
Neutral W(V) Complexes Featuring the W2O2(µ-O)2 Core and Amino Acids or EDTA Derivatives as Ligands: Synthesis and Structural Characterization

 ,
,  , ,
, ,
Abstract
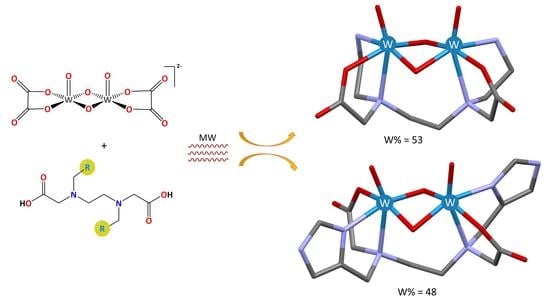
1. Introduction

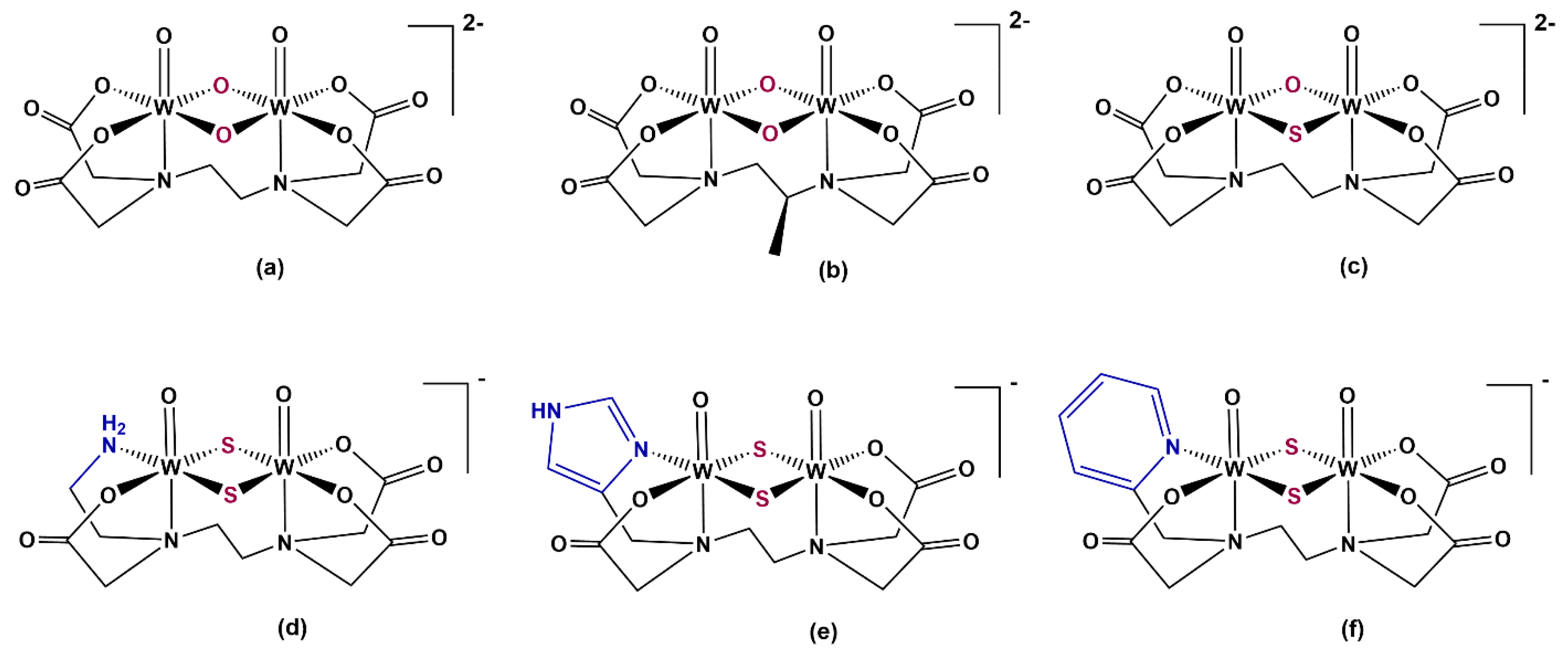
2. Results and Discussion
2.1. Synthesis
2.2. Characterization
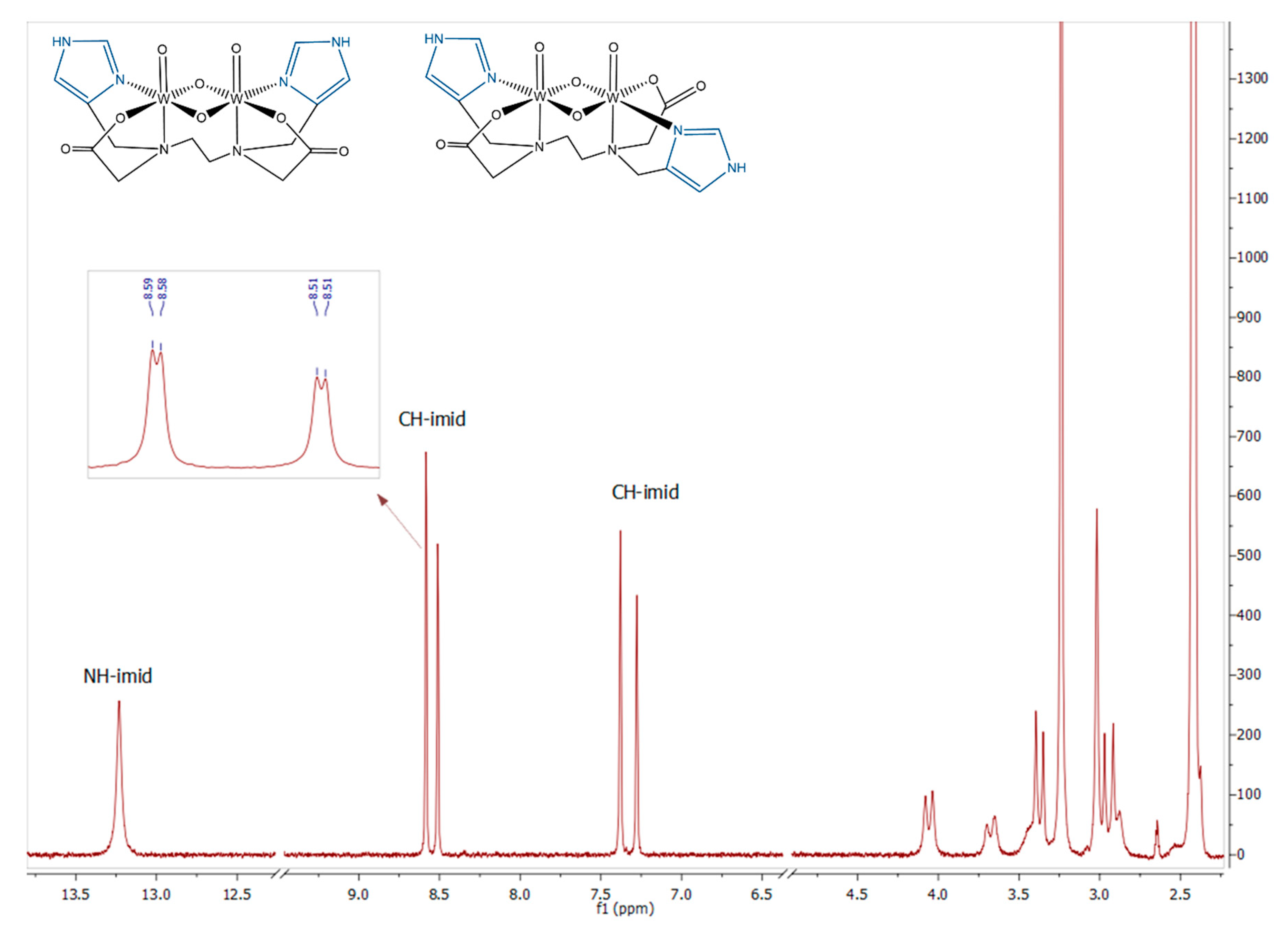
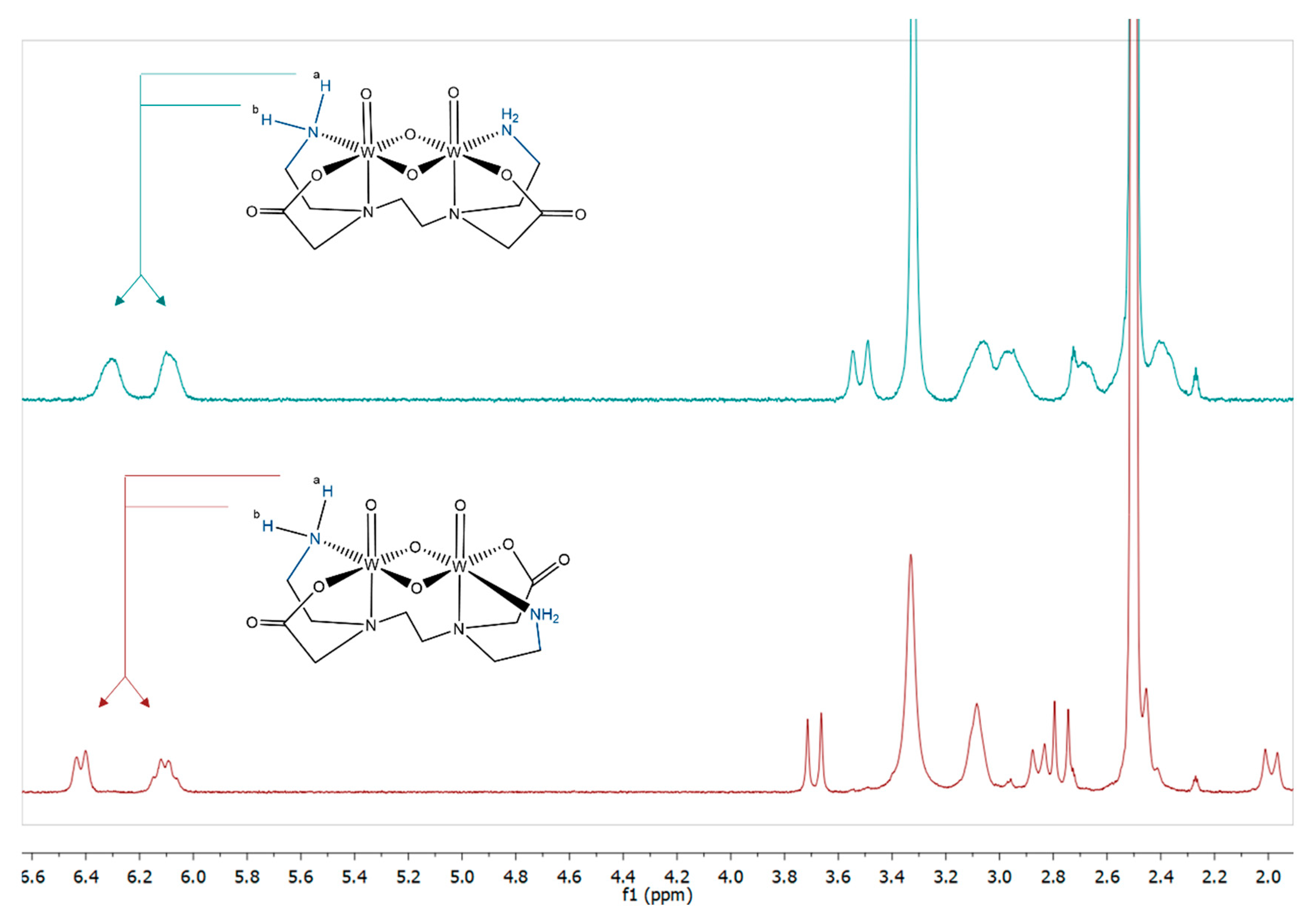
2.2.1. ESI-MS and NMR Spectroscopy
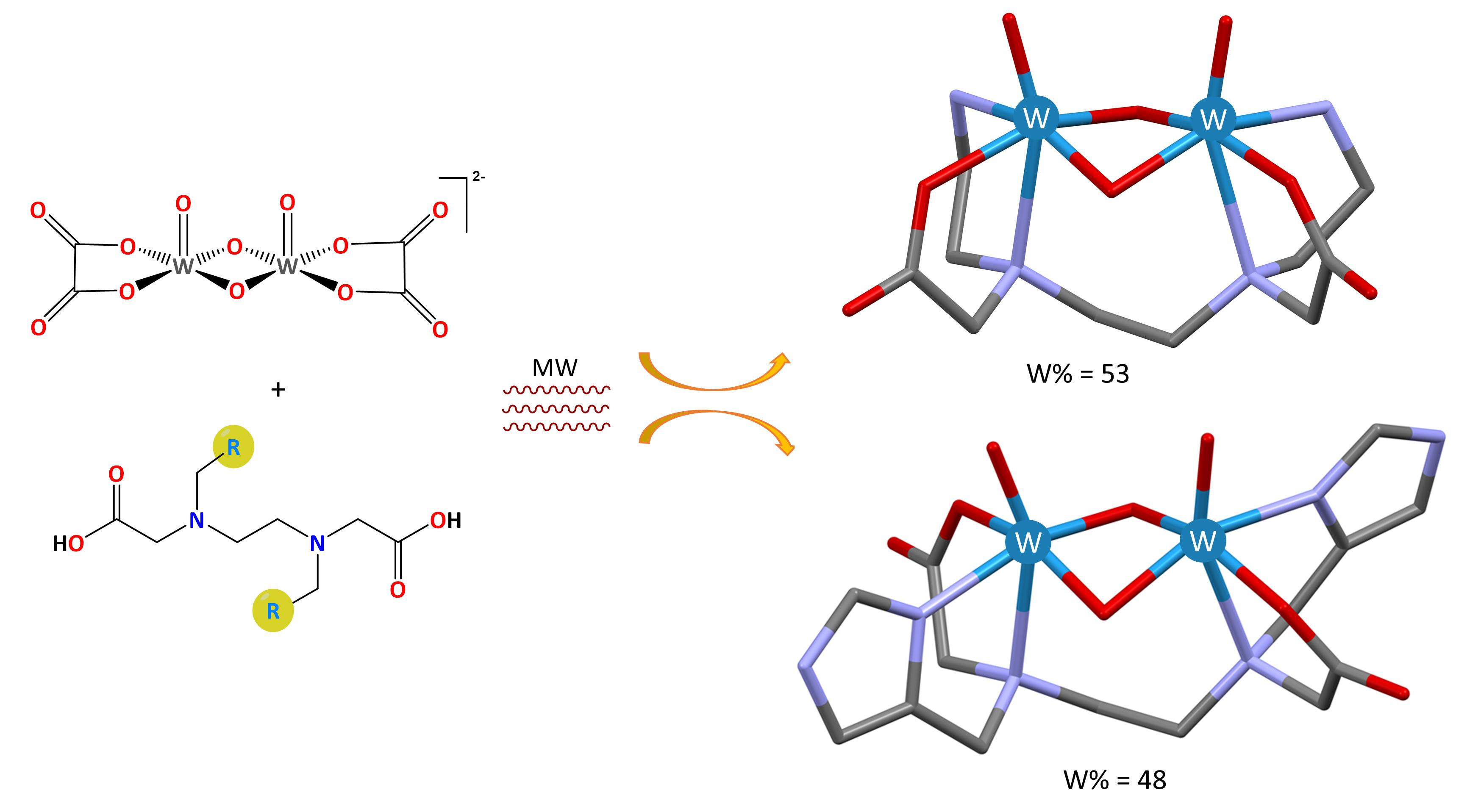
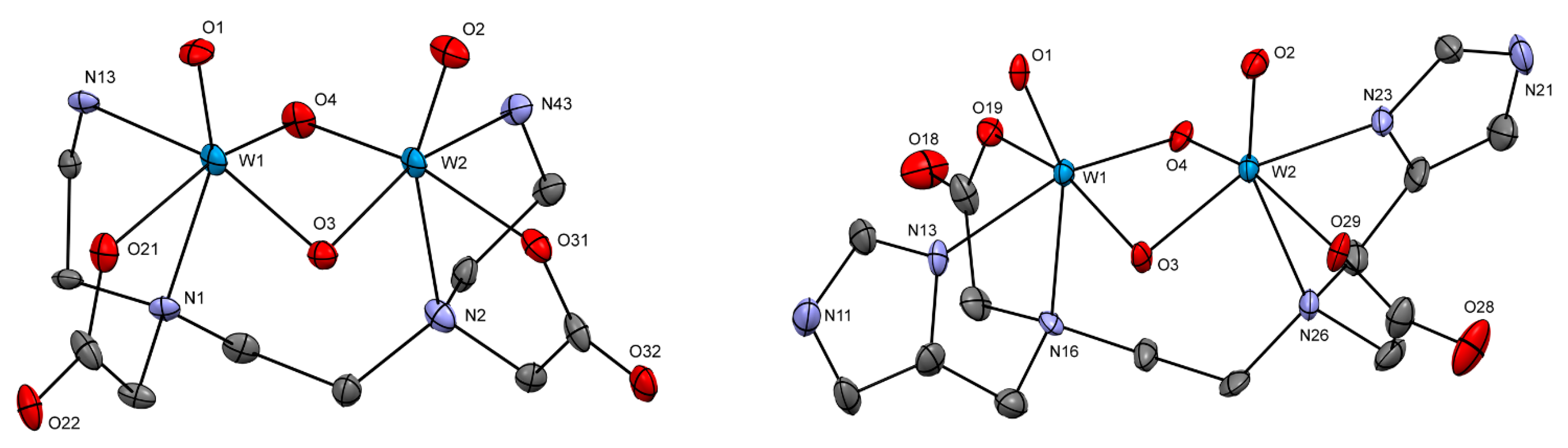
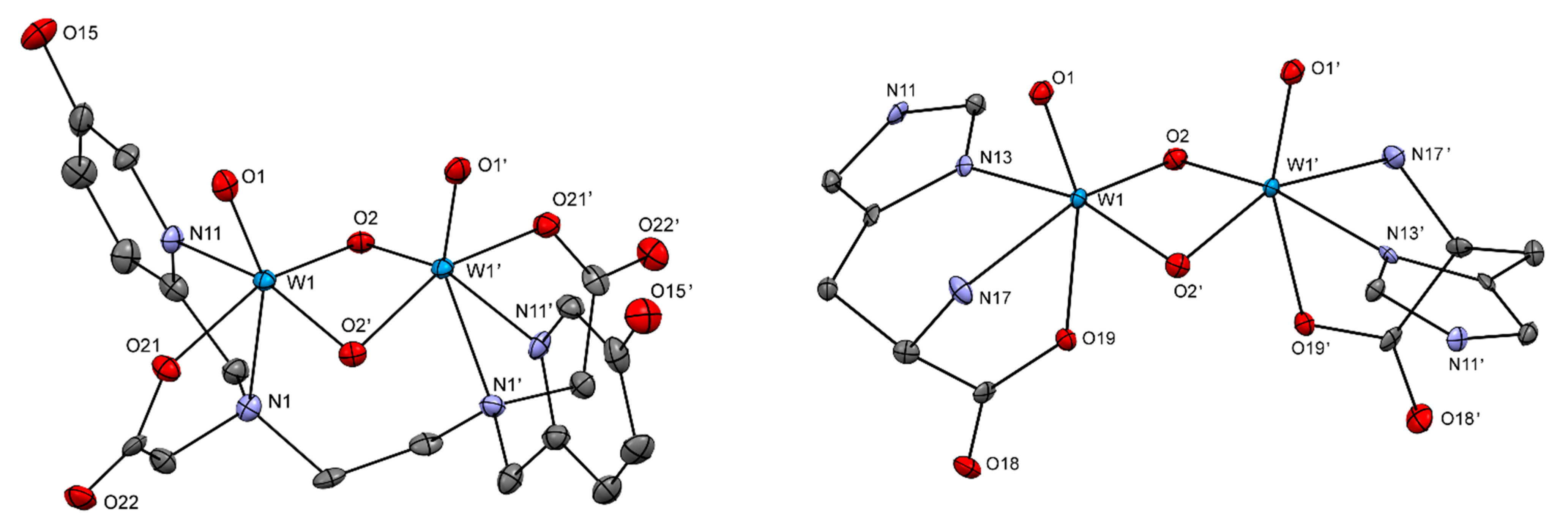
2.2.2. Crystal Structures
2.3. Solubility and Stability
3. Materials and Methods
3.1. Synthesis
General Procedure for the Synthesis of Neutral Complexes of the Type [W2O4(L)], L = L2–4, and [W2O4(His)2]
3.2. Crystallographic Structure Determination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dori, Z. The Coordination Chemistry of Tungsten. In Progress in Inorganic Chemistry; John Wiley & Sons, Ltd.: New York, NY, USA, 1981; Volume 28, pp. 239–307. [Google Scholar] [CrossRef]
- Salonen, P.; Peuronen, A.; Lehtonen, A. Bioinspired Mo, W and V Complexes Bearing a Highly Hydroxyl-Functionalized Schiff Base Ligand. Inorg. Chim. Acta 2020, 503, 119414. [Google Scholar] [CrossRef]
- Schachner, J.A.; Mösch-Zanetti, N.C.; Peuronen, A.; Lehtonen, A. Dioxidomolybdenum(VI) and –Tungsten(VI) Complexes with Tetradentate Amino Bisphenolates as Catalysts for Epoxidation. Polyhedron 2017, 134, 73–78. [Google Scholar] [CrossRef]
- Peuronen, A.; Lehtonen, A. Dioxomolybdenum(VI) and -Tungsten(VI) Amino Bisphenolates as Epoxidation Catalysts. Top. Catal. 2016, 59, 1132–1137. [Google Scholar] [CrossRef]
- Wong, Y.-L.; Ma, J.-F.; Law, W.-F.; Yan, Y.; Wong, W.-T.; Zhang, Z.-Y.; Mak, T.C.W.; Ng, D.K.P. Synthesis, Electrochemistry, and Oxygen-Atom Transfer Reactions of Dioxotungsten(VI) and -Molybdenum(VI) Complexes with N2O2 and N2S2 Tetradentate Ligands. Eur. J. Inorg. Chem. 1999, 1999, 313–321. [Google Scholar] [CrossRef]
- Ward, J.P.; Lim, P.J.; Evans, D.J.; White, J.M.; Young, C.G. Tungsten Ligand-Based Sulfur-Atom-Transfer Catalysts: Synthesis, Characterization, Sustained Anaerobic Catalysis, and Mode of Aerial Deactivation. Inorg. Chem. 2020, 59, 16824–16828. [Google Scholar] [CrossRef]
- Sokolov, M.N.; Adonin, S.A.; Abramov, P.A.; Mainichev, D.A.; Zakharchuk, N.F.; Fedin, V.P. Self-Assembly of Polyoxotungstate with Tetrarhodium-Oxo Core: Synthesis, Structure and 183W NMR Studies. Chem. Commun. 2012, 48, 6666–6668. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.B.; Watson, A.D. Metal-Based X-ray Contrast Media. Chem. Rev. 1999, 99, 2353–2377. [Google Scholar] [CrossRef]
- Yu, S.-B.; Droege, M.; Segal, B.; Kim, S.-H.; Sanderson, T.; Watson, A.D. Cuboidal W3S4 Cluster Complexes as New Generation X-ray Contrast Agents. Inorg. Chem. 2000, 39, 1325–1328. [Google Scholar] [CrossRef]
- Sülzle, D.; Bauser, M.; Frenzel, T.; Jost, G.; Pietsch, H.; Schäfer, M.; Berger, M.; Hassfeld, J.; Schmitt-Willich, H. New Tungsten Cluster Based Contrast Agents for X-ray Computed Tomography. J. Clust. Sci. 2015, 26, 111–118. [Google Scholar] [CrossRef]
- Yu, S.-B.; Droege, M.; Downey, S.; Segal, B.; Newcomb, W.; Sanderson, T.; Crofts, S.; Suravajjala, S.; Bacon, E.; Earley, W.; et al. Dimeric W3SO3 Cluster Complexes: Synthesis, Characterization, and Potential Applications as X-ray Contrast Agents. Inorg. Chem. 2001, 40, 1576–1581. [Google Scholar] [CrossRef]
- Gassan, A.D.; Ivanov, A.A.; Pozmogova, T.N.; Eltsov, I.V.; Kuratieva, N.V.; Mironov, Y.V.; Shestopalov, M.A. Water-Soluble Chalcogenide W6-Clusters: On the Way to Biomedical Applications. Int. J. Mol. Sci. 2022, 23, 8734. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-García, M.; Ortega-Zúñiga, C.; Meléndez, E. New Tungstenocenes Containing 3-Hydroxy-4-Pyrone Ligands: Antiproliferative Activity on HT-29 and MCF-7 Cell Lines and Binding to Human Serum Albumin Studied by Fluorescence Spectroscopy and Molecular Modeling Methods. J. Biol. Inorg. Chem. 2013, 18, 195–209. [Google Scholar] [CrossRef]
- Ehweiner, M.A.; Vidovič, C.; Belaj, F.; Mösch-Zanetti, N.C. Bioinspired Tungsten Complexes Employing a Thioether Scorpionate Ligand. Inorg. Chem. 2019, 58, 8179–8187. [Google Scholar] [CrossRef] [PubMed]
- Novák, J.; Podlaha, J. Tungsten(V) Complexes of Ethylenediaminetetraacetic Acid. J. Inorg. Nucl. Chem. 1974, 36, 1061–1065. [Google Scholar] [CrossRef]
- Soares, A.B.; Taylor, R.C.; Sykes, A.G. Studies on the Μ-(NN′)-Ethylenediaminetetra-Acetato-Di-µ-Oxo-Bis-[Oxotungstate(V)], [W2O4(Edta)] 2–, Complex in Aqueous Solutions. Formation of an Aquo-Ion and Redox Properties. J. Chem. Soc. Dalton Trans. 1980, 7, 1101–1104. [Google Scholar] [CrossRef]
- Ikari, S.; Sasaki, Y.; Ito, T. Asymmetric Distortion of Bis(µ-Oxo)Bis(Oxotungstate(V)) Complexes. Crystal Structure of Bis(µ-Oxo)(µ-N,N′-(R)-Propylenediaminetetraacetato)Bis(Oxotungstate(V)). Inorg. Chem. 1990, 29, 53–56. [Google Scholar] [CrossRef]
- Ikari, S.; Sasaki, Y.; Ito, T. (Μ-Ethylenediaminetetraacetato)(µ-Oxo)(µ-Sulfido)Bis(Oxotungstate(V)): The First Crystallographically Characterized Complex Containing the W2(O)2(µ-O)(µ-S) Unit. Inorg. Chem. 1989, 28, 447–451. [Google Scholar] [CrossRef]
- Yamasaki, M.; Shibahara, T. Syntheses of Sulfur-Bridged Tungsten(V) Complexes with Syn-W2O2S2 Cores. X-ray Structures of K2[W2O2S2(Cys)2]·5H2O and NaNH4[W2O2S2(Edta)]·2H2O. Inorg. Chim. Acta 1993, 205, 45–51. [Google Scholar] [CrossRef]
- Varbanov, H.P.; Glasnov, T.; Belaj, F.; Herbert, S.; Brumby, T.; Mösch-Zanetti, N.C. New Strategies towards Advanced CT Contrast Agents. Development of Neutral and Monoanionic Sulfur-Bridged W(V) Dimeric Complexes. Dalton Trans. 2022, 51, 11086–11097. [Google Scholar] [CrossRef]
- Collenberg, O. Zur Chemie Des 5-Wertigen Wolframs. Z. Anorg. Allg. Chem. 1918, 102, 247–276. [Google Scholar] [CrossRef]
- Mattes, R.; Mennemann, K. Mehrkernige Oxofluoroanionen Des Wolframs(V) Und Wolframs(IV) Die Kristallstrukturen von K3H[W2O4F6] Und (NH4)5[W3O4F9].NH4F.H2O. Z. Anorg. Allg. Chem. 1977, 437, 175–182. [Google Scholar] [CrossRef]
- Sharp, C.; Hills, E.F.; Sykes, A.G. Preparation and Properties of the Tungsten(V) Aqua Dimer [W2O4(H2O)6]2+: Kinetic Studies on the 1:1 Reaction with NCS? J. Chem. Soc. Dalton Trans. 1987, 10, 2293. [Google Scholar] [CrossRef]
- Baba, N.; Yoshino, T. Electrolytic Synthesis and Properties of Potassium Oxalatotungstate (V). J. Appl. Electrochem. 1982, 12, 607–612. [Google Scholar] [CrossRef]
- Ikari, S.; Sasaki, Y.; Nagasawa, A.; Kabuto, C.; Ito, T. A New Mixed Molybdenum-Tungsten Dinuclear Complex, Bis(µ-Oxo)(µ-Ethylenediaminetetraacetato-N,N′)Oxomolybdenum(V)Oxotungstate(V). Inorg. Chem. 1989, 28, 1248–1254. [Google Scholar] [CrossRef]
- Khalil, S.; Sheldrick, B. Barium μ-N,N′-Ethylenediaminetetraacetato-Di-μ-Oxo-Dioxoditungstate(V) Hydrate. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1978, 34, 3751–3753. [Google Scholar] [CrossRef]
- Modec, B.; Bukovec, P. Solid State Structures of Dinuclear and Trinuclear Tungsten and Molybdenum Complexes with Single Metal–Metal Bonds. Inorg. Chim. Acta 2015, 424, 226–234. [Google Scholar] [CrossRef]
- Hazama, R.; Umakoshi, K.; Ichimura, A.; Ikari, S.; Sasaki, Y.; Ito, T. Dinuclear Oxomolybdenum(V) and Oxotungsten(V) Complexes of the Hexadentate Ligands N,N,N′,N′-Tetrakis(2-Pyridylmethyl)Ethylenediamine and Its Propylenediamine Analog. Bull. Chem. Soc. Jpn. 1995, 68, 456–468. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
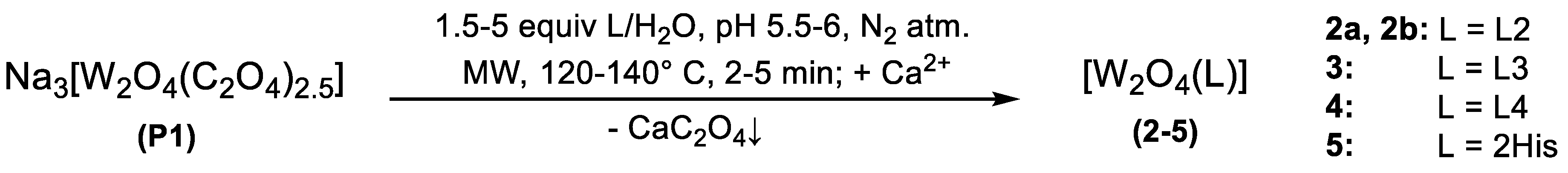{kind=link}
| 2a | 3 | 4 | Ba[W2O4(EDTA)] | |
|---|---|---|---|---|
| W…W | 2.5657(10) | 2.5555(5) | 2.5606(6) | 2.542(2)–2.557(3) |
| W=O | 1.709(8)–1.722(9) | 1.712(6)–1.735(6) | 1.716(8) | 1.696(46)–1.746(51) |
| W-Oµ | 1.938(8)–1.960(9) | 1.932(5)–1.946(6) | 1.946(6)–1.960(6) | 1.883(30)–1.957(30) |
| W-N (W-NC2H4N) | 2.391(9)–2.418(11) | 2.389(7)–2.412(8) | 2.362(8) | 2.469(40)–2.480(28) |
| W-O (W-OCO) | 2.072(9)–2.119(8) | 2.081(6)–2.066(6) | 2.086(7) | 2.032(31)–2.107(29) |
| W-N | 2.185(6)–2.224(5) | 2.171(8)–2.156(8) | 2.212(8) | - |
| O=W-N (W-NC2H4N) | 156.5(4)–157.6(4) | 155.4(3)–157.7(3) | 154.6(3) | 151.8(3)–161.5(3) |
| Oµ-W-Oµ | 92.0(4)–92.3(4) | 91.8(2)–92.1(3) | 92.84 | 89.0(4)–93.4(8) |
| W-Oµ-W | 81.8(3)–82.6(3) | 82.1(2)–82.6(2) | 81.9(2) | 81.4(2)–85.6(5) |
| Oµ-W-Oµ-W | 25.0(4) | 25.3(4) | 24.1(5) | 23.2(7)–25.0(7) |
| [W2O4(His)2] (5) | [W2O2S2(His)2] | |
|---|---|---|
| W=O | 1.737(4) | 1.692(11)–1.722(10) |
| W-Oµ/W-Sµ | 1.946(4) | 2.313(4)–2.348(4) |
| W-O (W-OCO) | 2.139(4) | 2.137(10)–2.203(10) |
| W-N (NH2) | 2.227(3) | 2.224(11)–2.239(12) |
| W-N (imid) | 2.184(5) | 2.218(11)–2.240(11) |
| O=W-O | 158.50(18) | 156.5(4)–161.9(5) |
| Oµ-W-Oµ/Sµ-W-Sµ | 95.40(17) | 102.15(14)–104.29(14) |
| W-Oµ-W/W-Sµ-W | 82.04(16) | 74.10(12)–75.27(13) |
| Oµ-W-Oµ-W/Sµ-W-Sµ-W | 17.14 | 12.08(16)–18.56(14) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varbanov, H.P.; Belaj, F.; Glasnov, T.; Herbert, S.; Brumby, T.; Mösch-Zanetti, N.C. Neutral W(V) Complexes Featuring the W2O2(µ-O)2 Core and Amino Acids or EDTA Derivatives as Ligands: Synthesis and Structural Characterization. Inorganics 2023, 11, 114. https://doi.org/10.3390/inorganics11030114
Varbanov HP, Belaj F, Glasnov T, Herbert S, Brumby T, Mösch-Zanetti NC. Neutral W(V) Complexes Featuring the W2O2(µ-O)2 Core and Amino Acids or EDTA Derivatives as Ligands: Synthesis and Structural Characterization. Inorganics. 2023; 11(3):114. https://doi.org/10.3390/inorganics11030114
Chicago/Turabian StyleVarbanov, Hristo P., Ferdinand Belaj, Toma Glasnov, Simon Herbert, Thomas Brumby, and Nadia C. Mösch-Zanetti. 2023. "Neutral W(V) Complexes Featuring the W2O2(µ-O)2 Core and Amino Acids or EDTA Derivatives as Ligands: Synthesis and Structural Characterization" Inorganics 11, no. 3: 114. https://doi.org/10.3390/inorganics11030114
APA StyleVarbanov, H. P., Belaj, F., Glasnov, T., Herbert, S., Brumby, T., & Mösch-Zanetti, N. C. (2023). Neutral W(V) Complexes Featuring the W2O2(µ-O)2 Core and Amino Acids or EDTA Derivatives as Ligands: Synthesis and Structural Characterization. Inorganics, 11(3), 114. https://doi.org/10.3390/inorganics11030114