wide and diverse product scope consisting of 73 different products with good to excellent yields (up to 94%). Investigation of the reaction mechanism by means of KIE studies led to the conclusion that C-H bond activation may not be the turnover-limiting step. Furthermore, authors were able to obtain chelated Co(III)-aryl complex
62, which was accurately characterized by NMR and HRMS analyses. The employment of Co(III) intermediate
62 as the reaction catalyst gave the desired product
61 in 42% yield, whereas under standard reaction conditions using CoCl
2 catalyst, it was obtained in 73% yield. From these results, the authors concluded that complex
62 was the key intermediate in the developed C-H bond acyloxylation reaction.
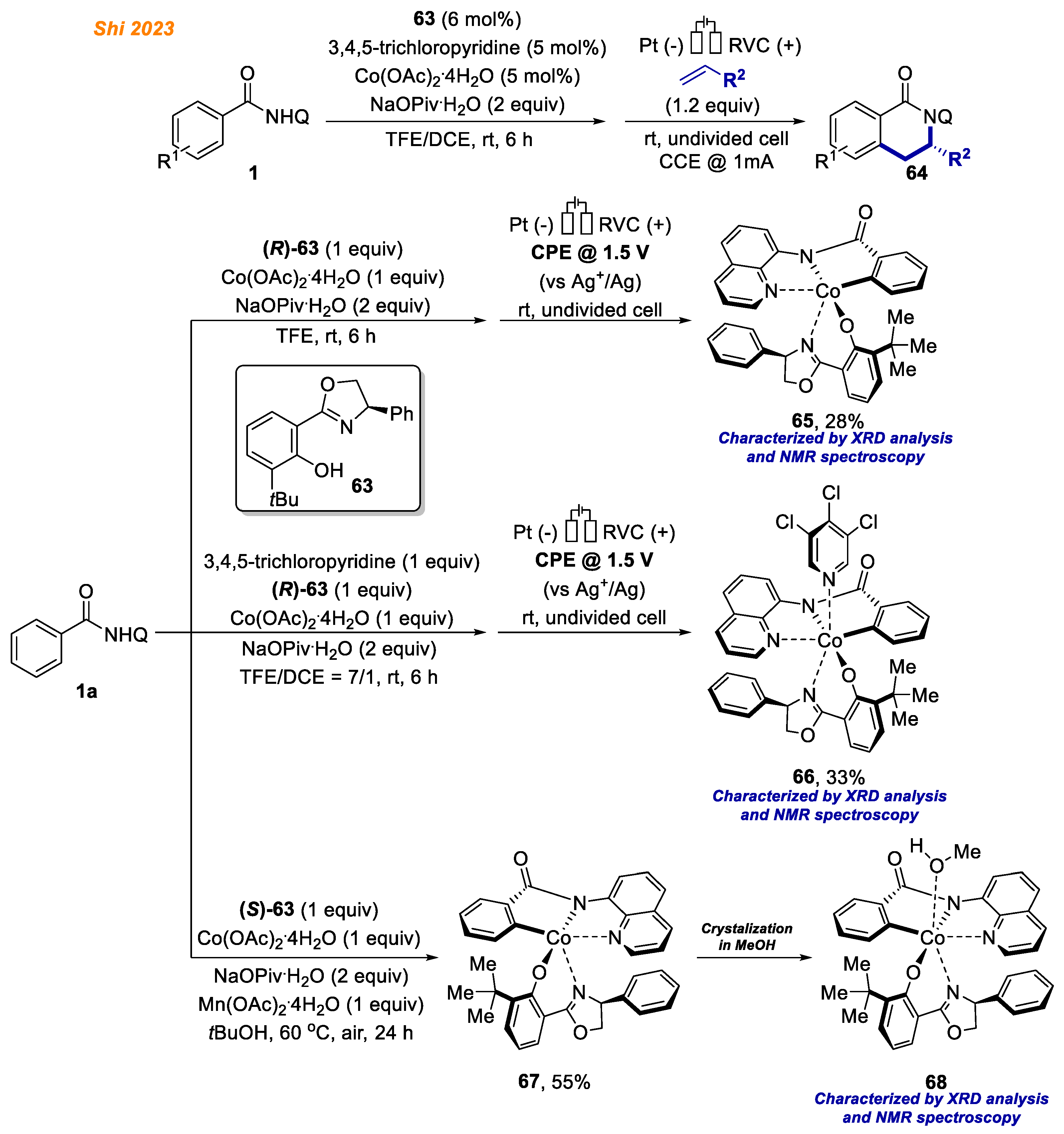
Very recently, an enantio- and regioselective electrooxidative cobalt-catalyzed C-H/N-H annulation reaction with alkenes was developed by Shi and co-workers (
Scheme 24) [
46]. In their report, π–π interactions between the phenyl ring in the oxazoline ligand
63 and the quinoline moiety of the benzamides
1 secured the chirality at cobalt, leaving chiral cave in one direction open for alkene coordination, facilitating the formation of annulation products
64 in high enantio- and regioselectivities (up to 99%
ee).
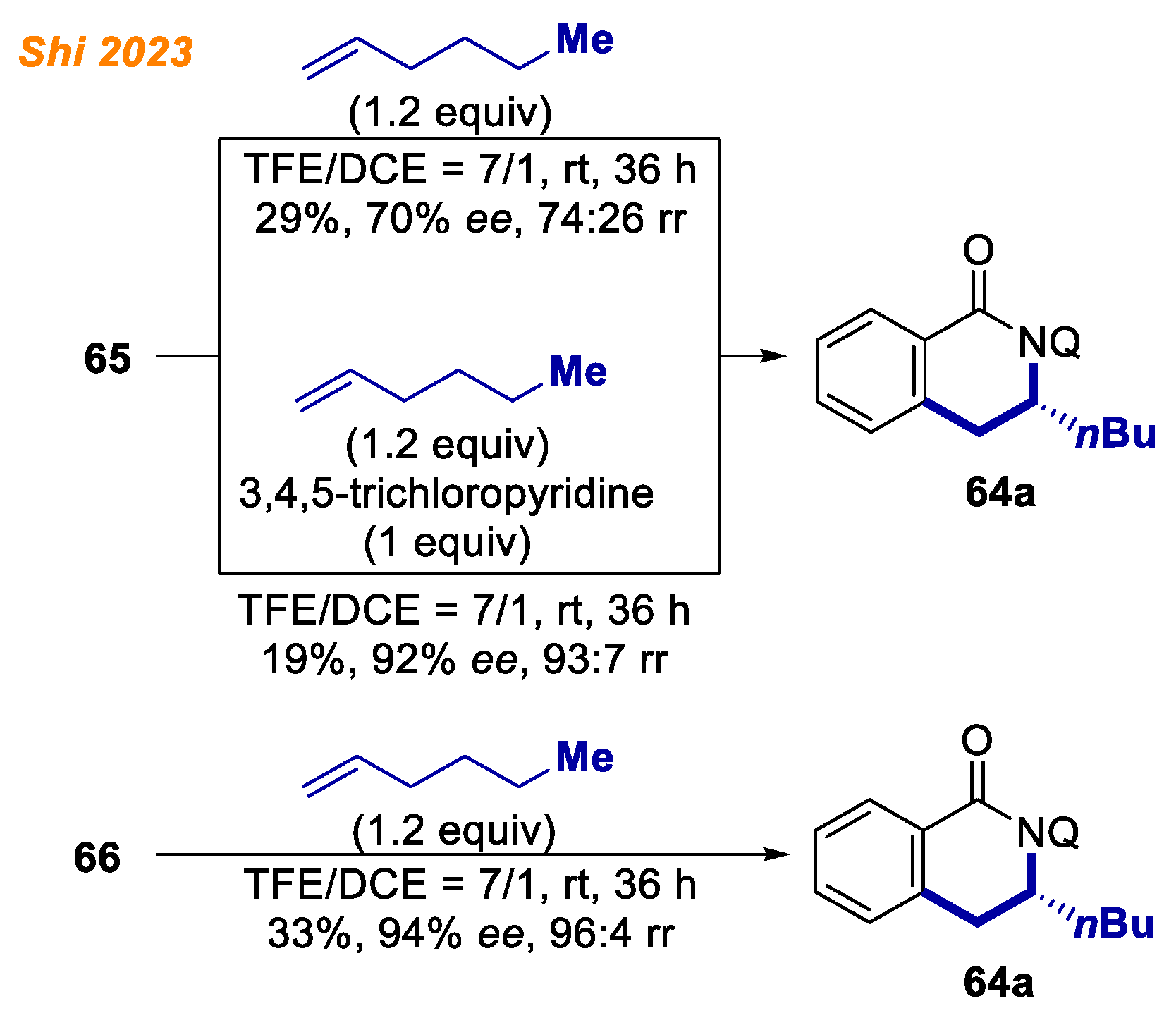
Additionally, octahedral Co(III) complexes were synthesized and characterized to understand the coordination fashion of cobalt catalyst and the mode of stereoinduction. Benzamide 1a in the reaction with an equimolar amount of Co(OAc)2·4H2O and oxazoline ligand (R)-63 under electrolysis conditions provided penta-coordinated Co(III)-aryl complex 65 in 28% yield. The addition of 3,4,5-trichloropyridine as a coordinative ligand under similar reaction conditions provided hexa-coordinated Co(III)-aryl complex 66 in 33% yield. The authors note that both 65 and 66 were stable at ambient temperature and were fully characterized by NMR and ESI-MS analyses. However, attempts to obtain single-crystals for X-ray diffraction analysis were unsuccessful. On the other hand, under thermal conditions, using oxazoline ligand (S)-63 analogous penta-coordinated Co(III)-aryl, complex 67 was obtained in 55% yield. Complex 67 crystallization from MeOH gave Co(III) complex 68, whose structure was unambiguously confirmed by XRD analysis.
Interestingly, the stoichiometric reaction of Co(III)-aryl complex
65 with hex-1-ene yielded product
64a with moderate enantioselectivity and poor regioselectivity, which was attributed to the lack of secondary bond interaction (
Scheme 25) [
46]. The authors found, that enantio- and regioselectivities reappeared with the addition of 1 equivalent of 3,4,5-trichloropyridine, which acted as a coordinative ligand. Moreover,
hexa-coordinated Co(III)-aryl complex
66 gave product
64a with excellent enantioselectivity and regioselectivity. These results led authors to conclusion that the combination of oxazoline and pyridine ligands is essential to ensure high enantio- and regio- selectivity for the developed methodology.



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}













