Abstract
This paper explores a dual inhibition of main protease (Mpro) and nonstructural protein 10/nonstructural protein 16 (NSP16) methyltransferase complex as the key targets for COVID-19 therapy. These are based on the new Schiff-base ligand that was obtained from the condensation of (4-chloro-3-methyl phenyl) hydrazine with 2-pyridine-carboxaldehyde and its novel Schiff-base metal complexes. These include Ni(II), Pd(II), Pt(II), Zn(II), and Hg(II). The newly synthesized compounds have been characterized using FT-IR, 1H NMR, 13C NMR, and elemental analysis. The results suggested that the Schiff-base ligand is coordinated as a bidentate ligand through the nitrogen atoms of the azomethine group and pyridyl ring. In addition, the biological activity of the prepared complexes was examined against Pseudomonas aeruginosa and Staphylococcus aureus, and the results showed that the Zn(II) complex has the highest activity compared with other compounds. The active sites were found by looking at the molecular electrostatic potential (MEP) maps of the above ligands and complexes. The activity of the compound and its Ni(II) and Zn(II) complexes against Mpro and NSP10/ NSP16 was investigated using a molecular docking approach. They showed excellent binding energies ranging from −5.9 to −7.2 kcal/mol and −5.8 to −7.2 for Mpro and NSP16, respectively. All conformers of the metal complexes were docked with the active site of the NSP16 receptor, showing a binding affinity of 100%. According to our knowledge, this was the first report of these metal complexes as dual inhibitors for Mpro and NSP16 of SARS-CoV-2.
1. Introduction
Coordination chemistry has made a big contribution to science through the many metal complexes used in different fields and for different purposes, such as catalysis and biological applications [1,2,3,4,5,6,7]. The biological and catalytic functions of transition metal complexes containing different Schiff bases are significantly impacted. Biologically active hydrazine ligands include those with nitrogen and oxygen donor atoms [8,9,10]. In addition to being employed as herbicides, insecticides, and rodenticides in agriculture, these substances have also been utilized to treat illnesses including leprosy, TB, and CNS problems [11,12]. Hydrazones exhibit a variety of metal coordination characteristics, and binding to d-metals often increases their biological activity [1,13]. Hydrazine-based Schiff bases have been studied in their design, synthesis, characterization, and structures; these structures will surely provide helpful information regarding their coordination capabilities. Researchers have used Schiff-base ligands of hydrazone molecules and many hydrazine-based Schiff bases to perform synthetic, structural, spectroscopic, theoretical, and biological studies. These structures will undoubtedly provide insightful data on their coordination characteristics [14,15,16,17].
SARS-CoV-2 is the causative agent of the COVID-19 pandemic, belonging to the coronaviridae family. Despite the significant decrease in the COVID-19 infections worldwide due to vaccination, there are still new cases, and it is possible that mutations will lead to vaccine-resistant mutant strains emerging. The main protease (Mpro) is an essential enzyme for viral replication and transcription. Therefore, it was thoroughly investigated as a key target to finding a suitable medication for the disease [18,19,20,21,22,23,24]. On another hand, the nonstructural protein 16 (NSP16) is S-adenosylmethionine-dependent, and methyltransferase forms a heterodimer with its cofactor NSP10 and activates the action of 2’-O-methyltransferase. NSP16 is involved in the RNA methylation of the first nucleotide transcribed at the (2′-O-Me) position of the ribose, a major step in evading immune responses triggered by viral RNAs by preventing host recognition [25,26,27].
In this work, a ligand and its Ni(II), Pd(IV), Pt(IV), Zn(II), and Hg(II) complexes were optimized using density functional theory (DFT), with the B3LYP functional, 6-31+G(d, p) basis set for the ligand atoms and LANL2DZ for the center metal ions. The experimental stretching frequencies were used to talk about how well the chosen levels for the complexes were working. The experimental stretching frequencies were used to discuss the acceptability of the levels chosen for the complexes described. The relationship between structure and activity was investigated using a number of quantum chemical identifiers, including the highest occupied molecular orbital (EHOMO), energy of the lowest unoccupied molecular orbital (ELUMO), energy gap between LUMO and HOMO (EGAP), absolute hardness (η), absolute softness (σ), absolute electronegativity (χ), chemical potential (μ), electrophilicity index (ω), and global softness (S). By looking at the molecular electrostatic potential (MEP) maps, the active parts of the molecules were found. Finally, we report for the first time the dual inhibition of Mpro and NSP16 of the virus of concern, SARS-CoV-2, with the synthetic ligand and its Ni(II) and Zn(II) complexes in addition to their antibacterial activity.
2. Results and Discussion
2.1. Synthesis
The condensation of (4-chloro-3-methylphenyl)hydrazine with 2-Pyridine-carboxaldehyde in present glacial acetic acid for 12 h afforded off-white powder as a solo product in high yield (87%) (Scheme 1). The physical properties such as color, melting point, and elemental analysis are listed in Table 1.

Scheme 1.
Synthesis of Schiff-base ligand (Cmpy).

Table 1.
Color, yield, M.p (°C), conductivity (ohm−1 cm2 mol−1), and CHN analysis of Cmpy and its complexes.
Treatment equivalent molar of (E)-2-((2-(4-chloro-3-methylphenyl) hydrazono) methyl) pyridine ligand (Cmpy) with chloride salts of Ni(II), Pd(II), Pt(II), Zn(II), and Hg(II) (Scheme 2) gave complexes of the formula [MCl2(Cmpy)], where MII = Ni(1), Pd(2), Pt(3), Zn(4), and Hg(5). The results suggest that the Cmpy ligand performs in a bidentate-chelating fashion through the nitrogen atoms of azomethine and pyridyl ring groups to afford a square planner around Ni(1), Pd(2), and Pt(3) ions or a tetrahedral geometry around Zn(4) and Hg(5) with two chloride ions. The synthesized complexes were characterized by elemental analysis, IR, NMR, and molar conductivity measurements (see Table 1, Table 2 and Table 3).
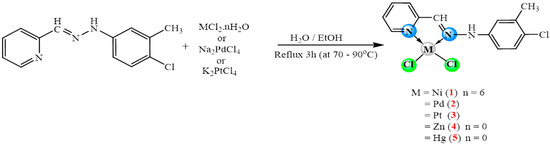
Scheme 2.
Preparation of complexes 1–5.
Table 2.
Experimental and calculated IR results, with determination of the correlation coefficient.
Table 3.
Chemical shifts (in ppm) of the Cmpy ligand and its complexes.
2.2. IR Spectra
The infrared spectrum of the Schiff-base (Cmpy) ligand (Figure S1) showed a band at 3240 cm−1 due to the stretching vibration of the NH group, and a new band displayed at 1606 cm−1 due to the ν(C=N) azomethine group, whereas the ν(C=N) of the pyridyl ring displayed at 1689 cm−1. Moreover, the IR spectrum showed the ν(C-H) stretching of the aliphatic and aromatic groups within the ranges 2918–2987 cm−1 and 2997–3055 cm−1, respectively. From the observation of the IR spectra Schiff-base complexes 1–5 (Figures S2–S6), the azomethine group was shifted towards a lower frequency (within the 1531–1550 cm−1 range) compared with the free ligand, indicating that the Schiff-base ligand was coordinated through the nitrogen atom of the azomethine group. As well as the υ(C=N) appearing within the 1595–1622 cm−1 range, this band was shifted to a lower frequency compared with the frequency of that in the free ligand. These data indicate that the ligand is coordinated to ions through the nitrogen atom of the pyridyl ring. This was confirmed by the emergence of bands belonging to the ν(M-N) bond, in the range of 447–511 cm−1. Other selected IR bands of the free ligand and their complexes are listed in Table 2.
2.3. NMR Spectra
The 1H NMR spectrum of the Cmpy ligand (Figure S7) showed four singlet peaks at δH = 11.25 ppm, 7.80 ppm, 7.65 ppm, and 2.38 ppm attributed to the protons of the NH, CH=N, H8, and CH3 group, respectively, and its integration indicates that it corresponds to the number of protons. The H5 and H3 displayed doublet peaks at δH = 8.62 ppm and 7.92 ppm, respectively. Moreover, the spectrum displayed a doublet of doublet peaks at δH = 7.56 ppm, and due to the proton in position H4, the complementarity of each of the three peaks of the pyridyl ring indicates that they correspond to one proton. Moreover, the spectrum showed the protons in positions H2, H11, and H12 as a multiplet peak with a δH = 7.25–7.33 ppm range. Scheme 3 shows the numbering of the atoms in the Cmpy ligand.
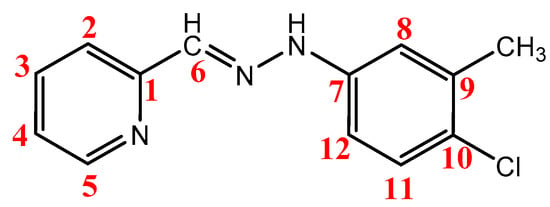
Scheme 3.
The numbering of the atoms in the Cmpy ligand.
In the 1H NMR spectra of the M(II) complexes 1–5 (Figures S8–S12), the azomethine proton signals at 7.80–7.85 ppm (s, 1H) for complexes 1–3 or with a multiplet peak for complexes 4–5. This peak does not shift, or a slight shift was noted in the M(II) complexes. The complexes showed the proton of NH signals at δ 10.89–11.09 (s, 1H) for complexes 1–5. A slight shift was distinguished in the proton signal of the NH group after being coordinated with metal ions. The protons of the methyl group displayed as a singlet peak within a δ 2.10–2.37 ppm range. Other aromatic proton signals of the phenyl and pyridyl rings are listed in Table 3.
In the 13C NMR spectrum (Figure S13), the signals due to (C5=N) and (CH=N) azomethine and CH3 are observed at 162.12, 155.75, and 22.02 ppm, respectively. Aromatic carbons give signals in overlapped areas of the spectrum with chemical shift values from 120.21 to 132.41 ppm. Comparison of 13C NMR spectra complexes with the free Schiff base demonstrates that those carbon atoms connected with bonding sites were slightly shifted to the downfield region in the M(II) complexes (Figures S14 and S15). The (C5=N) and (CH=N) azomethine showed within 159.76–161.14 and 151.30–153.45, respectively. Other carbon signals of the phenyl and pyridyl rings are listed in Table 4.
Table 4.
Carbon chemical shifts (in ppm) of the Cmpy ligand and their complexes.
2.4. Antibacterial Studies
The development of novel bacterial strains that are resistant to modern antibiotics is a serious issue for public health. Therefore, it is important to discover substitute substances that have drug-like properties. Recently, scientists have examined the production of novel metal complexes with novel chemical ligands and evaluated their antibacterial efficacy.
In this study, we measured the diameter of the zone of inhibition (DIZ) and the activity index of the prepared compounds’ antibacterial activity against two pathogenic bacteria (S. aureus and P. aeruginosa) and compared it with the standard drug, which was the commercial medication amoxicillin (10−3M in DMSO), as a positive control and a DMSO (the solvent in 30%) as a negative control (which had no antimicrobial properties by itself). The standard error for the test was ±0.03 %, and the tests were three frequent periods at similar conditions (Table 5).
Table 5.
Diameter inhibition zone (DIZ in mm) and activity index (A.I. in %) of the prepared compounds.
The findings showed that complexes 1–5 have better antibacterial activity than Cmpy, and their activity was in the following order: 4 > 1 > 2 = 3 > 5 > Cmpy against S. aureus, whereas against P. aeruginosa their activity was in the following order: 4 > 1 > 2 = 5 > 3 > Cmpy. The solubility of a compound in the cell membrane is thought to play a significant role in antibacterial activity because it allows for the passage of only soluble materials in lipids, according to the overtone concept [28]. Alternatively, Tweedy’s chelation theory [29] contends that the polarity of a metal ion is greatly reduced due to the ligand orbital overlap and the positive charge division of the central metallic ion with the donor atoms of the ligand. The activity index values of the synthesized compounds were recoded as the following equation:
2.5. DFT Studies
2.5.1. DFT Calculations Studies
In order to identify some quantum parameters, such as the partial charge of the atoms, the energy molecular orbitals (HOMO and LUMO), the chemical potential, etc., we incorporated theoretical research using the theorem “DFT” based on computation B3LYP/6-311G (d, p) into the practical portion. By perhaps exposing the favorable sites, they can cause a decline in antioxidant activity.
The results of the theoretical DFT calculations (Figure 1 and Figure 2) showed different geometrical structures for the ligand and its metal complexes which agree with the experimental results. Ni, Pd, and Pt complexes have square planner geometry while Zn and Hg complexes have tetrahedral geometry in good agreement with the experimental data of electronic spectra and magnetic moment measurements. The approximate DFT calculations for the electronic energy, heat capacity, entropy (S), thermal energy, polarizability, and dipole moment of the ligand and its complexes are summarized in Table 6.
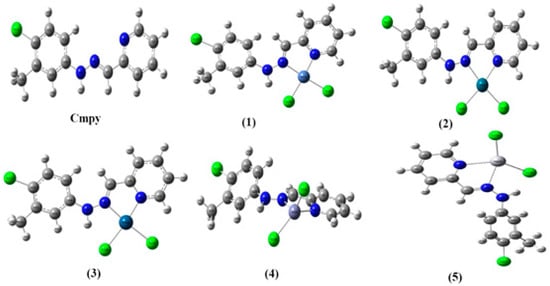
Figure 1.
Optimized geometrical structures of Cmpy and its complexes with atomic numbering.
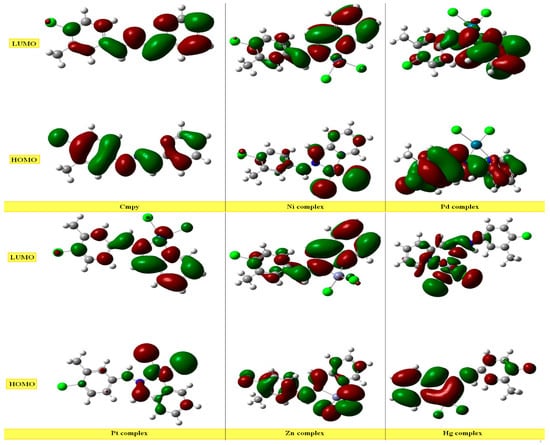
Figure 2.
Occupied higher energy and unoccupied lower energy molecular orbitals of the ligand and its complexes.
Table 6.
Electronic energy (Hartree/particle), thermal energy (kcal/mol), heat capacity (Cv), entropy (S) (cal/mol-kelvin), polarizability α (a.u.), and dipole moment (Debye) of Cmpy, Ni, Pd, Pt, Zn, and Hg.
Since dipole moments are used to express a molecule’s polarity, it is clear that the more polar molecules dissolve more readily than less polar ones, in the order of Cmpy < 5 < 4 < 1 < 3 < 2.
Pd is the highest dipole moment where Cmpy is the lowest one. The degree to which a substance is polarizable is determined by the extent to which a charge approach alters the resistance of the electron cloud in a molecular system. It also depends on the structure of the chemicals and the size of the molecules. Chemicals with bigger molecules are more polarizable. Although Cmpy is the smallest and least polarizable (211.62 a.u.), the Pt complex is projected to have the highest polarizability because it is the most complicated (270.78 a.u.).
Of all the key thermodynamic variables identified for proteins, heat capacity has the most intricate web of ideas and the widest range of consequences for protein folding and binding. Entropy and enthalpy are given a temperature dependency, changing their signs and determining which one will dominate. The order by heat capacity Cv is Cmpy < 2 < 3 < 1 < 4 < 5, which results in maximum stability and frequent cold denaturation for an unfolding protein.
2.5.2. Study of Frontier Orbitals
The energy of the highest occupied (HOMO) and the lowest unoccupied molecular orbital (LUMO), which are related to the ionization potential and electron affinity, defines the electron transfer process as the electron donor or acceptor unit. The gap energy, or ΔE, is the absolute energy difference between the boundary molecular orbitals, which expresses the reactivity of the compounds. When energy deficits are small, this activity becomes crucial [30,31].
The charge density distribution of the HOMO and LUMO levels for the studied molecules is shown in Figure 2. HOMOs completely covert whole molecule, except the methyl group, with LUMOs being mainly located throughout the molecular structure of L, except for the methyl group and two carbon atoms in the ring. In case of metal complexes, different locations of HOMO and LUMO are present. The lowest EGAP energy is calculated at 4.489 ev and illustrates the highest reactivity of the HMHP molecule, which agrees well with the biological experimental data, except for the H2O2 scavenging activity, where HIN is the best at this activity.
The results of the FMO energy analysis revealed that the energies of HOMOs of Zn are higher compared with other complexes and the ligand. However, the destabilization of the LUMO level is found to be higher in Zn than the others. Consequently, the energy gap is in the order of Pd < Pt < Ni < Zn < Hg< Cmpy.
According to the FMOs theory, the HOMO and LUMO energy levels have the greatest effects on the bioactivities of tiny structural medicines. The LUMOs receive electrons, although it is mostly HOMOs that do. Evidently, the energy of HOMOs varies for each chemical under study. Pd had the most HOMOs compared to the other compounds, suggesting that it would be a better medication for electron donation. It is interesting to note that there are a number of hydrophilic interactions at the biggest energy gap, ΔE = 3.71 eV, that might help molecules connect to receptors. This implies that such hydrophilic interactions have a significant impact on how well tiny medicines bind to receptors. During the binding process, the HOMO of a certain medication and the LUMO of the nearby residues may share orbital interactions (Table 7).
Table 7.
Calculated EHOMO (EH), ELUMO (EL), energy band gap (EH–EL), chemical potential (μ), electronegativity (χ), global hardness (η), global softness (S), global electrophilicity index (ω), and softness (σ) for the ligand and its complexes.
2.5.3. Chemical Reactivity Descriptors
Calculations, such as EHOMO and ELUMO, obtain the quantium chemical parameters of organic compounds. Additional parameters, such as (ΔE), absolute electro-negativities (v), chemical potentials (Pi), absolute hardness (g), absolute softness (r), global electrophilicity (x), and global softness (S) were calculated by Equations (1–6) [32,33].
χ = −1/2 (ELUMO + EHOMO)
µ = −χ = 1/2 (ELUMO + EHOMO)
η = 1/2 (ELUMO − EHOMO)
S = 1/2 η
ω = µ2/2 η
The inverse value of the global hardness is designed as the softness (σ), as follows:
σ = 1/ η
For predicting biological activity, some quantum chemical metrics derived from optimized molecule structures are helpful. There is a remarkable correlation between estimated quantum chemical parameters and experimental inhibitory activities in much recent research. Since HOMO is the electron-containing orbital with the highest energy, it serves as an electron donor orbital. EHOMO increases, which facilitates inhibitor electron emission and boosts inhibitory activity. The capacity of the inhibitor to interact rises with decreased ELUMO energy, which results in an increase in inhibitory activity. When comparing the inhibitory actions of different chemical species, the chemical identifier absolute electronegativity (χ) is taken into account. Low electronegativity values enable facile electron donation, which results in high inhibitory activity for these inhibitors. Electronegativity is the exact opposite of chemical potential (μ). Because of this, an increase in chemical potential causes an increase in inhibitory activity. The global electrophilic force of a molecule is represented numerically by the electrophilicity index (ω). The ability to accept electrons is gauged by the electrophilicity index, which measures chemical reactivity. These indices were discussed in the section on the calculating process. They assert that a decrease in the electrophilicity index causes an increase in biological reactivity. The rising value of global softness suggests that the compound’s biological activity is rising. In this case, Table 7 shows the parameters that were used to evaluate the compounds listed above.
2.5.4. Molecular Electrostatic Potential (MEP)
The electrostatic potential is a meaningful statistic for describing electrophilic attack sites, nucleophilic reactions, and hydrogen bonding interactions. The electrostatic potential maps were created to analyze a molecule’s structural characteristics. The significance of MESP lines shows the color grading scheme’s form, size, negative, positive, and neutral electrostatic potential areas. The most positive electrostatic potential of the molecules is shown by the blue color-coding region (signifying a severely electron-deficient region), while the most electronegative potential is represented by the red color-coding region (signifying an electron-rich region). The negative and positive regions of MEP are related to electrophilic and nucleophilic reactivity [34]. The MEP energy map (Figure 3) shows that the distribution of negative potential sites of the ligand molecule (Cmpy) is shown only on the nitrogen atoms N4 and N8, while the positive potential sites are located around the hydrogen atoms. In case of Ni, Pd, Pt, Zn, and Hg complexes, distribution of negative potential sites is shown only on the chlorine atoms, while the positive potential sites are located around the hydrogen atoms. It can be considered that the positive sites play an essential role in donating hydrogen atoms and electrons to the oxidizing agents during reduction processes. The variance in the binding affinities of the chemical may be mostly caused by differences in how the electrostatic potential around the compound is mapped.
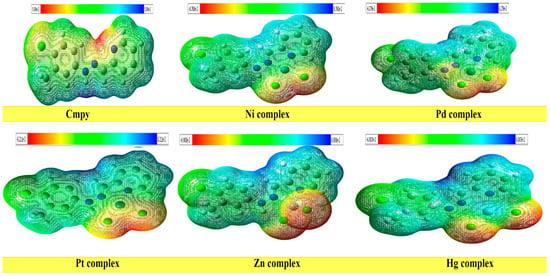
Figure 3.
Molecular electrostatic potentials (MEP) of the Cmpy and its complexes.
2.5.5. Mulliken Atomic Charges
Mulliken atomic charge tends to produce qualitative results at best; it is very useful for estimating the partial atomic charges of molecular systems. The DFT determined that the nitrogen atoms of the ligand Cmpy have the highest distribution of negative charges at N4 nitrogen of the azomethine group (−0.02) and N8 nitrogen of the pyridine ring (−0.01). This coordination of the ligand to the metal ions through both nitrogen atoms is suggested by this conclusion, which is consistent with the experimental findings. This shows that the nitrogen atoms in the azomethine group and the N8 nitrogen of the pyridine ring are reactive sites for metal attack. As a result of our discovery that the N4 and N8 change from a negative to a positive ionic character during chemical processes, they are directed toward the loss of electrons. These findings help characterize the molecular structure’s most reactive regions. Table S1 lists the Mulliken atomic charges in numerical order.
2.5.6. Analytical Study of FT-IR spectra
All normal vibrational modes have been assigned using the computed vibrational modes, and related parameters are obtained from the Gauss View of the Gaussian 09 program package [35]. Table 2 provides a summary of all the theoretical and experimental data for the primary wavenumber functions examined. The theoretical calculations are attributed to the gaseous phase of the molecule, but the observed results are true for the solid phase of the molecule, which helps to explain the disparities between the experimental and theoretical aspects. Additionally, the calculated findings and the experimental results agree with R2, or the coefficient of linear correlation, with a value between 0.9965 and 0.9978. The evaluation of the peaks unique to each ultimate structure’s spectrum as revealed by practical and theoretical data exposed by the processing of spectral IR, as well as a helpful correlation discovered therein, is summarized in Table 2.
2.6. Molecular Docking
The interaction between the Cmpy and its Ni(II) and Zn(II) complexes and the active sites of the target receptors of Mpro and NSP16 was observed in order to assess the validity of the compounds as potential inhibitors. The residues of ASP 6928, ASP 6897, ASP 6912, and ASP 6913 were determined as the active site in NSP16 [27,36]. His 41 and Cys 145 are the catalytic dyads of Mpro, and the important residue of Glu 166 was given extra attention. The ligand and its Ni(II) and Zn(II) complexes were docked with the NSP16 enzyme. They showed a low score energy and high binding affinity percentage, as shown in Table 8 and depicted in Figure 4. They all interact with the NSP16 enzyme pocket active sites forming a hydrogen bond to ASP 6928. Zn (II) complex was additionally forming a hydrogen bond with ASP 6897. Their binding affinity was significantly improved by adding a metal complex to the ligand. The complexation makes the interaction to the residues of ASP 6928 and ASP 6912 become more accessible and improved the score energies. Interestingly, this interaction was comparable to sinefungin, which docked as the reference inhibitor for NSP16.
Table 8.
The binding affinity of the Schiff-base ligand and its complexes docked with the active site of the nonstructural protein 16 of SARS-CoV-2.
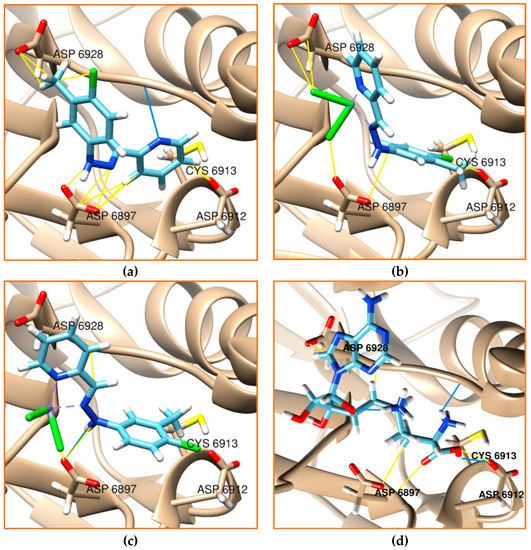
Figure 4.
Schiff-base ligand and its complexes docked with the active site of the nonstructural protein 16 of SARS-CoV-2; (a) ligand, (b) nickel complex, (c) zinc complex, and (d) sinefungin. The hydrocarbon skeleton of the ligand is cyan, nitrogen is blue, and oxygen is red. Hydrogen bonds were showed in blue lines and van der Waals in yellow.
The Cmpy ligand does not dock to the active sites of Mpro, unlike the complexes. The Zn (II) complex showed the lowest binding energy value up to −7.2 kcal/mol (Table 9); further, it has shown the ability to form hydrogen bonds to LEU 287 and LEU 75 (Figure 5). Thus, these complexes may offer potential Mpro and NSP16/NSP10 methyltransferase complex inhibitors. Based on the Mpro organometal inhibitor reported in the literature [37], we can predict the mechanisms of inhibition of Mpro using these complexes. As it is known, the electrophilicity of Ni and Zn will be attacked by the thoil of Cys 145 forming the Mpro adduct (Figure 5c). It may explain why the ligand free from metal (Cmpy) dose docked with the active sites of Mpro and highlighted the importance of organo-metal inhibitors in the inhibition of Mpro of SARS-CoV-2.
Table 9.
The binding affinity of the Schiff-base ligand complexes docked with the active site of the main protease of SARS-CoV-2.
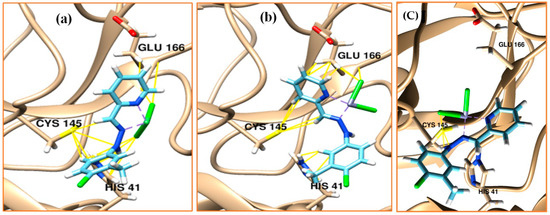
Figure 5.
Schiff-base ligand complexes docked with the active site of the main protease of SARS-CoV-2; (a) nickel complex, (b) and (c) zinc complex. The hydrocarbon skeleton of the ligand is cyan, nitrogen is blue, and oxygen is red. Hydrogen bonds were showed in blue lines and van der Waals in yellow.
Previous reports have shown the ability of the natural product sinefungin to inhibit NSP16 [35,38], bioactive compounds from tea, Ertapenem [39], mexicanin E [40], CID 135566620 compound from PubChem [41], dihydroagarofuran and myristicin [42], and other natural and synthetics compounds [43,44,45,46,47], whereas the Mpro was inhibited by caffeine [48,49], Methylxanthines [50], natural product isolates [51], Glycyrrhizin [52], ML188 [53] Schiff-base ligands, pyrimidonic and pyridonic pharmaceuticals [54], hepatitis C virus protease drugs [55], and other inhibitors [56,57,58,59,60,61,62]. Our results from molecular docking may represent for the first time the role of metalorganic in the inhibition of Mpro and NSP16.
2.7. In Silico ADME Predictions
Critical physiochemical parameters were established using an in silico ADME (absorption, distribution, metabolism, and excretion) analysis of the ligand and its metal complexes using the Swiss ADME web tool [63,64,65]. Table 4, Table 5 and Table 6 provide the physiochemical properties determined using an in silico ADME (absorption, distribution, metabolism, and excretion) analysis of the ligand and its metal complexes using the Swiss ADME web tool [63,64,65]. Tables S2–S4 provide the physiochemical properties (lipophilicity, water solubility, pharmacokinetics, and drug similarity values) for the ligand and its metal complexes. All the complexes and the ligand are present with good membrane permeability (BBB) and strong gastrointestinal absorption (GI). All compounds adhere to and satisfy the Lipinski rule, and their pharmacophore or drug-like characteristics demonstrate that all of their features fall within an acceptable range. The substances that, in this in silico ADME prediction, satisfied Lipinski’s rule of five conditions without deviating from any of them would make good candidates for oral medications.
3. Materials and Methods
3.1. Materials and Instrumental
All materials and solvents of analytical grade were supplied and used without purification. The FT-IR spectra were recorded in 4000–400 cm−1 as KBr pellets on a SHIMADZU FT-IR instrument. The NMR spectra were specified on Bruker 400 MHz spectrometer using DMSO-d6 as a solvent. UV–vis data were measured within 800–200 nm by Agilent Cary-60 spectrophotometer.
3.2. Synthesis of Schiff-Base Ligand
The Schiff-base ligands were synthesized by the method described by S. Parvarinezhad and M. Salehi (2020) [66].
3.3. Synthesis of Complex [NiCl2(Cmpy)] (1)
A solution of (E)-2-((2-(4-chloro-3-methylphenyl) hydrazono)methyl)pyridine (Cmpy) (0.100 g, 0.407 mole) in EtOH (10 mL) was added to an aqueous solution of nickel chloride (NiCl2·6H2O) (0.097 g, 0.407 mole) in 10 mL with stirring. A green ppt. was formed. The mixture was refluxed for 3 h and then filtered off, washed with distilled water, and dried under vacuum. The product recrystallized from DMSO/EtOH (2:1, V:V) to afford a green solid powder (yield: 0.156 g, 91%. m.p (°C): 230 (decompose)).
The following complexes, [PdCl2(Cmpy)] (2), [PtCl2(Cmpy)] (3), [ZnCl2(Cmpy)] (4), and [HgCl2(Cmpy)] (5), were prepared and isolated in similar method.
3.4. Antibacterial Studies
The microbial activity effects of the compounds were screened against Pseudomonas aeruginosa and Staphylococcus aureus using diffusion method agar nutrient describe by Baurer et al. [38]. Briefly, in 0.001M of DMSO solution of synthesized compounds, the results were compared with amoxicillin as the standard drug. Then, the zone of inhibition instead of established inhibition region was recorded. The activities of the free Cmpy and their metal complexes were established by calculating the activity index (AI).
3.5. DFT Study
For the ligand, Ni(II), Pd(IV), Pt(IV), Zn(II), and Hg(II) complexes, geometry optimization calculations and vibrational analysis were first carried out. The software Gaussian 09 was used for all calculations [35]. The ligand and its metal complexes were optimized using density functional theory (DFT), with the B3LYP functional, 6-31+G(d, p) basis set for the ligand atoms and LANL2DZ for the center metal ions. The B3LYP functional is said to have assisted researchers in achieving satisfactory transition metal complex geometries at low computational costs [67,68,69,70]. B3LYP/LANL2DZ+6-31+G(d, p) has previously been used successfully, and its applicability to metal complexes has been clearly stated [71].
3.6. Molecular Docking
The ligand and its Ni(II) and Zn(II) complexes were prepared as described in the previous section. The crystal structure of the Mpro (PDB ID: 6Y2E) and the NSP16 (PDB ID: 6WKQ) of SARS-CoV-2 were downloaded from the Protein Data Bank database. Then, water residues were removed, their net charge was computed using antechamber [72], and its energy was minimized utilizing the Molecular Modeling Toolkit plugin UCSF Chimera using 1000 steepest descent steps of 0.02 Å and 20 conjugate gradient steps of 0.02 Å, as described previously [73,74,75]. Molecular docking was accomplished using the AutoDock Vina using grid box size of (35.0, 65.0, 65.0) Å, centered at (−16.0 × −24.0 × 17.0) Å and (43.87, 42.76, 45.74) Å, centered at (91.7 × 18.82 × −2.96) Å for Mpro and NSP16, respectively. UCSF Chimera has been used for images processing and bonds interactions visualization [75,76,77,78].
4. Conclusions
To sum up, the coordination complexes of Hg(II), Pd(II), Pt(II), Ni(II), and Zn(II) with 4-chloro-3-methyl phenyl hydrazine Schiff-base ligand were synthesized and characterized using thermal and elemental analyses, magnetic properties, and different spectroscopic techniques including FT-IR and 13C and 1H NMR. Their properties were investigated using DFT. The synthesized complexes were investigated against bacteria (E. coli, P. aeruginosa, and St. aureus). Furthermore, the ligand and its Ni(II) and Zn(II) complexes were docked with Mpro and NSP16/NSP10 methyltransferase complex active sites with excellent binding affinity and binding energy. In this way, these Ni(II) and Zn(II) complexes highlighted the role of metalorganics in the inhibition of Mpro and NSP16 and may offer new hope for COVID-19 therapy.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/inorganics11020063/s1, Figure S1: IR spectrum of complex Cmpy ligand; Figure S2: IR spectrum of complex 1; Figure S3: IR spectrum of complex 2; Figure S4: IR spectrum of complex 3; Figure S5: IR spectrum of complex 4; Figure S6: IR spectrum of complex 5; Figure S7: 1H NMR spectrum of Cmpy; Figure S8: 1H NMR spectrum of complex 1; Figure S9: 1H NMR spectrum of complex 2; Figure S10: 1H NMR spectrum of complex 3; Figure S11: 1H NMR spectrum of complex 4; Figure S12: 1H NMR spectrum of complex 5; Figure S13: 13C NMR spectrum of Cmpy; Figure S14: 13C NMR spectrum of complex 1; Figure S15: 13C NMR spectrum of complex 2. Table S1: Mulliken atomic charges of Cmpy and its complexes determined by DFT using 6-311G-based B3LYP (d, p).; Table S2: Physiochemical properties of synthesized compounds.; Table S3: Lipophilicity and water solubility of synthesized compounds.; Table S4: Pharmacokinetics and druglikeness of synthesized compounds.
Author Contributions
A.S.M.A.-J.: conceptualization; data curation; formal analysis; methodology; visualization; writing—original draft; writing—review and editing. A.O.E.: conceptualization; data curation; formal analysis; methodology; visualization; writing—original draft; writing—review and editing. M.M.A.-K.: data curation; formal analysis; methodology; visualization; writing—original draft; writing—review and editing. T.A.Y.: conceptualization; funding acquisition; data curation; formal analysis; methodology; visualization; writing—original draft; writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Deanship of Scientific Research, Imam Mohammad Ibn Saud Islamic University (IMSIU), Saudi Arabia, grant no. 21-13-18-065.
Data Availability Statement
In the supporting information of this article, you will find the data that support the findings of this study.
Conflicts of Interest
The authors declare that there is no conflict of interest to be reported.
References
- Turan, N.; Buldurun, K.; Bursal, E.; Mahmoudi, G. Pd (II)-Schiff base complexes: Synthesis, characterization, Suzuki-Miyaura and Mizoroki-Heck cross-coupling reactions, enzyme inhibition and antioxidant activities. J. Organomet. Chem. 2022, 970, 122370. [Google Scholar] [CrossRef]
- Alfonso-Herrera, L.A.; Rosete-Luna, S.; Hernández-Romero, D.; Rivera-Villanueva, J.M.; Olivares-Romero, J.L.; Cruz-Navarro, J.A.; Soto-Contreras, A.; Corona, A.A.; Morales-Morales, D.; Colorado-Peralta, R. Transition metal complexes with tridentate Schiff bases (O N O and O N N) derived from salicylaldehyde: An analysis of their potential anticancer activity. ChemMedChem. 2022, 17, e202200367. [Google Scholar] [CrossRef] [PubMed]
- Fnfoon, D.Y.; Al-Adilee, K.J. Synthesis and spectral characterization of some metal complexes with new heterocyclic azo imidazole dye ligand and study biological activity as anticancer. J. Mol. Struct. 2022, 1271, 134089. [Google Scholar] [CrossRef]
- Ahmetali, E.; Yıldız, B.; Ahi, E.E.; Durmuş, M.; Şener, M.K. Synthesis, Photophysical and Photochemical Properties of Unsymmetrical Zinc (II) Phthalocyanines Bearing 8-Hydroxyquinoline Unit. Polyhedron 2022, 226, 116111. [Google Scholar] [CrossRef]
- Hassan, A.M.; Said, A.O.; Heakal, B.H.; Younis, A.; Aboulthana, W.M.; Mady, M.F. Green Synthesis, Characterization, Antimicrobial and Anticancer Screening of New Metal Complexes Incorporating Schiff Base. ACS Omega 2022, 7, 32418–32431. [Google Scholar] [CrossRef] [PubMed]
- Odularu, A.T. Manganese Schiff Base Complexes, Crystallographic Studies, Anticancer Activities, and Molecular Docking. J. Chem. 2022, 2022, 7062912. [Google Scholar] [CrossRef]
- Frei, A.; Zuegg, J.; Elliott, A.G.; Baker, M.; Braese, S.; Brown, C.; Chen, F.; Dowson, C.G.; Dujardin, G.; Jung, N. Metal complexes as a promising source for new antibiotics. Chem. Sci. 2020, 11, 2627–2639. [Google Scholar] [CrossRef]
- Liu, X.; Hamon, J.-R. Recent developments in penta-, hexa-and heptadentate Schiff base ligands and their metal complexes. Coord. Chem. Rev. 2019, 389, 94–118. [Google Scholar] [CrossRef]
- Catalano, A.; Sinicropi, M.S.; Iacopetta, D.; Ceramella, J.; Mariconda, A.; Rosano, C.; Scali, E.; Saturnino, C.; Longo, P. A review on the advancements in the field of metal complexes with Schiff bases as antiproliferative agents. Appl. Sci. 2021, 11, 6027. [Google Scholar] [CrossRef]
- Ghanghas, P.; Choudhary, A.; Kumar, D.; Poonia, K. Coordination metal complexes with Schiff bases: Useful pharmacophores with comprehensive biological applications. Inorg. Chem. Commun. 2021, 130, 108710. [Google Scholar] [CrossRef]
- Jamil, W.; Solangi, S.; Ali, M.; Khan, K.M.; Taha, M.; Khuhawar, M.Y. Syntheses, characterization, in vitro antiglycation and DPPH radical scavenging activities of isatin salicylhydrazidehydrazone and its Mn (II), Co (II), Ni (II), Cu (II), and Zn (II) metal complexes. Arab. J. Chem. 2019, 12, 2262–2269. [Google Scholar] [CrossRef]
- El-Tabl, A.S.; Aly, F.A.; Shakdofa, M.M.; Shakdofa, A.M. Synthesis, characterization, and biological activity of metal complexes of azohydrazone ligand. J. Coord. Chem. 2010, 63, 700–712. [Google Scholar] [CrossRef]
- Manimaran, P.; Balasubramaniyan, S.; Azam, M.; Rajadurai, D.; Al-Resayes, S.I.; Mathubala, G.; Manikandan, A.; Muthupandi, S.; Tabassum, Z.; Khan, I. Synthesis, spectral characterization and biological activities of Co (II) and Ni (II) mixed ligand complexes. Molecules 2021, 26, 823. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Chowdhury, D.; Gomila, R.M.; Chattopadhyay, S. Recent advances on the tetrel bonding interaction in the solid state structure of lead complexes with hydrazine based bis-pyridine Schiff base ligands. Polyhedron 2022, 216, 115670. [Google Scholar] [CrossRef]
- Parvarinezhad, S.; Salehi, M. Synthesis, characterization, anti-proliferative activity and chemistry computation of DFT theoretical methods of hydrazine-based Schiff bases derived from methyl acetoacetate and α-hydroxyacetophenone. J. Mol. Struct. 2021, 1225, 129086. [Google Scholar] [CrossRef]
- Dalapati, S.; Alam, M.A.; Jana, S.; Karmakar, S.; Guchhait, N. “Test kit” for detection of biologically important anions: A salicylidene-hydrazine based Schiff base. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2013, 102, 314–318. [Google Scholar] [CrossRef]
- Jana, S.; Dalapati, S.; Alam, M.A.; Guchhait, N. Spectroscopic, colorimetric and theoretical investigation of Salicylidene hydrazine based reduced Schiff base and its application towards biologically important anions. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2012, 92, 131–136. [Google Scholar] [CrossRef]
- Elkaeed, E.B.; Eissa, I.H.; Elkady, H.; Abdelalim, A.; Alqaisi, A.M.; Alsfouk, A.A.; Elwan, A.; Metwaly, A.M. A Multistage In Silico Study of Natural Potential Inhibitors Targeting SARS-CoV-2 Main Protease. Int. J. Mol. Sci. 2022, 23, 8407. [Google Scholar] [CrossRef]
- Lopes, A.J.O.; Calado, G.P.; Fróes, Y.N.; Araújo, S.A.d.; França, L.M.; Paes, A.M.d.A.; Morais, S.V.d.; Rocha, C.Q.d.; Vasconcelos, C.C. Plant Metabolites as SARS-CoV-2 Inhibitors Candidates: In Silico and In Vitro Studies. Pharmaceuticals 2022, 15, 1045. [Google Scholar] [CrossRef]
- Bhullar, K.S.; Nael, M.A.; Elokely, K.M.; Drews, S.J.; Wu, J. Structurally Modified Bioactive Peptide Inhibits SARS-CoV-2 Lentiviral Particles Expression. Pharmaceutics 2022, 14, 2045. [Google Scholar] [CrossRef]
- Narayanan, A.; Narwal, M.; Majowicz, S.A.; Varricchio, C.; Toner, S.A.; Ballatore, C.; Brancale, A.; Murakami, K.S.; Jose, J. Identification of SARS-CoV-2 inhibitors targeting Mpro and PLpro using in-cell-protease assay. Commun. Biol. 2022, 5, 169. [Google Scholar] [CrossRef] [PubMed]
- Kneller, D.W.; Li, H.; Phillips, G.; Weiss, K.L.; Zhang, Q.; Arnould, M.A.; Jonsson, C.B.; Surendranathan, S.; Parvathareddy, J.; Blakeley, M.P. Covalent narlaprevir-and boceprevir-derived hybrid inhibitors of SARS-CoV-2 main protease. Nat. Commun. 2022, 13, 2268. [Google Scholar] [CrossRef]
- Kisakov, D.N.; Kisakova, L.A.; Borgoyakova, M.B.; Starostina, E.V.; Taranov, O.S.; Ivleva, E.K.; Pyankov, O.V.; Zaykovskaya, A.V.; Shcherbakov, D.N.; Rudometov, A.P.; et al. Optimization of In Vivo Electroporation Conditions and Delivery of DNA Vaccine Encoding SARS-CoV-2 RBD Using the Determined Protocol. Pharmaceutics 2022, 14, 2259. [Google Scholar] [CrossRef] [PubMed]
- Fadlalla, M.; Ahmed, M.; Ali, M.; Elshiekh, A.A.; Yousef, B.A. Molecular docking as a potential approach in repurposing drugs against COVID-19: A systematic review and novel pharmacophore models. Curr. Pharmacol. Rep. 2022, 8, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Bhardwaj, V.K.; Sharma, J.; Purohit, R.; Kumar, S. In-silico evaluation of bioactive compounds from tea as potential SARS-CoV-2 nonstructural protein 16 inhibitors. J. Tradit. Complement. Med. 2022, 12, 35–43. [Google Scholar] [CrossRef]
- Krafcikova, P.; Silhan, J.; Nencka, R.; Boura, E. Structural analysis of the SARS-CoV-2 methyltransferase complex involved in RNA cap creation bound to sinefungin. Nat. Commun. 2020, 11, 3717. [Google Scholar] [CrossRef]
- Rosas-Lemus, M.; Minasov, G.; Shuvalova, L.; Inniss, N.L.; Kiryukhina, O.; Brunzelle, J.; Satchell, K.J. High-resolution structures of the SARS-CoV-2 2′-O-methyltransferase reveal strategies for structure-based inhibitor design. Sci. Signal. 2020, 13, eabe1202. [Google Scholar] [CrossRef]
- Tweedy, B.G. Plant Extracts with Metal Ions as Potential Antimicrobial Agents. Phytopathology 1964, 55, 910–918. [Google Scholar]
- Thakur, M.L.; Coss, R.; Howell, R.; Vassileva-Belnikolovska, D.; Liu, J.; Rao, S.P.; Spana, G.; Wachsberger, P.; Leeper, D.L. Role of lipid-soluble complexes in targeted tumor therapy. J. Nucl. Med. 2003, 44, 1293–1300. [Google Scholar]
- Lewis, D.; Ioannides, C.; Parke, D. Interaction of a series of nitriles with the alcohol-inducible isoform of P450: Computer analysis of structure—Activity relationships. Xenobiotica 1994, 24, 401–408. [Google Scholar] [CrossRef]
- Shahidha, R.; Muthu, S.; Raja, M.; Muhamed, R.R.; Narayana, B.; Nayak, P.S.; Sarojini, B. Spectroscopic (FT-IR, FT-Raman), first order hyperpolarizabilities, NBO, Fukui function and molecular docking study of N-(4-Chloro-3-methylphenyl)-2-phenylacetamide. Optik 2017, 140, 1127–1142. [Google Scholar] [CrossRef]
- Fukui, K. Role of Frontier Orbitals in Chemical Reactions. Science 1982, 218, 747–754. [Google Scholar] [CrossRef]
- Yang, W.; Parr, R.G. Hardness, Softness, and the Fukui Function in the Electronic Theory of Metals and Catalysis. Proc. Natl. Acad. Sci. USA 1985, 82, 6723–6726. [Google Scholar] [CrossRef] [PubMed]
- Secretan, P.-H.; Annereau, M.; Kini-Matondo, W.; Prost, B.; Prudhomme, J.; Bournane, L.; Paul, M.; Yagoubi, N.; Sadou-Yayé, H.; Do, B. Unequal Behaviour between Hydrolysable Functions of Nirmatrelvir under Stress Conditions: Structural and Theoretical Approaches in Support of Preformulation Studies. Pharmaceutics 2022, 14, 1720. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.,: Wallingford, CT, USA, 2009. [Google Scholar]
- Shi, L.; Wen, Z.; Song, Y.; Wang, J.; Yu, D. Computational investigation of potent inhibitors against SARS-CoV-2 2′-O-methyltransferase (NSP16): Structure-based pharmacophore modeling, molecular docking, molecular dynamics simulations and binding free energy calculations. J. Mol. Graph. Model. 2022, 117, 108306. [Google Scholar] [CrossRef] [PubMed]
- Amporndanai, K.; Meng, X.; Shang, W.; Jin, Z.; Rogers, M.; Zhao, Y.; Rao, Z.; Liu, Z.J.; Yang, H.; Zhang, L.; et al. Inhibition mechanism of SARS-CoV-2 main protease by ebselen and its derivatives. Nat. Commun. 2021, 12, 3061. [Google Scholar] [CrossRef]
- Bauer, A.W.; PERRY, D.M.; KIRBY, W.M. Single-disk antibiotic-sensitivity testing of staphylococci: An analysis of technique and results. AMA Arch. Intern. Med. 1959, 104, 208–216. [Google Scholar] [CrossRef]
- Mahalapbutr, P.; Kongtaworn, N.; Rungrotmongkol, T. Structural insight into the recognition of S-adenosyl-L-homocysteine and sinefungin in SARS-CoV-2 NSP16/NSP10 RNA cap 2′-O-Methyltransferase. Comput. Struct. Biotechnol. J. 2020, 18, 2757–2765. [Google Scholar] [CrossRef]
- Eissa, I.H.; Alesawy, M.S.; Saleh, A.M.; Elkaeed, E.B.; Alsfouk, B.A.; El-Attar, A.-A.M.; Metwaly, A.M. Ligand and structure-based in silico determination of the most promising SARS-CoV-2 NSP16-NSP10 2′-o-Methyltransferase complex inhibitors among 3009 FDA approved drugs. Molecules 2022, 27, 2287. [Google Scholar] [CrossRef]
- Faletrov, Y.V.; Staravoitava, V.A.; Dudko, A.R.; Shkumatov, V.M. Application of docking-based inverse high throughput virtual screening to found phytochemical covalent inhibitors of SARS-CoV-2 main protease, NSP12 and NSP16. 2022. Available online: https://europepmc.org/article/ppr/ppr473072 (accessed on 17 December 2022).
- Nguyen, H.L.; Thai, N.Q.; Li, M.S. Identifying inhibitors of NSP16-NSP10 of SARS-CoV-2 from large databases. J. Biomol. Struct. Dyn. 2022, 40, 1–10. [Google Scholar] [CrossRef]
- Alves Borges Leal, A.L.; Fonseca Bezerra, C.; Ferreira e Silva, A.K.; Everson da Silva, L.; Bezerra, L.L.; Almeida-Neto, F.W.; Marinho, E.M.; Celedonio Fernandes, C.F.; Nunes da Rocha, M.; Marinho, M.M. Seasonal variation of the composition of essential oils from Piper cernuum Vell and Piper rivinoides Kunth, ADMET study, DFT calculations, molecular docking and dynamics studies of major components as potent inhibitors of the heterodimer methyltransferase complex NSP16-NSP10 SARS COV-2 protein. J. Biomol. Struct. Dyn. 2022, 40, 1–19. [Google Scholar]
- Gomes, J.P.A.; de Oliveira Rocha, L.; Leal, C.E.Y.; de Alencar Filho, E.B. Virtual screening of molecular databases for potential inhibitors of the NSP16/NSP10 methyltransferase from SARS-CoV-2. J. Mol. Struct. 2022, 1261, 132951. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, A.; Sharma, S.; Singh, S.K. Molecular docking studies to identify promising natural inhibitors targeting SARS-CoV-2 NSP10-NSP16 protein complex. Turk. J. Pharm. Sci. 2022, 19, 93. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Mulpuru, V.; Mishra, N. Identification of SARS-CoV-2 inhibitors through phylogenetics and drug repurposing. Struct. Chem. 2022, 33, 1789–1797. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Pitsillou, E.; Burbury, L.; Hung, A.; Karagiannis, T.C. In silico investigation of potential small molecule inhibitors of the SARS-CoV-2 NSP10-NSP16 methyltransferase complex. Chem. Phys. Lett. 2021, 774, 138618. [Google Scholar] [CrossRef]
- El Hassab, M.A.; Ibrahim, T.M.; Al-Rashood, S.T.; Alharbi, A.; Eskandrani, R.O.; Eldehna, W.M. In silico identification of novel SARS-COV-2 2′-O-methyltransferase (NSP16) inhibitors: Structure-based virtual screening, molecular dynamics simulation and MM-PBSA approaches. J. Enzym. Inhib. Med. Chem. 2021, 36, 727–736. [Google Scholar] [CrossRef]
- Romero-Martínez, B.S.; Montaño, L.M.; Solís-Chagoyán, H.; Sommer, B.; Ramírez-Salinas, G.L.; Pérez-Figueroa, G.E.; Flores-Soto, E. Possible beneficial actions of caffeine in SARS-CoV-2. Int. J. Mol. Sci. 2021, 22, 5460. [Google Scholar] [CrossRef]
- Elzupir, A.O. Caffeine and caffeine-containing pharmaceuticals as promising inhibitors for 3-chymotrypsin-like protease of SARS-CoV-2. J. Biomol. Struct. Dyn. 2022, 40, 2113–2120. [Google Scholar] [CrossRef]
- Rolta, R.; Salaria, D.; Sharma, B.; Awofisayo, O.; Fadare, O.A.; Sharma, S.; Patel, C.N.; Kumar, V.; Sourirajan, A.; Baumler, D.J. Methylxanthines as Potential Inhibitor of SARS-CoV-2: An in Silico Approach. Curr. Pharmacol. Rep. 2022, 8, 149–170. [Google Scholar] [CrossRef]
- Abdallah, H.M.; El-Halawany, A.M.; Sirwi, A.; El-Araby, A.M.; Mohamed, G.A.; Ibrahim, S.R.; Koshak, A.E.; Asfour, H.Z.; Awan, Z.A.; Elfaky, M.A. Repurposing of some natural product isolates as SARS-COV-2 main protease inhibitors via in vitro cell free and cell-based antiviral assessments and molecular modeling approaches. Pharmaceuticals 2021, 14, 213. [Google Scholar] [CrossRef]
- van de Sand, L.; Bormann, M.; Alt, M.; Schipper, L.; Heilingloh, C.S.; Steinmann, E.; Todt, D.; Dittmer, U.; Elsner, C.; Witzke, O. Glycyrrhizin effectively inhibits SARS-CoV-2 replication by inhibiting the viral main protease. Viruses 2021, 13, 609. [Google Scholar] [CrossRef] [PubMed]
- Lockbaum, G.J.; Reyes, A.C.; Lee, J.M.; Tilvawala, R.; Nalivaika, E.A.; Ali, A.; Kurt Yilmaz, N.; Thompson, P.R.; Schiffer, C.A. Crystal structure of SARS-CoV-2 main protease in complex with the non-covalent inhibitor ML188. Viruses 2021, 13, 174. [Google Scholar] [CrossRef] [PubMed]
- Elzupir, A.O. Molecular Docking and Dynamics Investigations for Identifying Potential Inhibitors of the 3-Chymotrypsin-like Protease of SARS-CoV-2: Repurposing of Approved Pyrimidonic Pharmaceuticals for COVID-19 Treatment. Molecules 2021, 26, 7458. [Google Scholar] [CrossRef]
- Gammeltoft, K.A.; Zhou, Y.; Duarte Hernandez, C.R.; Galli, A.; Offersgaard, A.; Costa, R.; Pham, L.V.; Fahnøe, U.; Feng, S.; Scheel, T.K. Hepatitis C virus protease inhibitors show differential efficacy and interactions with remdesivir for treatment of SARS-CoV-2 in vitro. Antimicrob. Agents Chemother. 2021, 65, e0268020. [Google Scholar] [CrossRef] [PubMed]
- Hamed, M.I.; Darwish, K.M.; Soltane, R.; Chrouda, A.; Mostafa, A.; Shama, N.M.A.; Elhady, S.S.; Abulkhair, H.S.; Khodir, A.E.; Elmaaty, A.A. β-Blockers bearing hydroxyethylamine and hydroxyethylene as potential SARS-CoV-2 Mpro inhibitors: Rational based design, in silico, in vitro, and SAR studies for lead optimization. Rsc. Adv. 2021, 11, 35536–35558. [Google Scholar] [CrossRef]
- Huang, Y.-H.; Yeh, C.-T.; Hsu, C.-W.; Lin, Y.-H. Reduction of ACE2 Serum Concentrations by Telbivudine in Chronic Hepatitis B Patients. Curr. Mol. Med. 2022, 23, 420–424. [Google Scholar] [CrossRef]
- Hussein, R.K.; Khouqeer, G.; Alkaoud, A.M.; El-Khayatt, A.M. Probing the Action of Screened Anticancer Triazole–Tetrazole Derivatives against COVID-19 Using Molecular Docking and DFT Investigations. Nat. Prod. Commun. 2022, 17, 1934578X221093915. [Google Scholar] [CrossRef]
- Baig, A.; Srinivasan, H. SARS-CoV-2 Inhibitors from Nigella Sativa. Appl. Biochem. Biotechnol. 2022, 194, 1051–1090. [Google Scholar] [CrossRef]
- Kashyap, P.; Bhardwaj, V.K.; Chauhan, M.; Chauhan, V.; Kumar, A.; Purohit, R.; Kumar, A.; Kumar, S. A ricin-based peptide BRIP from Hordeum vulgare inhibits Mpro of SARS-CoV-2. Sci. Rep. 2022, 12, 12802. [Google Scholar] [CrossRef]
- Mercorelli, B.; Desantis, J.; Celegato, M.; Bazzacco, A.; Siragusa, L.; Benedetti, P.; Eleuteri, M.; Croci, F.; Cruciani, G.; Goracci, L. Discovery of novel SARS-CoV-2 inhibitors targeting the main protease Mpro by virtual screenings and hit optimization. Antivir. Res. 2022, 204, 105350. [Google Scholar] [CrossRef]
- Jatczak, M.; Muylaert, K.; De Coen, L.M.; Keemink, J.; Wuy, B.; Augusti-Jins, P.; Stevens, C.V. Straightforward entry to pyrido [2, 3-d] pyrimidine-2, 4 (1H, 3H)-diones and their ADME properties. Bioorg. Med. Chem. 2014, 22, 3947–3956. [Google Scholar] [CrossRef]
- Arif, R. Design, synthesize and antiurease activity of novel thiazole derivatives: Machine learning, molecular docking and biological investigation. J. Mol. Struct. 2020, 1208, 127905. [Google Scholar] [CrossRef]
- Yousef, T.A.; Khairy, M. Synthesis, Characterization, Optical, DFT, TD DFT Studies and in Silico ADME Predictions of Thiosemicarbazone Ligand and its Au(III) Complex Orient. J. Chem. 2022, 38, 537–546. [Google Scholar] [CrossRef]
- Parvarinezhad, S.; Salehi, M. Synthesis, characterization, crystal structures, Hirshfeld surface analysis and DFT computational studies of new Schiff Bases derived from Phenylhydrazine. J. Mol. Struct. 2020, 1222, 128780. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Rassolov, V.A.; Pople, J.A.; Ratner, M.A.; Windus, T.L. 6-31G* basis set for atoms K through Zn. J. Chem. Phys. 1998, 109, 1223–1229. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Elzupir, A.O. Inhibition of SARS-CoV-2 main protease 3CLpro by means of α-ketoamide and pyridone-containing pharmaceuticals using in silico molecular docking. J. Mol. Struct. 2020, 1222, 128878. [Google Scholar] [CrossRef] [PubMed]
- Al-Janabi, A.S.; Elzupir, A.O.; Yousef, T.A. Synthesis, anti-bacterial evaluation, DFT study and molecular docking as a potential 3-chymotrypsin-like protease (3CLpro) of SARS-CoV-2 inhibitors of a novel Schiff bases. J. Mol. Struct. 2021, 1228, 129454. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Shapovalov, M.V.; Dunbrack, R.L., Jr. A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure 2011, 19, 844–858. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).