
The Electronic Nature of Cationic Group 10 Ylidyne Complexes


Abstract
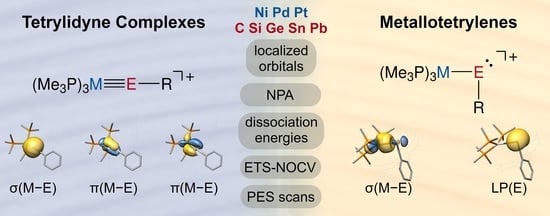
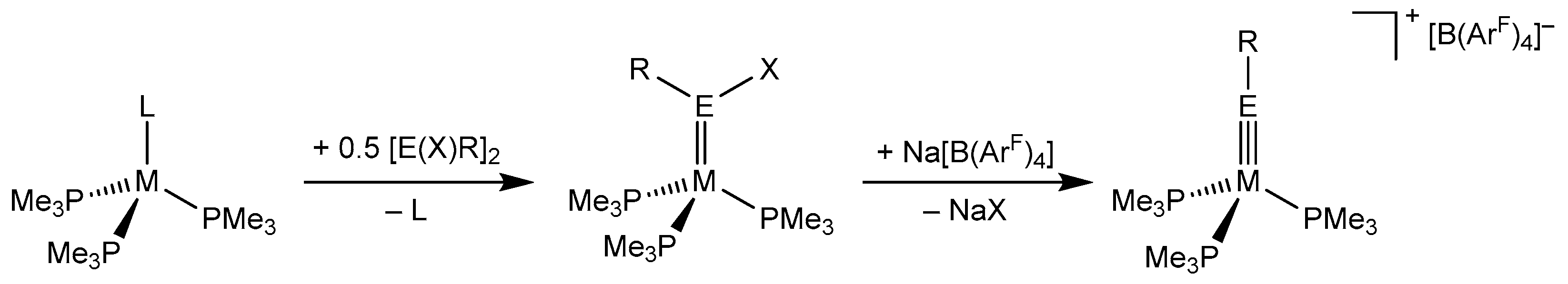
1. Introduction
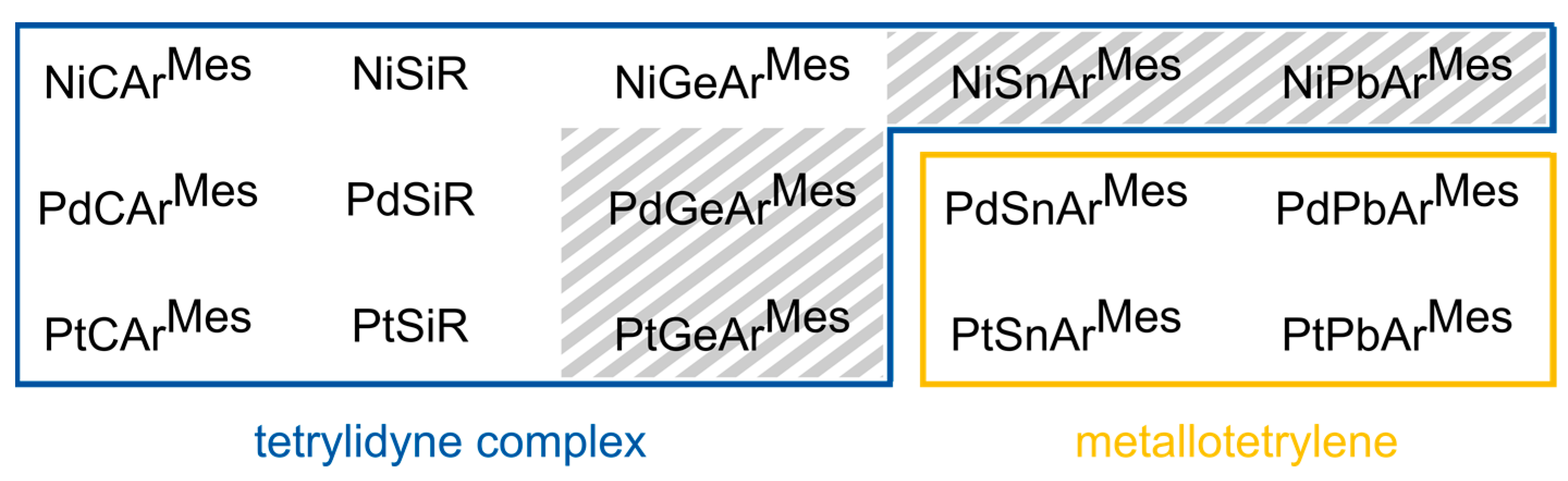
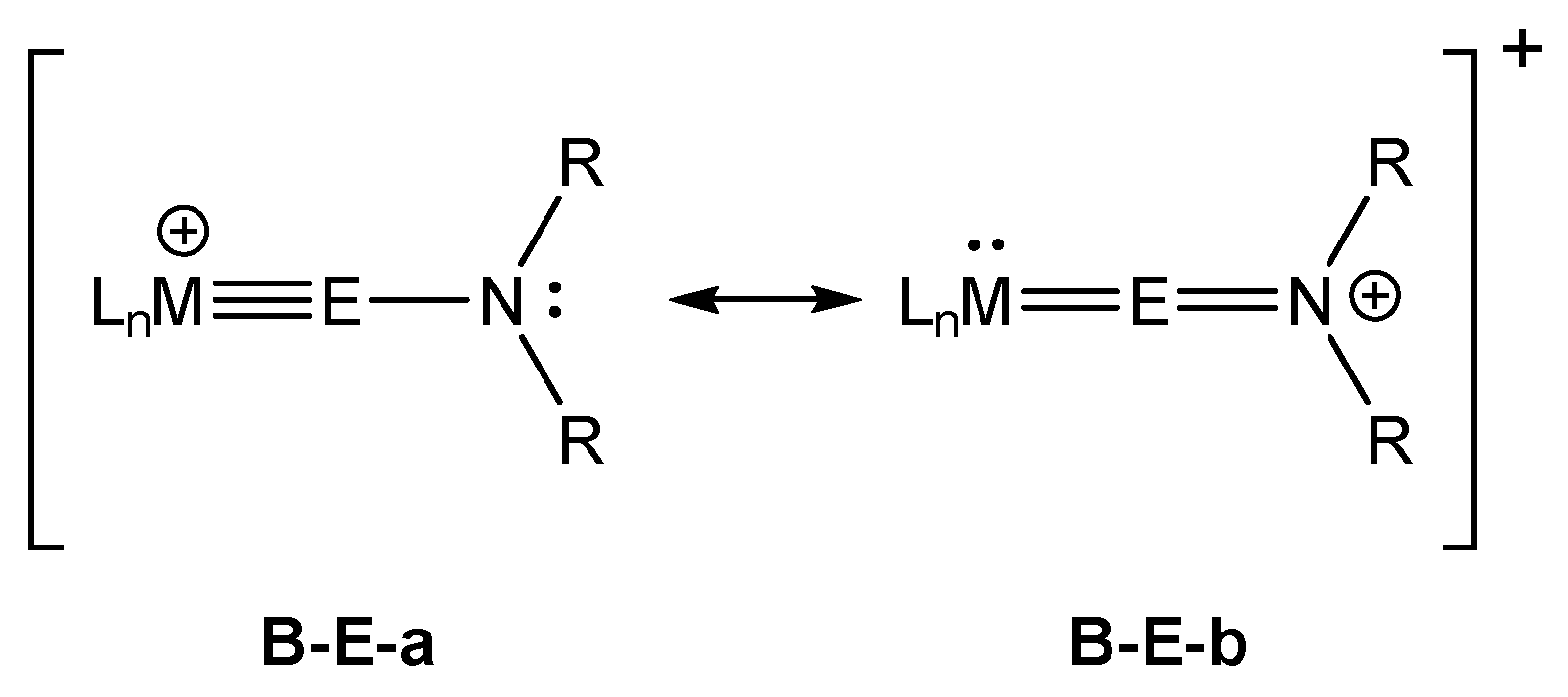
2. Results and Discussion
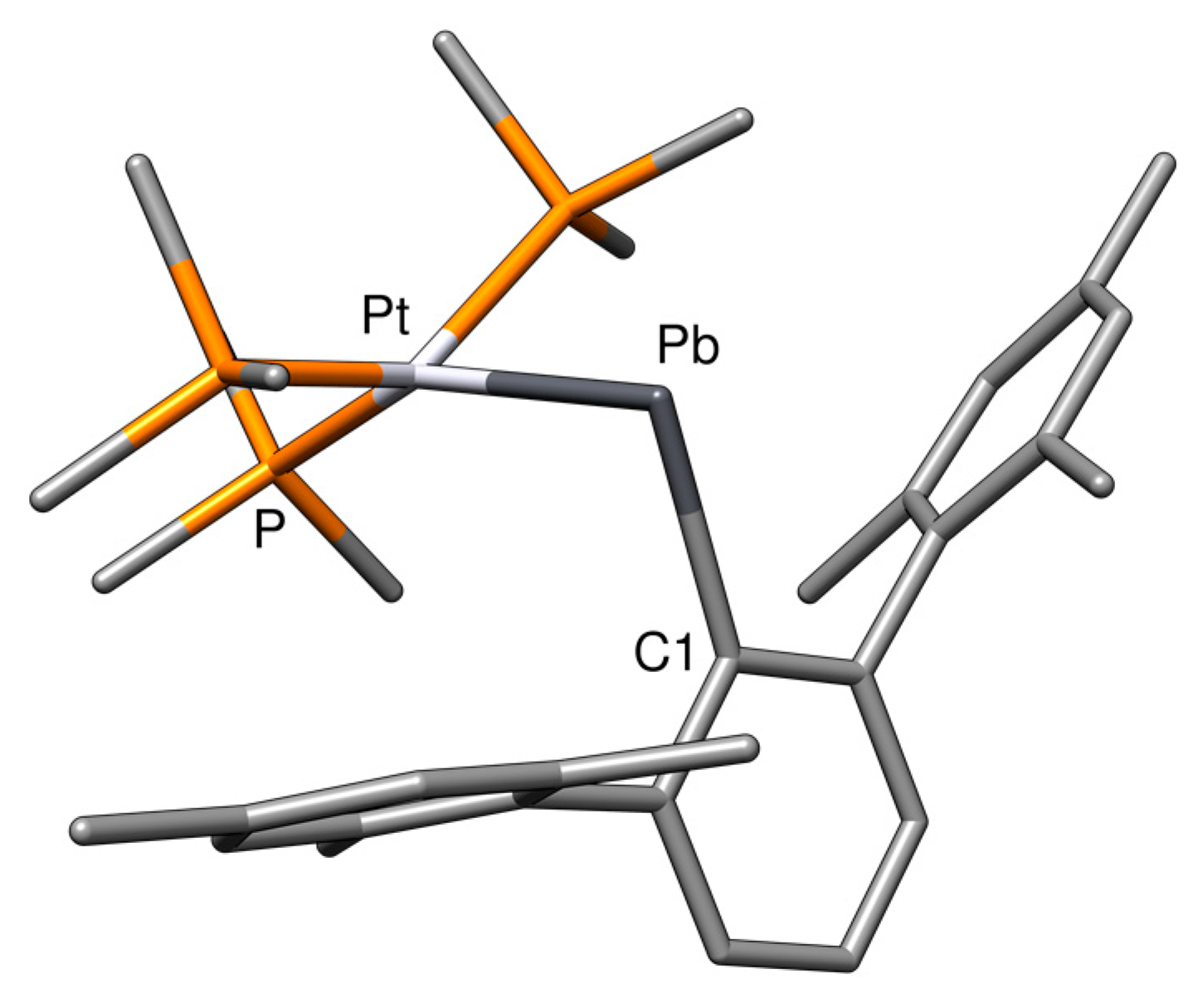
2.1. Structural Results
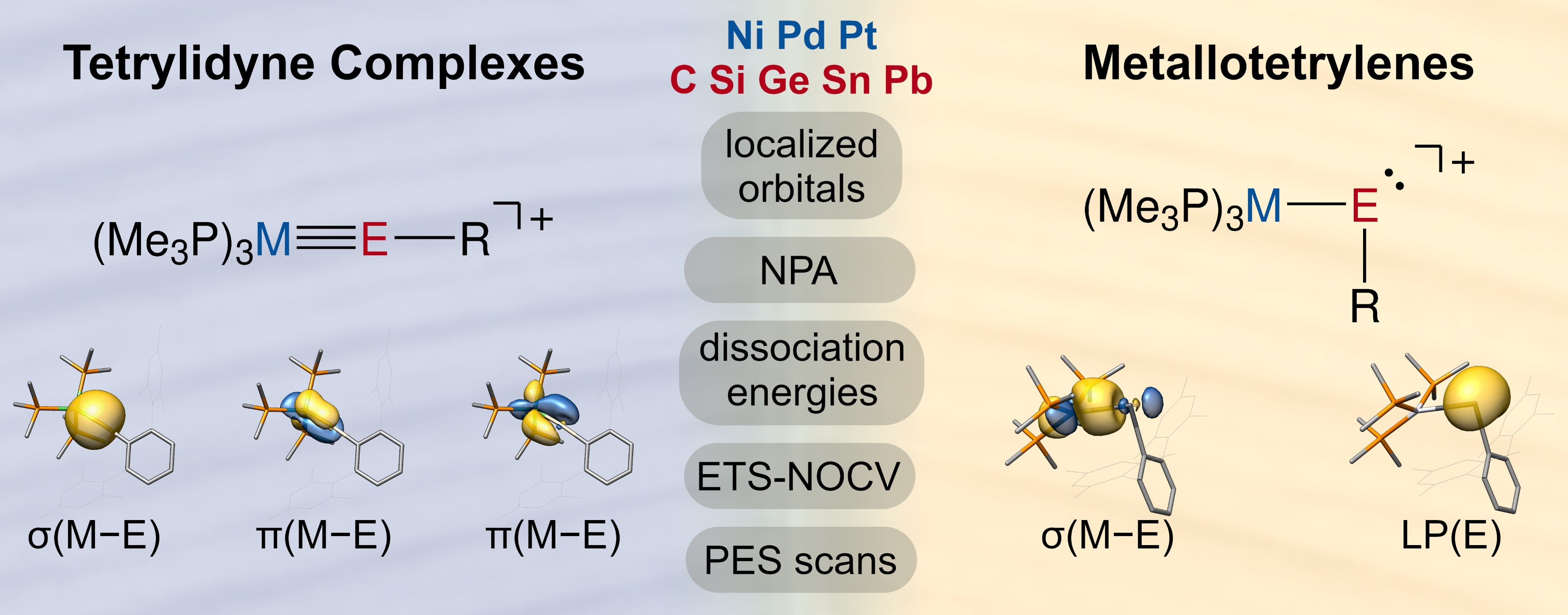
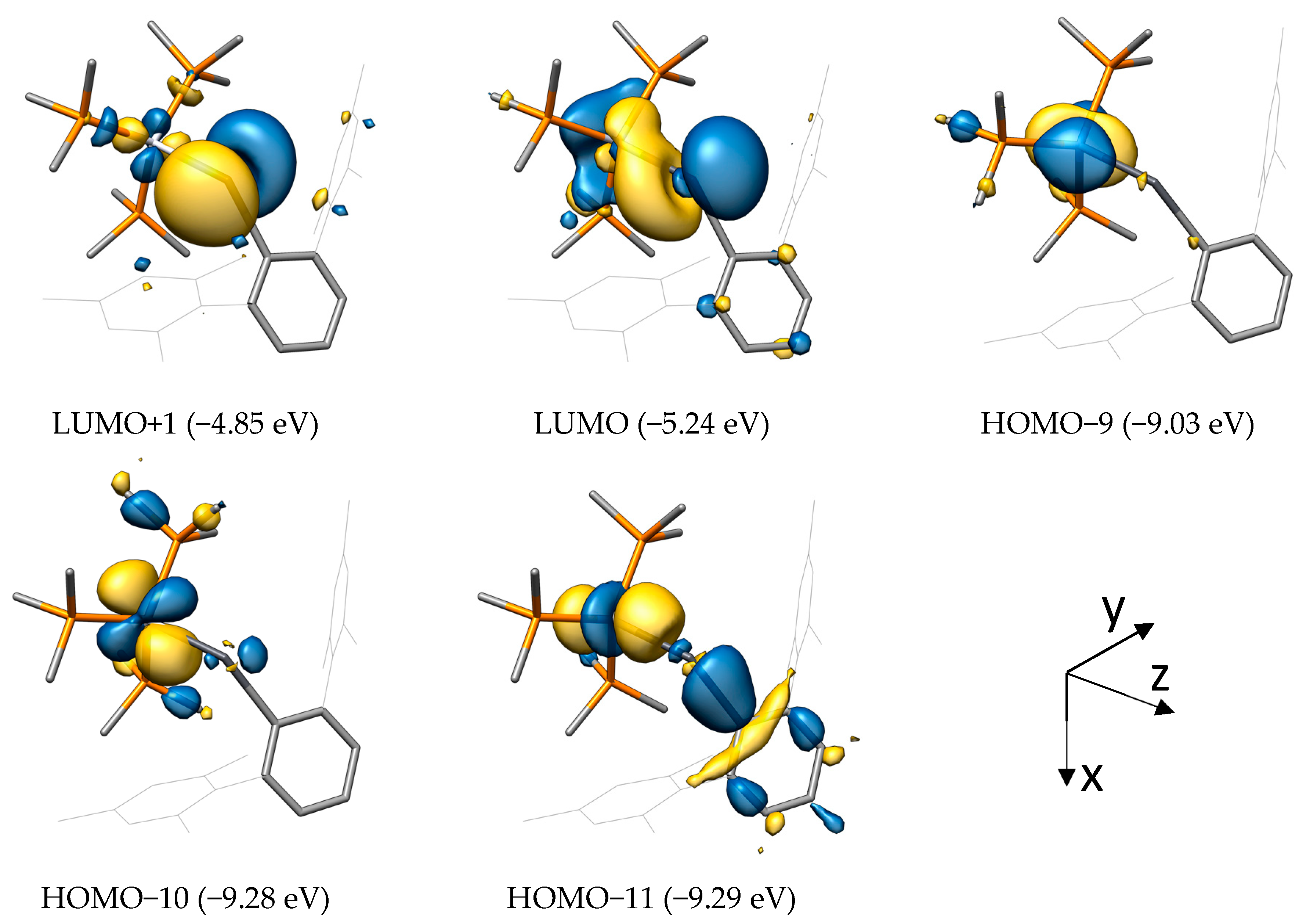
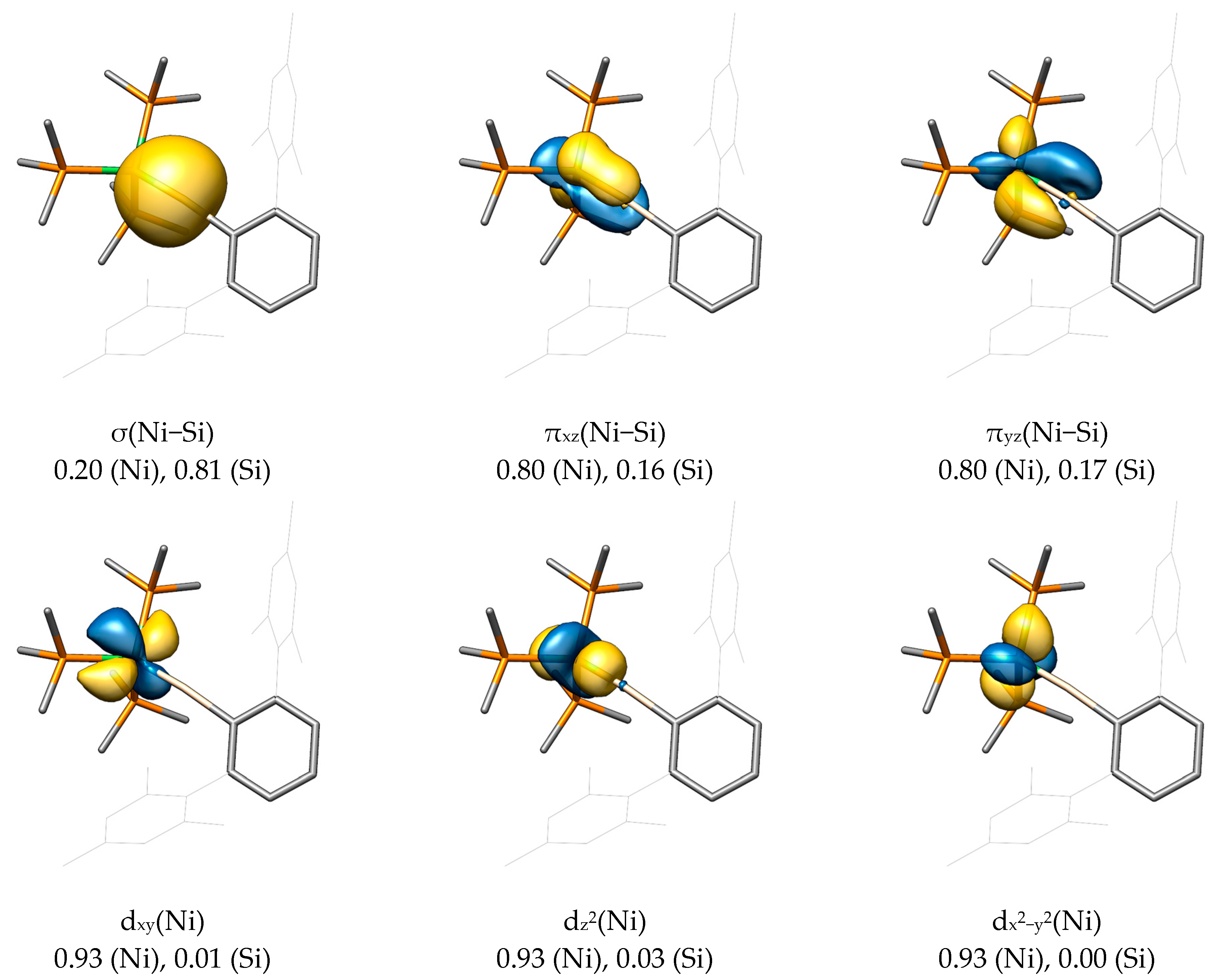
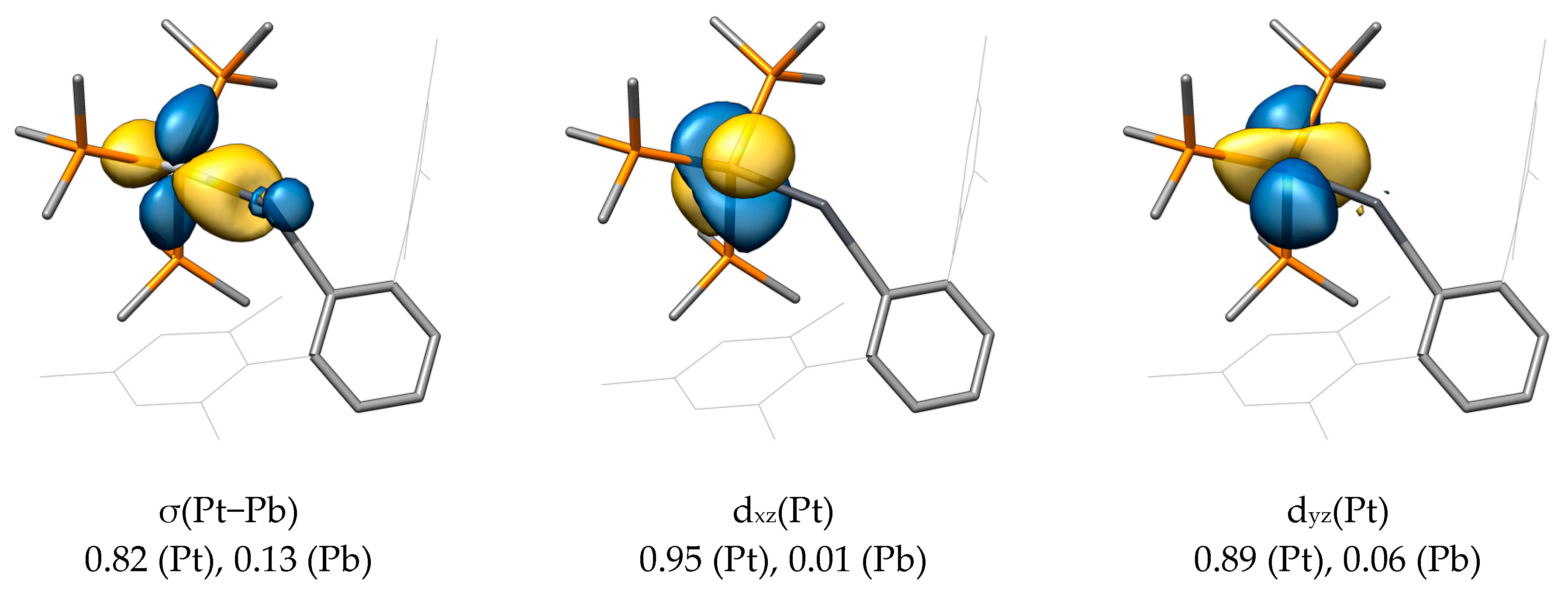
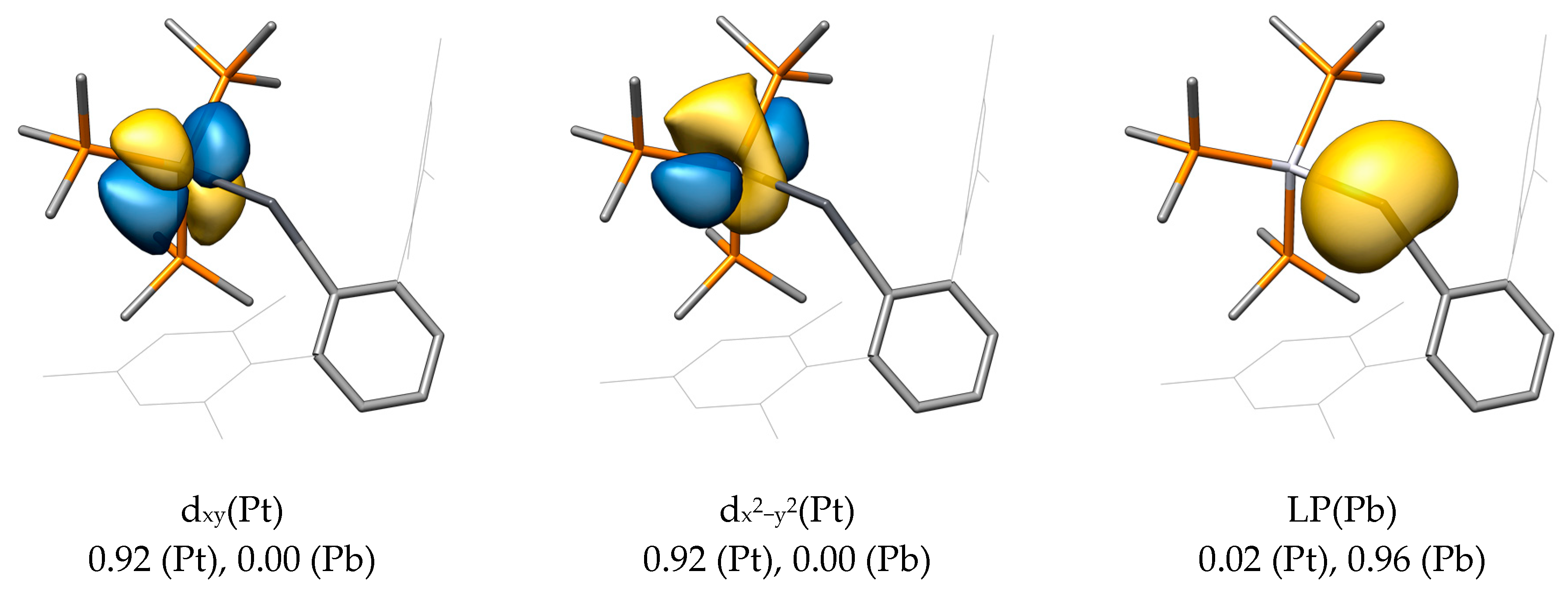
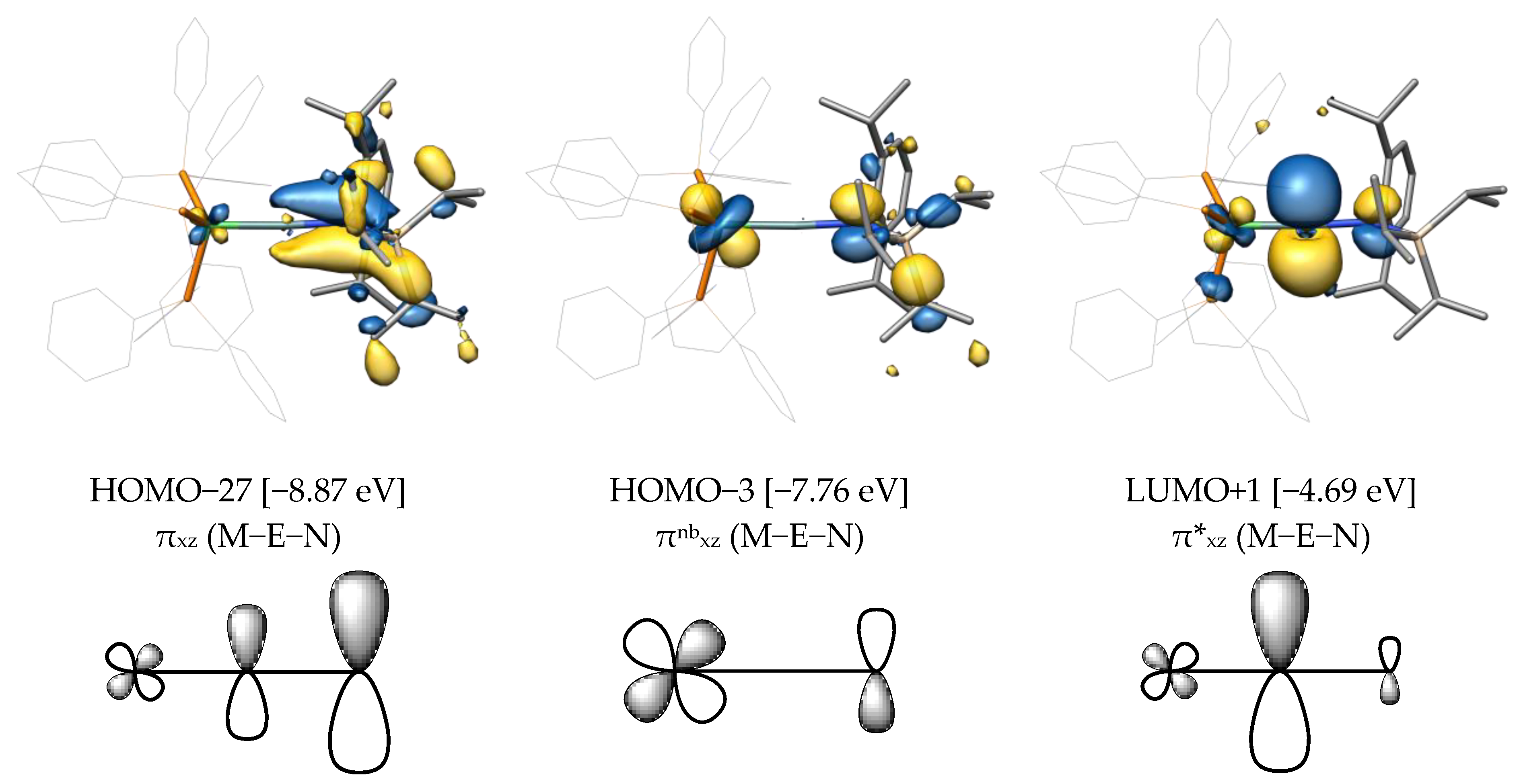
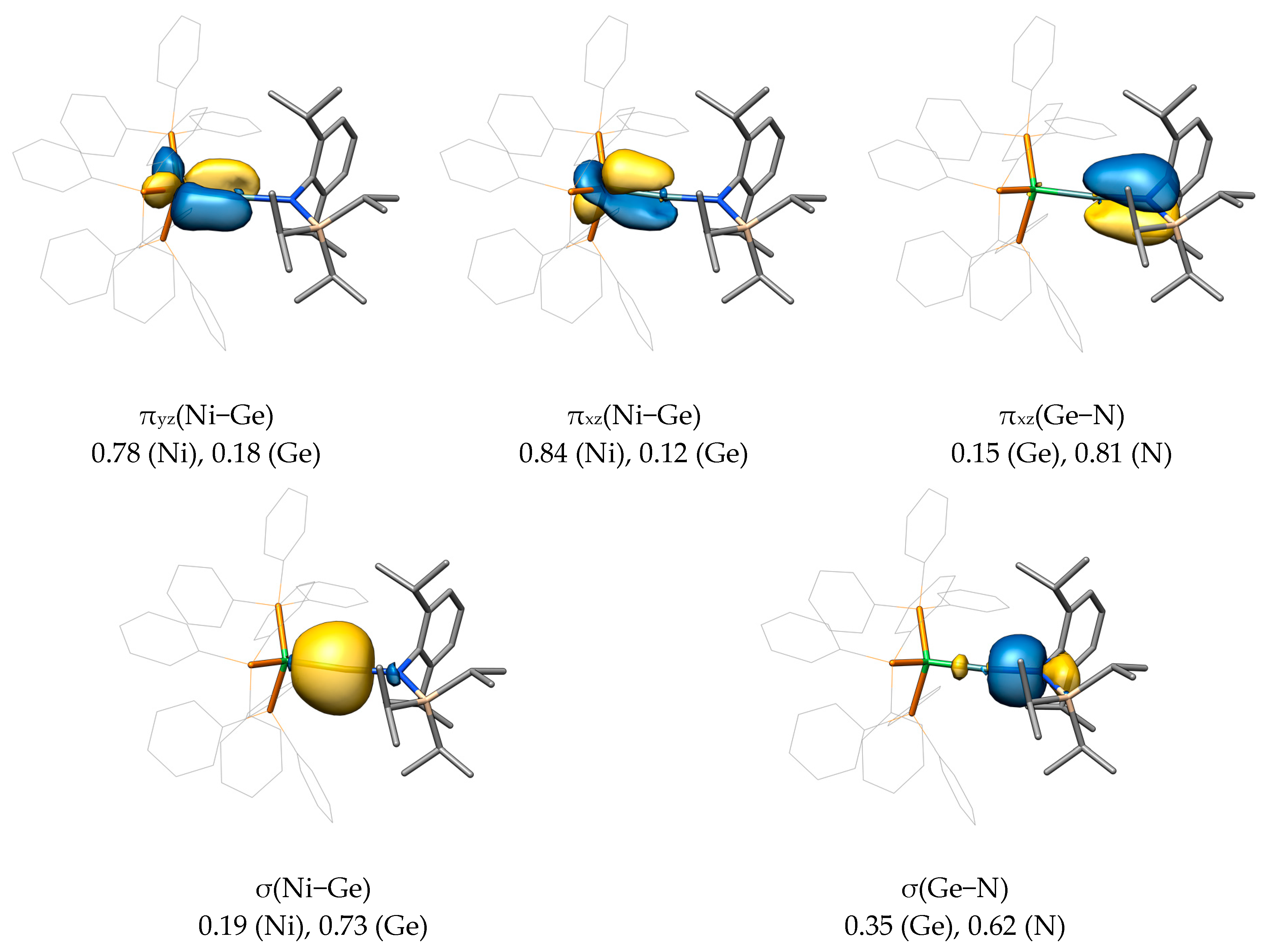
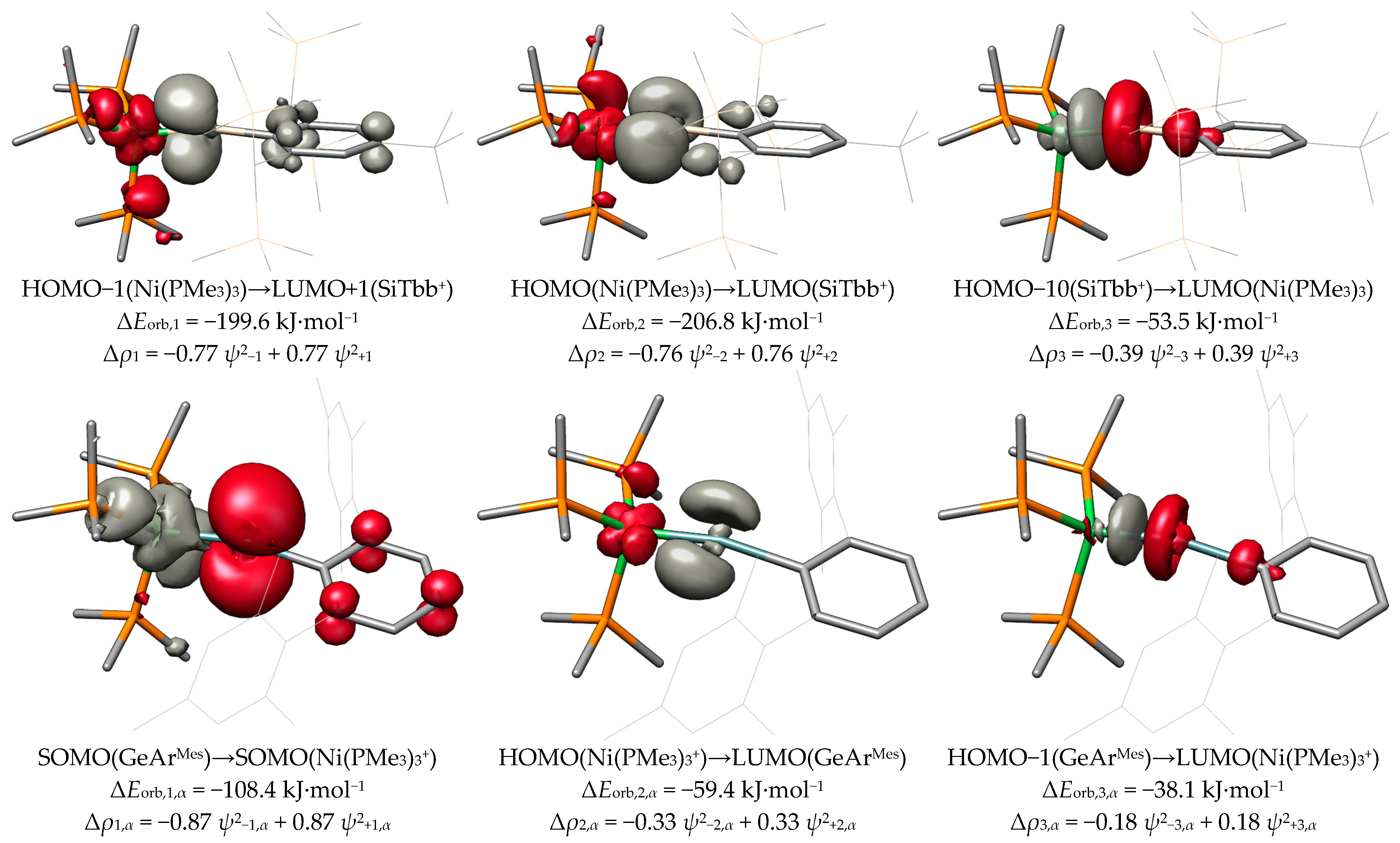
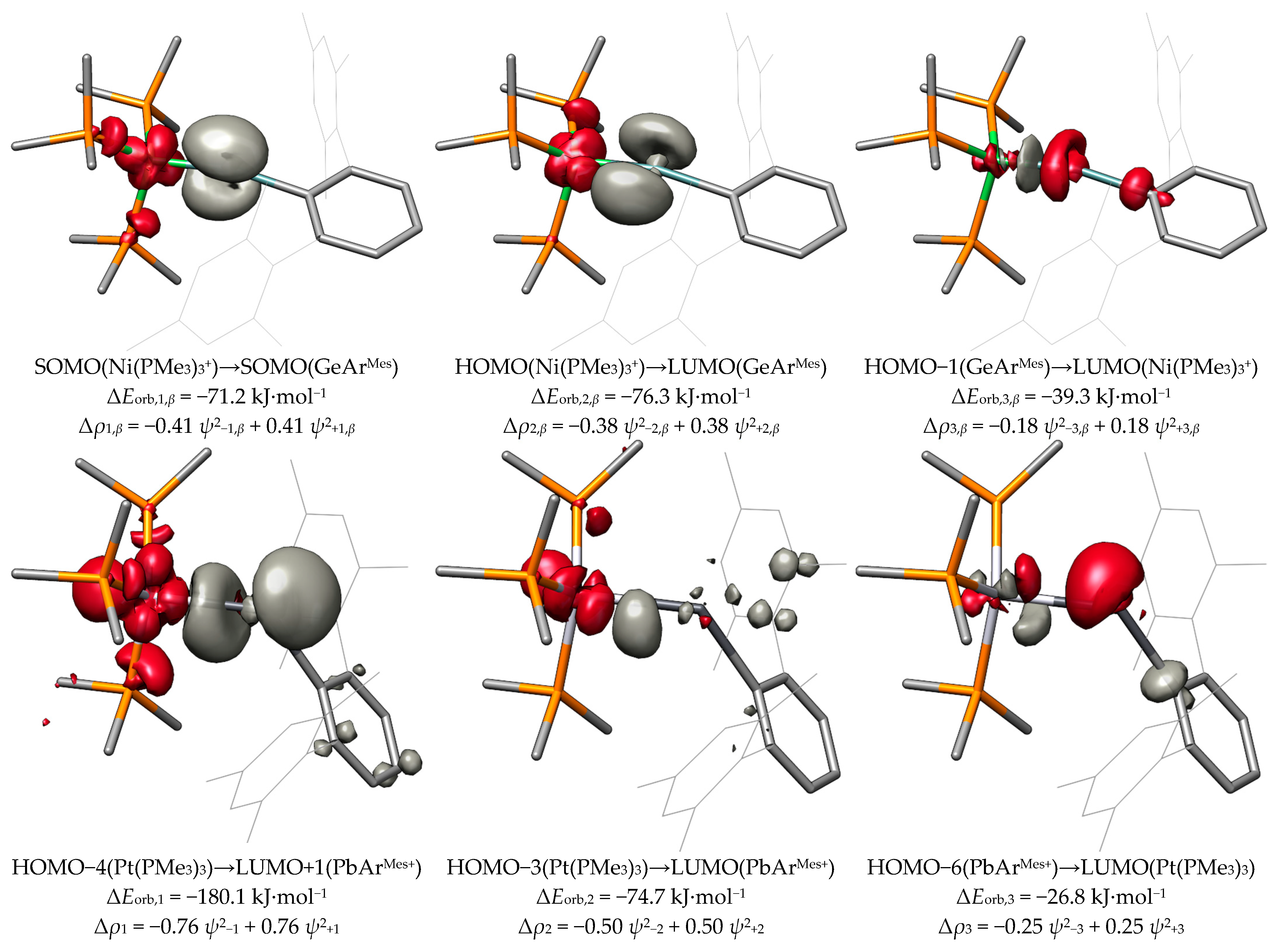
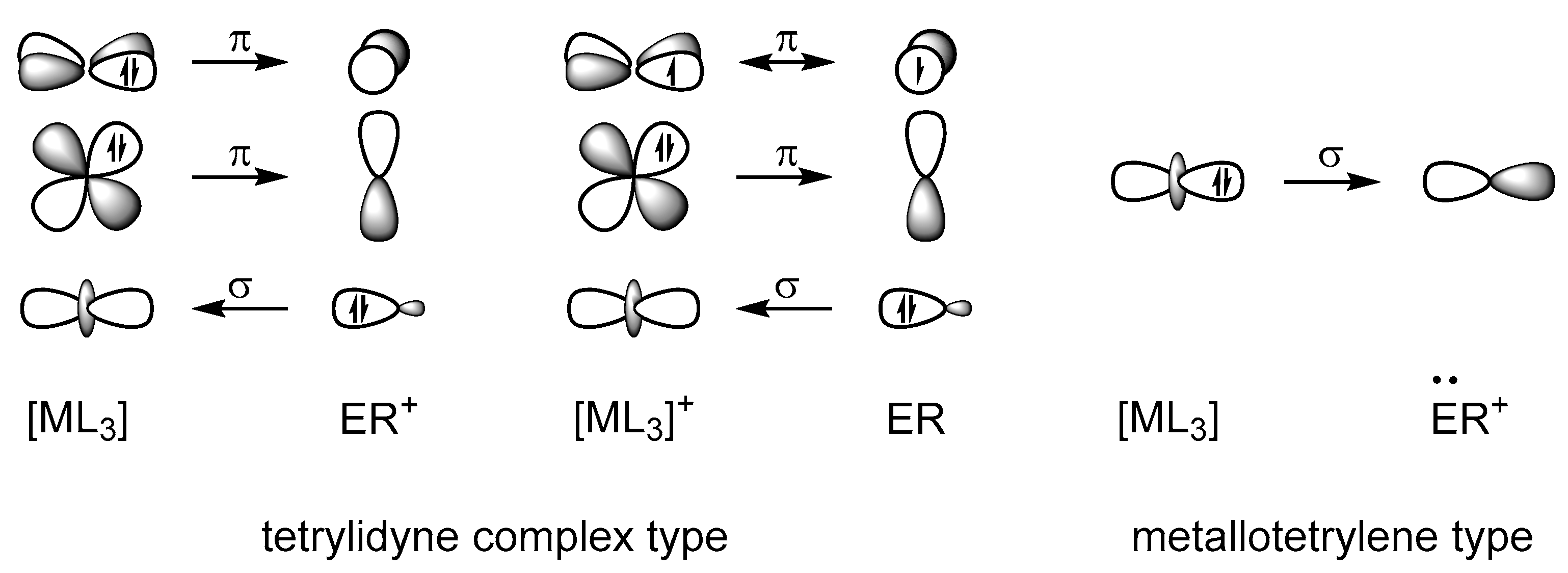
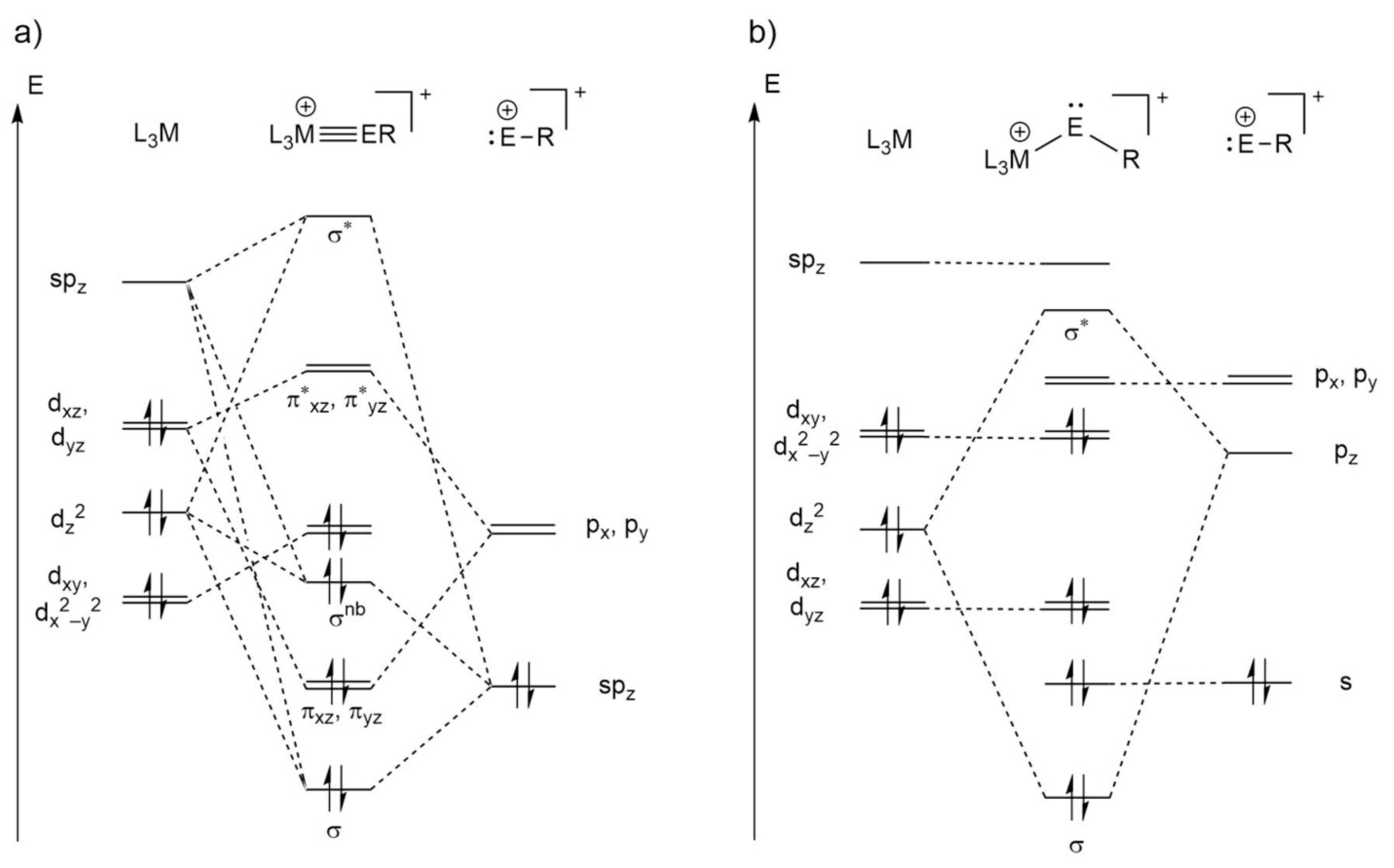
2.2. Molecular Orbital Analysis
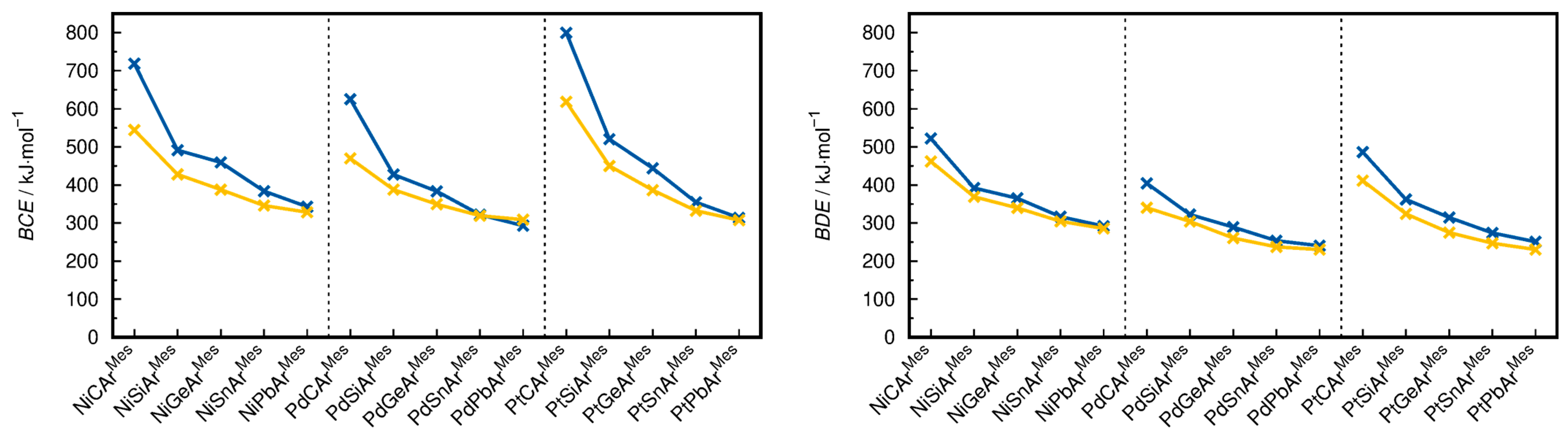
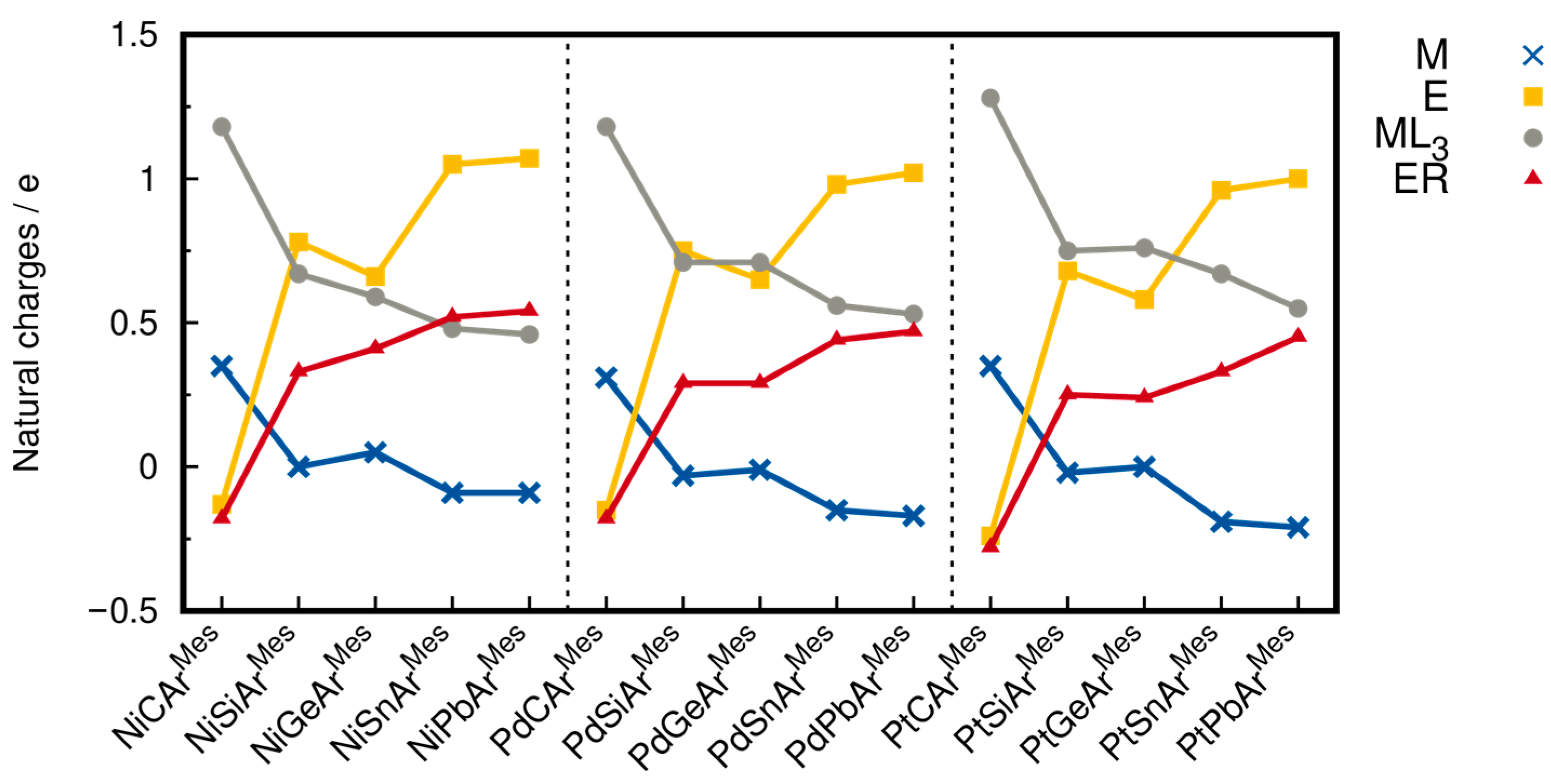
2.3. Bond Dissociation Energies and Natural Population Analysis
- [(PMe3)3ME−R]+ → [M(PMe3)3] + [E−R]+: An investigation of the electronic states of the fragments [ML3] and [E−R]+ revealed that all fragments have a singlet ground state, with the only exception being the structurally relaxed [C−ArMes]+ fragment, which is stabilized in the triplet state after activation of the Mes substituent (see the calculated structure file). The singlet−triplet excitation energies range from 191.4 kJ·mol−1 ([Ni(PMe3)3]) to 214.3 kJ·mol−1 ([Pd(PMe3)3]) for the metal fragments and from 140.8 kJ·mol−1 ([Si−Tbb]+) to 174.7 kJ·mol−1 ([Sn−ArMes]+) for the tetryliumylidene ions.
- [(PMe3)3ME−R]+→[M(PMe3)3]+ + [E−R]: This fragmentation scheme involves an interaction of two open-shell fragments either in their doublet or quartet states. The doublet state is preferred by all fragments with doublet−quartet excitation energies ranging from 232.5 kJ·mol−1 ([Ni(PMe3)3]+) to 284.0 kJ·mol−1 ([Pd(PMe3)3]+) for the metal fragments and from 120.3 kJ·mol−1 ([C−ArMes]) to 218.7 kJ·mol−1 ([Pb−ArMes]) for the tetrylidyne fragments (see the SI for details).
- (a)
- Concerning the BCEs, the fragmentation into the [ML3]+ and [ER] fragments is favored for all compounds by 2.4 kJ·mol−1 (PdSnArMes) to 181.0 kJ·mol−1 (PtCArMes), except for PdPbArMes, for which the cleavage into the [ML3] and [ER]+ fragments is favored by 15.7 kJ·mol−1.
- (b)
- The BDEs are lower for the dissociation into the [ML3]+ and [ER] fragments in all cases, and the energetic differences between the two fragmentation schemes are lower than for the BCEs in most cases, ranging from 6.1 kJ·mol−1 (NiPbArMes) to 74.7 kJ·mol−1 (PtCArMes).
- (c)
- When comparing the energetic differences ΔBCE and ΔBDE between the two fragmentation schemes in dependence on the transition metal, the observed trend is Ni ≈ Pt > Pd for the ΔBCEs and Pt > Pd > Ni for the ΔBDEs (ΔBCE = BCE(i) − BCE(ii) and ΔBDE = BDE(i) − BDE(ii), where i and ii denote the fragmentation schemes).
- (d)
- The energetic difference between the two fragmentation schemes (ΔBCE and ΔBDE) follows the order C >> Si > Ge > Sn > Pb regarding the tetrel for both the ΔBCE and ΔBDE. The substituent effect (ArMes vs. Tbb) in the silylidyne complexes on the BCEs and BDEs is minute. However, because the BCEs of MSiTbb are slightly lower for the fragmentation into the ML3 + ER+ fragments than those of MSiArMes but slightly higher for the fragmentation into the ML3+ + ER fragments, the ∆BCEs of the MSiTbb complexes are lower than the ∆BCEs of the MSiArMes and MGeArMes complexes. For ΔBDE, the difference between Si and Ge is negligible.
- (e)
- The BCEs, if ordered by transition metal, follow the order Pt > Ni > Pd for E = C and Si and the order Ni > Pt > Pd for E = Ge, Sn and Pb. In comparison, the BDEs, if ordered by the transition metal, follow the order Ni > Pt >Pd for all tetrels. The reason for this difference is a significantly higher structural relaxation energy of the Pt(PMe3)3 fragment (avg. 104.4 kJ·mol−1) followed by the Pd(PMe3)3 (avg. 63.9 kJ·mol−1) and Ni(PMe3)3 fragments (avg. 54.8 kJ·mol−1), which lowers the BDEs of the PtER complexes more than the BDEs of the PdER and NiER complexes in comparison with the respective BCEs.
- (f)
- If ordered by tetrel, the BCEs and BDEs follow the trend C >> Si > Ge > Sn ≈ Pb. This means that the M−E bonds of the carbyne complexes are, as expected, the strongest. However, the heavier ylidyne complexes exhibit considerable BCEs and BDEs. These are lower than those of the carbyne complexes, with the difference, though, being considerably smaller than those of the ditetrylynes. For example, the experimental dissociation enthalpy ∆H° of acetylene of 964.8 ± 2.9 kJ·mol−1 [81] (∆H°calc(HCCH) = 953.0 kJ·mol−1 at the level of theory I and 970.2 kJ·mol−1 at the level of theory II) is ca. 13 times larger than that of the distannyne ArDippSnSnArDipp (∆Hexp = 72.0 ± 7.1 kJ·mol−1) [82]. Similarly, a calculation of the gas-phase dissociation enthalpy ∆H°calc of ArDippSnSnArDipp at the level of theory I leads to a value of 160.7 kJ·mol−1, which is still only a small fraction of that of the analogous acetylene derivative ArDippC≡CArDipp (∆H°calc = 721.7 kJ·mol−1). In comparison, the BDE of NiSnArMes is still 63 % and 66 % of the BDE of NiCArMes on the level theory I and II, respectively, illustrating the considerable bond strength of the M≡E triple bonds. An important implication of this comparison is that C≡C bonds are stronger than M≡C bonds, whereas the opposite is true for the heavier group 14 elements Si–Pb (i.e., the BDEs of the E−E bonds in E2R2 are smaller than those of the M≡E bonds).
- (g)
- The choice of tetrel generally has a larger influence on the BCEs and BDEs than the choice of the transition metal. For example, the BCEs of NiGeArMes, PdGeArMes, and PtGeArMes are within 40 kJ·mol−1 of each other, whereas the BDEs of NiSiArMes and NiPbArMes differ by 83.3 kJ·mol−1.
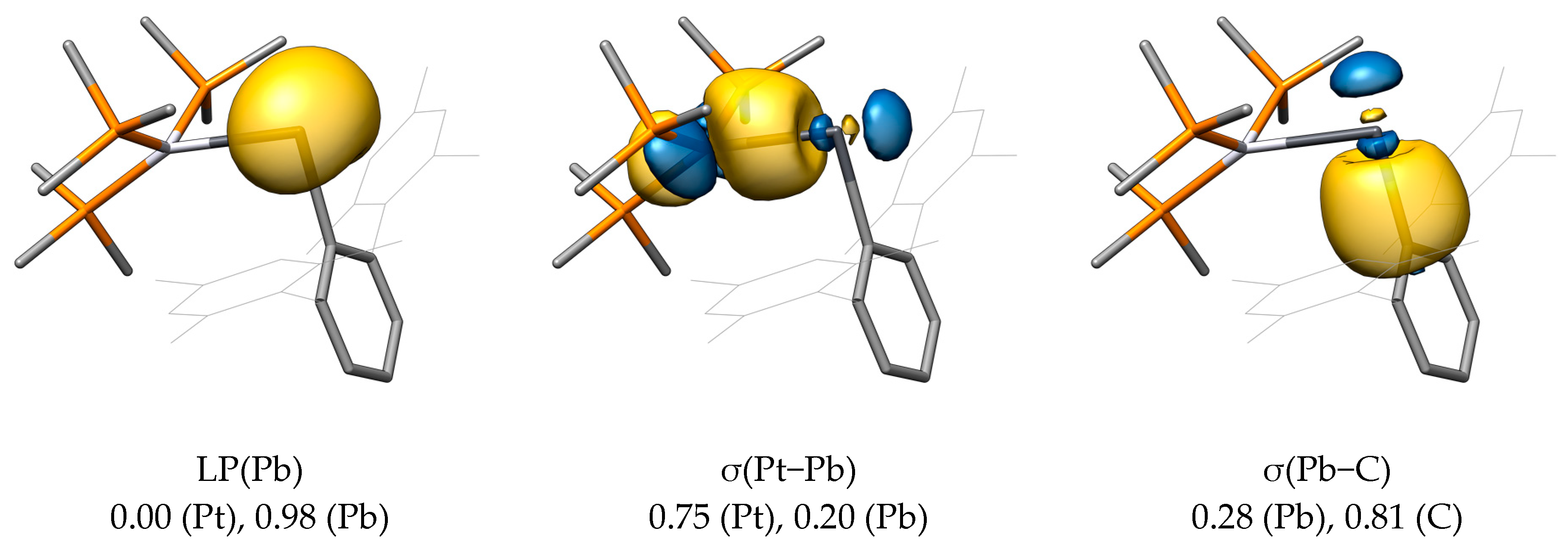
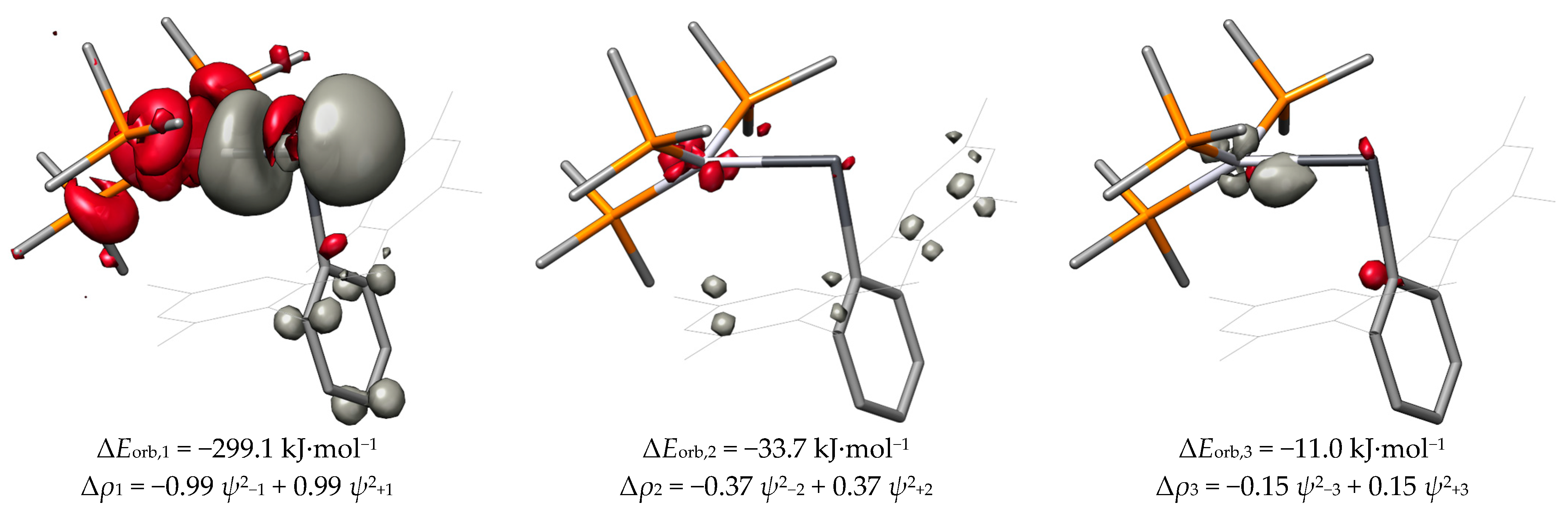
2.4. ETS-NOCV and EDA
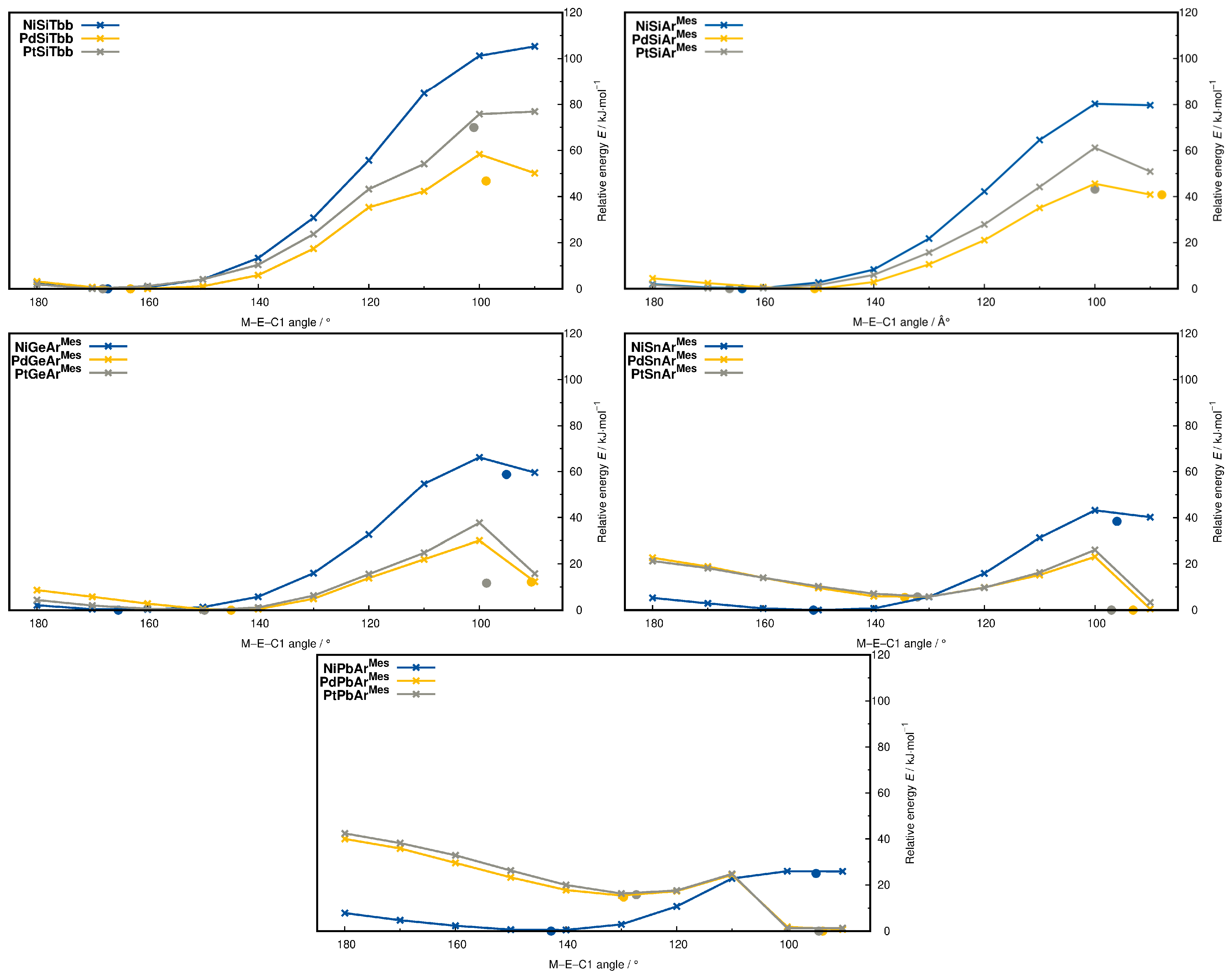
2.5. Metallotetrylene Isomers by PES Scans
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References and Notes
- Hashimoto, H.; Tobita, H. Recent Advances in the Chemistry of Transition Metal–Silicon/Germanium Triple-Bonded Complexes. Coord. Chem. Rev. 2018, 355, 362–379. [Google Scholar] [CrossRef]
- Saini, S.; Agarwal, A.; Bose, S.K. Transition Metal Chemistry of Heavier Group 14 Congener Triple-Bonded Complexes: Syntheses and Reactivity. Dalton Trans. 2020, 49, 17055–17075. [Google Scholar] [CrossRef]
- Fischer, E.O.; Kreis, G.; Kreiter, C.G.; Müller, J.; Huttner, G.; Lorenz, H. trans-Halogeno[alkyl(aryl)carbyne]tetracarbonyl Complexes of Chromium, Molybdenum, and Tungsten—A New Class of Compounds Having a Transition Metal-Carbon Triple Bond. Angew. Chem. Int. Ed. Engl. 1973, 12, 564–565. [Google Scholar] [CrossRef]
- Fischer, E.O. Auf dem Weg zu Carben- und Carbin-Komplexen (Nobel-Vortrag). Angew. Chem. 1974, 86, 651–663. [Google Scholar] [CrossRef]
- Kim, H.P.; Angelici, R.J. Transition Metal Complexes with Terminal Carbyne Ligands. In Advances in Organometallic Chemistry; Elsevier: Amsterdam, The Netherlands, 1987; Volume 27, pp. 51–111. ISBN 978-0-12-031127-9. [Google Scholar]
- Fischer, H.; Hofmann, P.; Kreissl, F.R.; Schrock, R.R.; Schubert, U.; Weiss, K. Carbyne Complexes; Wiley-VCH: Weinheim, Germany, 1988. [Google Scholar]
- Mayr, A.; Hoffmeister, H. Recent Advances in the Chemistry of Metal-Carbon Triple Bonds. In Advances in Organometallic Chemistry; Elsevier: Amsterdam, The Netherlands, 1991; Volume 32, pp. 227–324. ISBN 978-0-12-031132-3. [Google Scholar]
- Simons, R.S.; Power, P.P. (η5-C5H5)(CO)2MoGeC6H3-2,6-Mes2: A Transition-Metal Germylyne Complex. J. Am. Chem. Soc. 1996, 118, 11966–11967. [Google Scholar] [CrossRef]
- Pu, L.; Twamley, B.; Haubrich, S.T.; Olmstead, M.M.; Mork, B.V.; Simons, R.S.; Power, P.P. Triple Bonding to Germanium: Characterization of the Transition Metal Germylynes (η5-C5H5)(CO)2M≡Ge-C6H3-2,6-Mes2 (M = Mo, W; Mes = C6H2-2,4,6-Me3) and (η5-C5H5)(CO)2M≡Ge-C6H3-2,6-Trip2 (M = Cr, Mo, W; Trip = C6H2-2,4,6-i-Pr3) and the Related Single Bonded Metallogermylenes (η5-C5H5)(CO)3M-Ge-C6H3-2,6-Trip2 (M = Cr, W). J. Am. Chem. Soc. 2000, 122, 650–656. [Google Scholar] [CrossRef]
- Filippou, A.C.; Philippopoulos, A.I.; Portius, P.; Neumann, D.U. Synthesis and Structure of the Germylyne Complexes trans-[X(dppe)2W≡Ge(η1-Cp*)] (X = Cl, Br, I) and Comparison of the W≡E Bonds (E = C, Ge) by Density Functional Calculations. Angew. Chem. Int. Ed. 2000, 39, 2778–2781. [Google Scholar] [CrossRef]
- Filippou, A.C.; Portius, P.; Philippopoulos, A.I. Molybdenum and Tungsten Germylyne Complexes of the General Formula trans-[X(dppe)2M≡Ge-(η1-Cp*)] (X = Cl, Br, I; dppe = Ph2PCH2CH2PPh2; Cp* = C5Me5): Syntheses, Molecular Structures, and Bonding Features of the Germylyne Ligand. Organometallics 2002, 21, 653–661. [Google Scholar] [CrossRef]
- Filippou, A.C.; Philippopoulos, A.I.; Portius, P.; Schnakenburg, G. Halide Substitution Reactions of the Germylidyne Complexes trans-[X(dppe)2W≡Ge(η1-Cp*)] (X = Cl, I; dppe = Ph2PCH2CH2PPh2; Cp* = C5Me5). Organometallics 2004, 23, 4503–4512. [Google Scholar] [CrossRef]
- Filippou, A.C.; Schnakenburg, G.; Philippopoulos, A.I.; Weidemann, N. Ge2 Trapped by Triple Bonds between Two Metal Centers: The Germylidyne Complexes trans,trans-[Cl(depe)2M≡Ge–Ge≡M(depe)2Cl] (M = Mo, W) and Bonding Analyses of the M≡Ge–Ge≡M Chain. Angew. Chem. Int. Ed. 2005, 44, 5979–5985. [Google Scholar] [CrossRef]
- Filippou, A.C.; Weidemann, N.; Philippopoulos, A.I.; Schnakenburg, G. Activation of Aryl Germanium(II) Chlorides by [Mo(PMe3)6] and [W(η2-CH2PMe2)H(PMe3)4]: A New Route to Metal-Germanium Triple Bonds. Angew. Chem. Int. Ed. 2006, 45, 5987–5991. [Google Scholar] [CrossRef]
- Filippou, A.C.; Portius, P.; Philippopoulos, A.I.; Rohde, H. Triple Bonding to Tin: Synthesis and Characterization of the Stannylyne Complex trans-[Cl(PMe3)4W≡Sn–C6H3-2,6-Mes2]. Angew. Chem. Int. Ed. 2003, 42, 445–447. [Google Scholar] [CrossRef] [PubMed]
- Filippou, A.C.; Philippopoulos, A.I.; Schnakenburg, G. Triple Bonding to Tin: Synthesis and Characterization of the Square-Pyramidal Stannylyne Complex Cation [(dppe)2W≡Sn-C6H3-2,6-Mes2]+ (dppe = Ph2PCH2CH2PPh2, Mes = C6H2-2,4,6-Me3). Organometallics 2003, 22, 3339–3341. [Google Scholar] [CrossRef]
- Filippou, A.C.; Weidemann, N.; Schnakenburg, G.; Rohde, H.; Philippopoulos, A.I. Tungsten-Lead Triple Bonds: Syntheses, Structures, and Coordination Chemistry of the Plumbylidyne Complexes trans-[X(PMe3)4W≡Pb(2,6-Trip2C6H3)]. Angew. Chem. Int. Ed. 2004, 43, 6512–6516. [Google Scholar] [CrossRef] [PubMed]
- Filippou, A.C.; Rohde, H.; Schnakenburg, G. Triple Bond to Lead: Synthesis and Characterization of the Plumbylidyne Complex trans-[Br(PMe3)4Mo≡Pb–C6H3-2,6-Trip2]. Angew. Chem. Int. Ed. 2004, 43, 2243–2247. [Google Scholar] [CrossRef]
- Filippou, A.C.; Weidermann, N.; Schnakenburg, G. Tungsten-Mediated Activation of a PbII-N Bond: A New Route to Tungsten-Lead Triple Bonds. Angew. Chem. Int. Ed. 2008, 47, 5799–5802. [Google Scholar] [CrossRef]
- Filippou, A.C.; Chernov, O.; Stumpf, K.W.; Schnakenburg, G. Metal-Silicon Triple Bonds: The Molybdenum Silylidyne Complex [Cp(CO)2Mo≡Si-R]. Angew. Chem. Int. Ed. 2010, 49, 3296–3300. [Google Scholar] [CrossRef]
- Filippou, A.C.; Baars, B.; Chernov, O.; Lebedev, Y.N.; Schnakenburg, G. Silicon-Oxygen Double Bonds: A Stable Silanone with a Trigonal-Planar Coordinated Silicon Center. Angew. Chem. Int. Ed. 2014, 53, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Filippou, A.C.; Hoffmann, D.; Schnakenburg, G. Triple Bonds of Niobium with Silicon, Germaniun and Tin: The Tetrylidyne Complexes [(κ3-tmps)(CO)2Nb≡E–R] (E = Si, Ge, Sn; tmps = MeSi(CH2PMe2)3; R = aryl). Chem. Sci. 2017, 8, 6290–6299. [Google Scholar] [CrossRef] [PubMed]
- Ghana, P.; Arz, M.I.; Chakraborty, U.; Schnakenburg, G.; Filippou, A.C. Linearly Two-Coordinated Silicon: Transition Metal Complexes with the Functional Groups M≡Si–M and M═Si═M. J. Am. Chem. Soc. 2018, 140, 7187–7198. [Google Scholar] [CrossRef]
- Ghana, P.; Arz, M.I.; Schnakenburg, G.; Straßmann, M.; Filippou, A.C. Metal–Silicon Triple Bonds: Access to [Si(η5-C5Me5)]+ from SiX2(NHC) and Its Conversion to the Silylidyne Complex [TpMe(CO)2MoSi(η3-C5Me5)] (TpMe = κ3-N,N′,N″-hydridotris(3,5-dimethyl-1-pyrazolyl)borate). Organometallics 2018, 37, 772–780. [Google Scholar] [CrossRef]
- Mork, B.V.; Tilley, T.D. Multiple Bonding Between Silicon and Molybdenum: A Transition-Metal Complex with Considerable Silylyne Character. Angew. Chem. Int. Ed. 2003, 42, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Hayes, P.G.; Xu, Z.; Beddie, C.; Keith, J.M.; Hall, M.B.; Tilley, T.D. The Osmium–Silicon Triple Bond: Synthesis, Characterization, and Reactivity of an Osmium Silylyne Complex. J. Am. Chem. Soc. 2013, 135, 11780–11783. [Google Scholar] [CrossRef]
- Fukuda, T.; Yoshimoto, T.; Hashimoto, H.; Tobita, H. Synthesis of a Tungsten–Silylyne Complex via Stepwise Proton and Hydride Abstraction from a Hydrido Hydrosilylene Complex. Organometallics 2016, 35, 921–924. [Google Scholar] [CrossRef]
- Yoshimoto, T.; Hashimoto, H.; Hayakawa, N.; Matsuo, T.; Tobita, H. A Silylyne Tungsten Complex Having an Eind Group on Silicon: Its Dimer–Monomer Equilibrium and Cycloaddition Reactions with Carbodiimide and Diaryl Ketones. Organometallics 2016, 35, 3444–3447. [Google Scholar] [CrossRef]
- Dübek, G.; Hanusch, F.; Munz, D.; Inoue, S. An Air-Stable Heterobimetallic Si2M2 Tetrahedral Cluster. Angew. Chem. Int. Ed. 2020, 59, 5823–5829. [Google Scholar] [CrossRef]
- Vyboishchikov, S.F.; Frenking, G. Structure and Bonding of Low-Valent (Fischer-Type) and High-Valent (Schrock-Type) Transition Metal Carbyne Complexes. Chem.—Eur. J. 1998, 4, 1439–1448. [Google Scholar]
- Lein, M.; Szabó, A.; Kovács, A.; Frenking, G. Energy Decomposition Analysis of the Chemical Bond in Main Group and Transition Metal Compounds. Faraday Discuss. 2003, 124, 365–378. [Google Scholar] [CrossRef]
- Pandey, K.K.; Lein, M.; Frenking, G. Metal Germylyne Complexes [M≡Ge–R] and Metallogermylenes [M–Ge–R]: DFT Analysis of the Systems [(Cp)(CO)nM≡GeMe] (M = Cr, Mo, W, Fe2+, n = 2; M = Fe, n = 1) and [(Cp)(CO)nM–GeMe] (M = Cr, Mo, W, n = 3; M = Fe, n = 2). J. Am. Chem. Soc. 2003, 125, 1660–1668. [Google Scholar] [CrossRef]
- Pandey, K.K.; Lledós, A. Linear M≡E—Me Versus Bent M—E—Me: Bonding Analysis in Heavier Metal-Ylidyne Complexes [(Cp)(CO)2M≡EMe] and Metallo-Ylidenes [(Cp)(CO)3M–EMe] (M = Cr, Mo, W; E = Si, Ge, Sn, Pb). Inorg. Chem. 2009, 48, 2748–2759. [Google Scholar] [CrossRef]
- Pandey, K.K.; Patidar, P. Insights into the Nature of M≡E Bonds in [(PMe3)4M≡E(Mes)]+ (M = Mo, W) and [(PMe3)5W≡E(Mes)]+: A Dispersion-Corrected DFT Study. RSC Adv. 2014, 4, 13034. [Google Scholar] [CrossRef]
- Dewar, M.J.S. A Review of the π-Complex Theory. Bull. Soc. Chim. Fr. 1951, 18, C71–C79. [Google Scholar]
- Chatt, J.; Duncanson, L.A. 586. Olefin Co-Ordination Compounds. Part III. Infra-Red Spectra and Structure: Attempted Preparation of Acetylene Complexes. J. Chem. Soc. (Resumed) 1953, 2939–2947. [Google Scholar] [CrossRef]
- Guggenberger, L.J.; Schrock, R.R. A Tantalum Carbyne Complex. J. Am. Chem. Soc. 1975, 97, 2935. [Google Scholar] [CrossRef]
- Schrock, R.R. Multiple Metal–Carbon Bonds for Catalytic Metathesis Reactions (Nobel Lecture). Angew. Chem. Int. Ed. 2006, 45, 3748–3759. [Google Scholar] [CrossRef] [PubMed]
- Filippou, A.C.; Barandov, A.; Schnakenburg, G.; Lewall, B.; van Gastel, M.; Marchanka, A. Open-Shell Complexes Containing Metal-Germanium Triple Bonds. Angew. Chem. Int. Ed. 2012, 51, 789–793. [Google Scholar] [CrossRef]
- Filippou, A.C.; Stumpf, K.W.; Chernov, O.; Schnakenburg, G. Metal Activation of a Germylenoid, a New Approach to Metal-Germanium Triple Bonds: Synthesis and Reactions of the Germylidyne Complexes [Cp(CO)2M≡Ge-C(SiMe3)3] (M = Mo, W). Organometallics 2012, 31, 748–755. [Google Scholar] [CrossRef]
- Hicks, J.; Hadlington, T.J.; Schenk, C.; Li, J.; Jones, C. Utilizing Steric Bulk to Stabilize Molybdenum Aminogermylyne and Aminogermylene Complexes. Organometallics 2013, 32, 323–329. [Google Scholar] [CrossRef]
- Fukuda, T.; Hashimoto, H.; Tobita, H. Reactions of a Tungsten-Germylyne Complex with Alcohols and Arylaldehydes. Chem. Commun. 2013, 49, 4232–4234. [Google Scholar] [CrossRef]
- Wienkenhöver, N. 1,2-Dibromodisilenes: A Rich Source for Titanium Silylidyne Complexes, Acyclic Silylenes and Disilyne Dianions. Dissertation (Dr. rer. nat.), University of Bonn, Bonn, Germany, 2017. [Google Scholar]
- Arizpe, L. Synthesis and Characterization of Complexes Featuring Tantalum-Germanium Multiple Bonds. Dissertation (Dr. rer. nat.), University of Bonn, Bonn, Germany, 2019. [Google Scholar]
- Filippou, A.C.; Chakraborty, U.; Schnakenburg, G. Rhenium-Germanium Triple Bonds: Syntheses and Reactions of the Germylidyne Complexes mer-[X2(PMe3)3Re≡Ge–R] (X = Cl, I, H; R = m-terphenyl). Chem.—Eur. J. 2013, 19, 5676–5686. [Google Scholar] [CrossRef]
- Chakraborty, U. Multiple Bonds between Group 7 Transition Metals and Heavier Tetrel Elements (Ge–Pb). Dissertation (Dr. rer. nat.), University of Bonn, Bonn, Germany, 2013. [Google Scholar]
- Filippou, A.C.; Ghana, P.; Chakraborty, U.; Schnakenburg, G. Manganese–Tin Triple Bonds: A New Synthetic Route to the Manganese Stannylidyne Complex Cation trans-[H(dmpe)2Mn≡Sn(C6H3-2,6-Mes2)]+ (dmpe = Me2PCH2CH2PMe2, Mes = 2,4,6-trimethylphenyl). J. Am. Chem. Soc. 2013, 135, 11525–11528. [Google Scholar] [CrossRef] [PubMed]
- Blom, B. Reactivity of Ylenes at Late Transition Metal Centers. Dissertation (Dr. rer. nat.), University of Bonn, Bonn, Germany, 2011. [Google Scholar]
- Lebedev, Y.N. Multiple Bonding of Low-Valent Silicon and Germanium to Group 6 and 9 Metals. Dissertation (Dr. rer. nat.), University of Bonn, Bonn, Germany, 2014. [Google Scholar]
- Widemann, M.; Eichele, K.; Schubert, H.; Sindlinger, C.P.; Klenner, S.; Pöttgen, R.; Wesemann, L. Synthesis and Hydrogenation of Heavy Homologues of Rhodium Carbynes: [(Me3P)2(Ph3P)Rh≡E-Ar*] (E = Sn, Pb). Angew. Chem. Int. Ed. 2021, 60, 5882–5889. [Google Scholar] [CrossRef]
- Fischer, E.O.; Schneider, J.R. Übergangsmetallcarbin-komplexe. J. Organomet. Chem. 1985, 295, c29–c34. [Google Scholar] [CrossRef]
- Papazoglou, I. Unprecedented Tetrylidyne Complexes of Group 6 and 10 Metals. Dissertation (Dr. rer. nat.), University of Bonn, Bonn, Germany, 2016. [Google Scholar]
- Keil, P.M.; Hadlington, T.J. Accessing Cationic Tetrylene-Nickel(0) Systems Featuring Donor–Acceptor E–Ni Triple Bonds (E = Ge, Sn). Chem. Commun. 2022, 58, 3011–3014. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, C.; Inagawa, Y.; Iwamoto, T.; Kira, M. Synthesis and Structures of (Dialkylsilylene)Bis(Phosphine)-Nickel, Palladium, and Platinum Complexes and (η6-Arene)(Dialkylsilylene)Nickel Complexes. Dalton Trans. 2010, 39, 9414. [Google Scholar] [CrossRef]
- A CSD survey (11.08.2022) gave 6 compounds with Ni=Si double bond lengths ranging from 209.4(1) pm to 222.41(5) pm with a median and mean value of 215.1 pm and 216.4 pm, respectively
- A CSD survey (11.08.2022) gave 5 compounds with Pt=Si double bond lengths ranging from 220.8(2) pm to 227.0(2) pm with a median and mean value of 221.2 pm and 223.1 pm, respectively
- A CSD survey (11.08.2022) gave 1 compound with a Ni=Sn double bond length of 238.7(2) pm
- Tao, J.M.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the Density Functional Ladder: Nonempirical Meta-Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Design of Density Functionals That Are Broadly Accurate for Thermochemistry, Thermochemical Kinetics, and Nonbonded Interactions. J. Phys. Chem. 2005, 109, 5656–5667. [Google Scholar] [CrossRef]
- Bühl, M.; Kabrede, H. Geometries of Transition-Metal Complexes from Density-Functional Theory. J. Chem. Theory Comput. 2006, 2, 1282–1290. [Google Scholar] [CrossRef]
- Waller, M.P.; Braun, H.; Hojdis, N.; Bühl, M. Geometries of Second-Row Transition-Metal Complexes from Density-Functional Theory. J. Chem. Theory Comput. 2007, 3, 2234–2242. [Google Scholar] [CrossRef] [PubMed]
- Bühl, M.; Reimann, C.; Pantazis, D.A.; Bredow, T.; Neese, F. Geometries of Third-Row Transition-Metal Complexes from Density-Functional Theory. J. Chem. Theory Comput. 2008, 4, 1449–1459. [Google Scholar] [CrossRef]
- Maurer, L.R.; Bursch, M.; Grimme, S.; Hansen, A. Assessing Density Functional Theory for Chemically Relevant Open-Shell Transition Metal Reactions. J. Chem. Theory Comput. 2021, 17, 6134–6151. [Google Scholar] [CrossRef] [PubMed]
- van Wüllen, C. Molecular Density Functional Calculations in the Regular Relativistic Approximation: Method, Application to Coinage Metal Diatomics, Hydrides, Fluorides and Chlorides, and Comparison with First-Order Relativistic Calculations. J. Chem. Phys. 1998, 109, 392–399. [Google Scholar] [CrossRef]
- Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-Adjusted Ab-Initio Pseudopotentials for the Second and Third Row Transition Elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Metz, B.; Stoll, H.; Dolg, M. Small-Core Multiconfiguration-Dirac–Hartree–Fock-Adjusted Pseudopotentials for Post-d Main Group Elements: Application to PbH and PbO. J. Chem. Phys. 2000, 113, 2563–2569. [Google Scholar] [CrossRef]
- Hong, G.; Dolg, M.; Li, L. A Comparison of Scalar-Relativistic ZORA and DKH Density Functional Schemes: Monohydrides, Monooxides and Monofluorides of La, Lu, Ac and Lr. Chem. Phys. Lett. 2001, 334, 396–402. [Google Scholar] [CrossRef]
- Pyykkö, P. Additive Covalent Radii for Single-, Double-, and Triple-Bonded Molecules and Tetrahedrally Bonded Crystals: A Summary. J. Phys. Chem. A 2015, 119, 2326–2337. [Google Scholar] [CrossRef] [PubMed]
- Bent, H.A. An Appraisal of Valence-Bond Structures and Hybridization in Compounds of the First-Row Elements. Chem. Rev. 1961, 61, 275–311. [Google Scholar] [CrossRef]
- Power, P.P. An Update on Multiple Bonding between Heavier Main Group Elements: The Importance of Pauli Repulsion, Charge-Shift Character, and London Dispersion Force Effects. Organometallics 2020, 39, 4127–4138. [Google Scholar] [CrossRef]
- Kutzelnigg, W. Chemical Bonding in Higher Main Group Elements. Angew. Chem. Int. Ed. Engl. 1984, 23, 272–295. [Google Scholar] [CrossRef]
- Desclaux, J.P. Relativistic Dirac-Fock Expectation Values for Atoms with Z = 1 to Z = 120. At. Data Nucl. Data Tables 1973, 12, 311–406. [Google Scholar] [CrossRef]
- Pandey, K.K.; Patidar, P.; Power, P.P. Structure and Bonding Energy Analysis of Cationic Metal–Ylyne Complexes of Molybdenum and Tungsten, [(MeCN)(PMe3)4M≡EMes]+ (M = Mo, W; E = Si, Ge, Sn, Pb): A Theoretical Study. Inorg. Chem. 2011, 50, 7080–7089. [Google Scholar] [CrossRef]
- Schubert, U.; Fischer, E.O.; Wittmann, D. Structure of [(CO)5CrNEt2]BF4, a Key Organometallic Compound; Reaction to Give the Carbene Complex(CO)5CrC(AsPh2)NEt2. Angew. Chem. Int. Ed. Engl. 1980, 19, 643–644. [Google Scholar] [CrossRef]
- Kostic, N.; Fenske, R. Molecular Orbital Calculations on Carbyne Complexes CpMn(CO)2CR+ and (CO)5CrCNEt2+. Frontier-Controlled Nucleophilic Addition to Metal-Carbon Triple Bond. J. Am. Chem. Soc. 1981, 103, 4677–4685. [Google Scholar] [CrossRef]
- Filippou, A.C.; Grünleitner, W.; Fischer, E.O.; Imhof, W.; Huttner, G. Übergangsmetall-Carbin-Komplexe: XCIX. Synthese und Röntgenstruktur von (η5-C5Me5)(CO)2Mo≡CNEt2, dem ersten niedervalenten Diethylaminocarbin-Komplex von Molybdän mit einem Pentamethylcyclopentadienyl-Liganden. J. Organomet. Chem. 1991, 413, 165–179. [Google Scholar] [CrossRef]
- Filippou, A.C.; Portius, P.; Jankowski, C. Aminomethylene Complexes of Molybdenum(IV) and Tungsten(IV) Bearing 1,2-Dithiolato Ligands. J. Organomet. Chem. 2001, 617–618, 656–670. [Google Scholar] [CrossRef]
- Biancalana, L.; Marchetti, F. Aminocarbyne Ligands in Organometallic Chemistry. Coord. Chem. Rev. 2021, 449, 214203. [Google Scholar] [CrossRef]
- Pipek, J.; Mezey, P.G. A Fast Intrinsic Localization Procedure Applicable for ab initio and Semiempirical Linear Combination of Atomic Orbital Wave Functions. J. Chem. Phys. 1989, 90, 4916–4926. [Google Scholar] [CrossRef]
- Weiss, K. Catalytic Reactions of Carbyne Complexes. In Carbyne Complexes; VCH Verlagsgesellschaft: Weinheim, Germany; VCH Publishers: New York, NY, USA, 1988; p. 205. [Google Scholar]
- Ervin, K.M.; Gronert, S.; Barlow, S.E.; Gilles, M.K.; Harrison, A.G.; Bierbaum, V.M.; DePuy, C.H.; Lineberger, W.C.; Ellison, G.B. Bond Strengths of Ethylene and Acetylene. J. Am. Chem. Soc. 1990, 112, 5750–5759. [Google Scholar] [CrossRef]
- Lai, T.Y.; Tao, L.; Britt, R.D.; Power, P.P. Reversible Sn–Sn Triple Bond Dissociation in a Distannyne: Support for Charge-Shift Bonding Character. J. Am. Chem. Soc. 2019, 141, 12527–12530. [Google Scholar] [CrossRef]
- Ziegler, T.; Rauk, A. On the Calculation of Bonding Energies by the Hartree Fock Slater Method: I. The Transition State Method. Theor. Chim. Acta 1977, 46, 1–10. [Google Scholar] [CrossRef]
- Michalak, A.; Mitoraj, M.; Ziegler, T. Bond Orbitals from Chemical Valence Theory. J. Phys. Chem. A 2008, 112, 1933–1939. [Google Scholar] [CrossRef]
- Mitoraj, M.P.; Michalak, A.; Ziegler, T. A Combined Charge and Energy Decomposition Scheme for Bond Analysis. J. Chem. Theory Comput. 2009, 5, 962–975. [Google Scholar] [CrossRef] [PubMed]
- Okuniewski, A.; Rosiak, D.; Chojnacki, J.; Becker, B. Coordination Polymers and Molecular Structures among Complexes of Mercury(II) Halides with Selected 1-Benzoylthioureas. Polyhedron 2015, 90, 47–57. [Google Scholar] [CrossRef]
- Neese, F. Software Update: The ORCA Program System—Version 5.0. WIREs Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Axilrod, B.M.; Teller, E. Interaction of the van Der Waals Type Between Three Atoms. J. Chem. Phys. 1943, 11, 299–300. [Google Scholar] [CrossRef]
- Muto, Y. Force between Nonpolar Molecules. Proc. Phys. Math. Soc. Jpn. 1943, 17, 629–631. [Google Scholar]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Goerigk, L.; Grimme, S. Efficient and Accurate Double-Hybrid-Meta-GGA Density Functionals—Evaluation with the Extended GMTKN30 Database for General Main Group Thermochemistry, Kinetics, and Noncovalent Interactions. J. Chem. Theory Comput. 2011, 7, 291–309. [Google Scholar] [CrossRef]
- Vahtras, O.; Almlöf, J.; Feyereisen, M.W. Integral Approximations for LCAO-SCF Calculations. Chem. Phys. Lett. 1993, 213, 514–518. [Google Scholar] [CrossRef]
- Weigend, F.; Kattannek, M.; Ahlrichs, R. Approximated Electron Repulsion Integrals: Cholesky Decomposition versus Resolution of the Identity Methods. J. Chem. Phys. 2009, 130, 164106. [Google Scholar] [CrossRef]
- Weigend, F. Hartree–Fock Exchange Fitting Basis Sets for H to Rn. J. Comput. Chem. 2008, 29, 167–175. [Google Scholar] [CrossRef]
- Hellweg, A.; Hättig, C.; Höfener, S.; Klopper, W. Optimized Accurate Auxiliary Basis Sets for RI-MP2 and RI-CC2 Calculations for the Atoms Rb to Rn. Theor. Chem. Acc. 2007, 117, 587–597. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Karafiloglou, P.; Landis, C.R.; Weinhold, F. NBO 7.0; 2018. [Google Scholar]
- te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Baerends, E.J.; Ziegler, T.; Atkins, A.J.; Autschbach, J.; Baseggio, O.; Bashford, D.; Bérces, A.; Bickelhaupt, F.M.; Bo, C.; Boerrigter, P.M.; et al. ADF; 2021. [Google Scholar]
- Van Lenthe, E.; Baerends, E.J. Optimized Slater-Type Basis Sets for the Elements 1-118. J. Comput. Chem. 2003, 24, 1142–1156. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
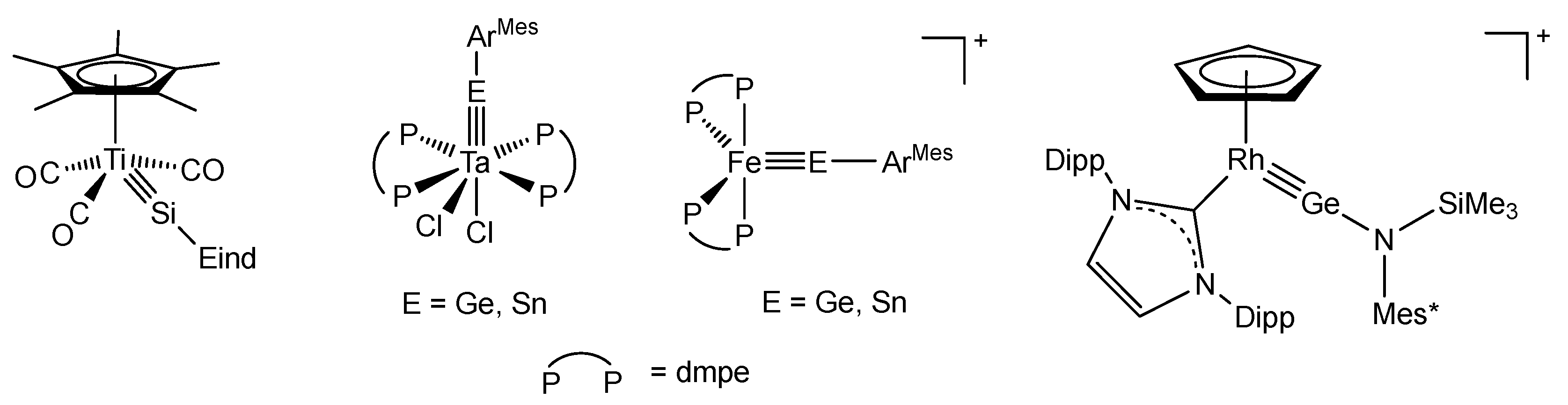{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | M−E | E−C1 | M−E−C1 |
| NiCArMes | 169.1 | 141.3 | 168.4 |
| NiSiTbb | 204.5 | 184.0 | 167.2 |
| NiSiTbbexp | 203.11(7) | 183.8(2) | 172.40(8) |
| NiSiArMes | 204.2 | 186.6 | 163.8 |
| NiGeArMes | 213.3 | 197.1 | 165.3 |
| NiGeArMesexp | 210.40(6) | 194.6(4) | 164.9(1) |
| 210.20(6) | 195.0(4) | 166.5(1) | |
| NiSnArMes | 235.1 | 219.4 | 150.9 |
| NiSnArMesexp | 228.08(9) | 214.0(5) | 165.1(2) |
| NiPbArMes | 244.9 | 229.7 | 142.7 |
| PdCArMes | 182.0 | 140.9 | 168.4 |
| PdSiTbb | 215.1 | 184.0 | 163.1 |
| PdSiArMes | 216.1 | 187.6 | 150.7 |
| PdGeArMes | 227.9 | 199.5 | 144.9 |
| PdSnArMes | 251.6 | 222.9 | 134.4 |
| PdPbArMes | 263.1 | 232.4 | 129.6 |
| PtCArMes | 179.9 | 141.2 | 175.1 |
| PtSiTbb | 215.8 | 183.8 | 168.1 |
| PtSiTbbexp | 213.43(7) | 184.2(3) | 173.83(9) |
| PtSiArMes | 215.7 | 186.3 | 166.1 |
| PtGeArMes | 228.4 | 198.9 | 149.7 |
| PtGeArMesexp | 222.42(7) | 194.7(7) | 161.8(2) |
| 222.69(8) | 195.2(7) | 163.3(2) | |
| PtSnArMes | 255.1 | 223.9 | 132.1 |
| PtPbArMes | 267.7 | 233.1 | 127.3 |
| compound | Ni−E | E−N | Ni−E−N |
| B-Ge | 218.3 | 186.4 | 173.4 |
| B-Geexp | 215.9(1) | 185.3(2) | 175.9(1) |
| B-Sn | 239.6 | 209.1 | 167.8 |
| B-Snexp | 235.5(1) | 206.6(6) | 173.6(2) |
| BCE | BDE | |||
|---|---|---|---|---|
| Compound | ML3 + ER+ | ML3+ + ER | ML3 + ER+ | ML3+ + ER |
| NiCArMes | 718.6 | 544.3 | 521.8 | 461.6 |
| NiSiTbb | 470.4 | 427.9 | 402.5 | 377.5 |
| NiSiArMes | 491.5 | 421.4 | 392.3 | 368.9 |
| NiGeArMes | 459.3 | 387.7 | 365.0 | 340.0 |
| NiSnArMes | 383.9 | 346.0 | 316.9 | 304.5 |
| NiPbArMes | 342.7 | 328.6 | 291.7 | 285.6 |
| PdCArMes | 625.1 | 469.7 | 404.2 | 340.1 |
| PdSiTbb | 415.3 | 395.0 | 333.0 | 304.1 |
| PdSiArMes | 427.4 | 387.9 | 322.5 | 295.3 |
| PdGeArMes | 383.2 | 349.2 | 289.4 | 260.5 |
| PdSnArMes | 321.9 | 319.5 | 253.6 | 237.3 |
| PdPbArMes | 293.3 | 309.0 | 240.4 | 230.4 |
| PtCArMes | 799.3 | 618.3 | 486.0 | 411.3 |
| PtSiTbb | 504.0 | 462.5 | 373.5 | 334.0 |
| PtSiArMes | 520.3 | 450.2 | 362.1 | 324.1 |
| PtGeArMes | 444.0 | 386.3 | 314.5 | 274.9 |
| PtSnArMes | 354.6 | 332.6 | 274.1 | 247.2 |
| PtPbArMes | 313.5 | 307.8 | 251.1 | 230.5 |
| Compound | M | E | ML3 | ER |
|---|---|---|---|---|
| NiCArMes | +0.35 | −0.13 | +1.18 | −0.18 |
| NiSiTbb | ±0.00 | +0.76 | +0.62 | +0.38 |
| NiSiArMes | ±0.00 | +0.78 | +0.67 | +0.33 |
| NiGeArMes | +0.05 | +0.66 | +0.59 | +0.41 |
| NiSnArMes | −0.09 | +1.05 | +0.48 | +0.52 |
| NiPbArMes | −0.09 | +1.07 | +0.46 | +0.54 |
| PdCArMes | +0.31 | −0.15 | +1.18 | −0.18 |
| PdSiTbb | −0.03 | +0.73 | +0.66 | +0.34 |
| PdSiArMes | −0.03 | +0.75 | +0.71 | +0.29 |
| PdGeArMes | −0.01 | +0.65 | +0.71 | +0.29 |
| PdSnArMes | −0.15 | +0.98 | +0.56 | +0.44 |
| PdPbArMes | −0.17 | +1.02 | +0.53 | +0.47 |
| PtCArMes | +0.35 | −0.24 | +1.28 | −0.28 |
| PtSiTbb | −0.02 | +0.65 | +0.71 | +0.29 |
| PtSiArMes | −0.02 | +0.68 | +0.75 | +0.25 |
| PtGeArMes | ±0.00 | +0.58 | +0.76 | +0.24 |
| PtSnArMes | −0.19 | +0.96 | +0.67 | +0.33 |
| PtPbArMes | −0.21 | +1.00 | +0.55 | +0.45 |
| ΔEorb | ||
|---|---|---|
| Compound | ML3 (s) + ER+ (s) | ML3+ (d) + ER (d) |
| NiCArMes | −848.6 | −645.3 |
| NiSiTbb | −498.1 | −522.8 |
| NiSiArMes | −521.9 | −486.2 |
| NiGeArMes | −461.1 | −443.5 |
| NiSnArMes | −372.7 | −377.6 |
| NiPbArMes | −331.9 | −370.1 |
| PdCArMes | −828.7 | −671.7 |
| PdSiTbb | −472.0 | −493.8 |
| PdSiArMes | −496.5 | −494.5 |
| PdGeArMes | −426.8 | −428.1 |
| PdSnArMes | −334.8 | −366.9 |
| PdPbArMes | −291.1 | −345.8 |
| PtCArMes | −1072.2 | −903.3 |
| PtSiTbb | −585.8 | −635.1 |
| PtSiArMes | −606.9 | −573.8 |
| PtGeArMes | −510.8 | −499.1 |
| PtSnArMes | −380.4 | −401.8 |
| PtPbArMes | −319.6 | −362.4 |
| Compound | ΔErel | M−E | E−C1 | M−E−C1 |
|---|---|---|---|---|
| PtPbArMes | +28.5 | 267.7 | 233.1 | 127.3 |
| PtPbArMes-2 | 0.0 | 281.9 | 233.7 | 94.3 |
| Compound | ΔEorb,1 | ΔEorb,2 | ΔEorb,3 | ΔEorb |
|---|---|---|---|---|
| PtPbArMes | −180.1 | −74.7 | −26.8 | −319.6 |
| PtPbArMes-2 | −299.1 | −33.7 | −11.0 | −386.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maurer, L.R.; Rump, J.; Filippou, A.C. The Electronic Nature of Cationic Group 10 Ylidyne Complexes. Inorganics 2023, 11, 129. https://doi.org/10.3390/inorganics11030129
Maurer LR, Rump J, Filippou AC. The Electronic Nature of Cationic Group 10 Ylidyne Complexes. Inorganics. 2023; 11(3):129. https://doi.org/10.3390/inorganics11030129
Chicago/Turabian StyleMaurer, Leonard R., Jens Rump, and Alexander C. Filippou. 2023. "The Electronic Nature of Cationic Group 10 Ylidyne Complexes" Inorganics 11, no. 3: 129. https://doi.org/10.3390/inorganics11030129
APA StyleMaurer, L. R., Rump, J., & Filippou, A. C. (2023). The Electronic Nature of Cationic Group 10 Ylidyne Complexes. Inorganics, 11(3), 129. https://doi.org/10.3390/inorganics11030129