
Synthesis, Structural, and Quantum Chemical Analysis of Neutral and Cationic Ruthenium(II) Complexes with Nicotinate-Polyethylene Glycol Ester Ligands





Abstract
:
1. Introduction
2. Results and Discussion
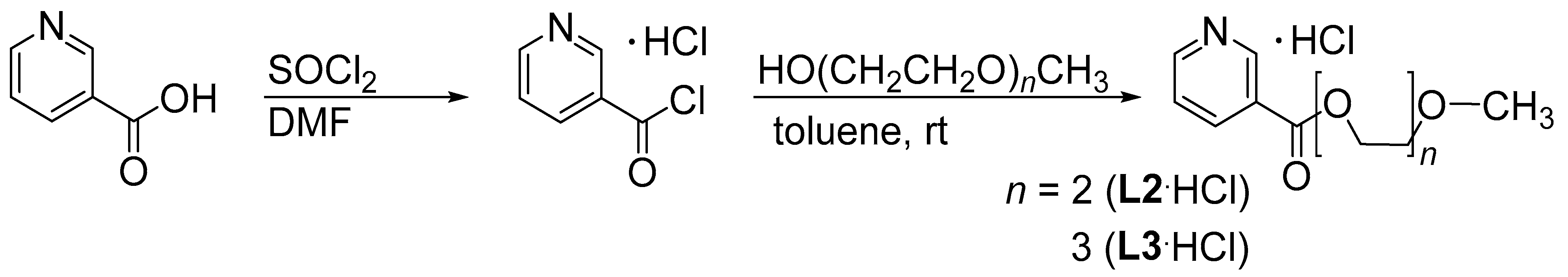
2.1. Synthesis and Characterization of Ligands
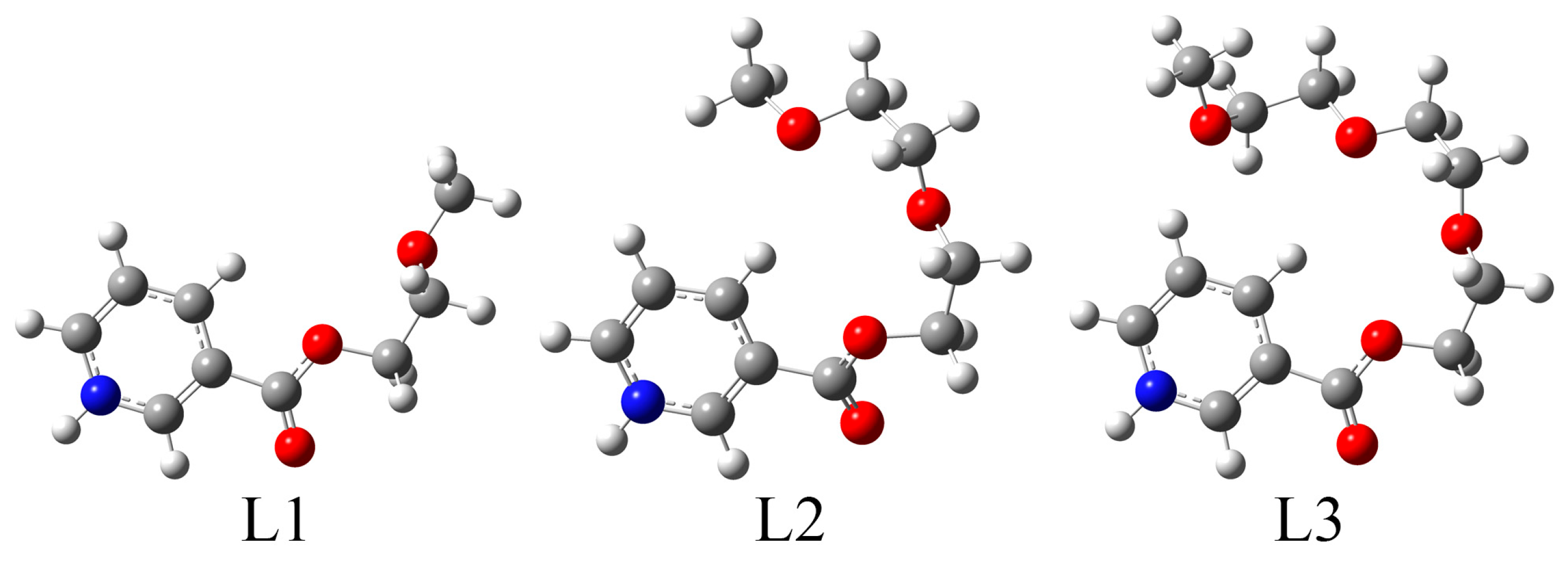
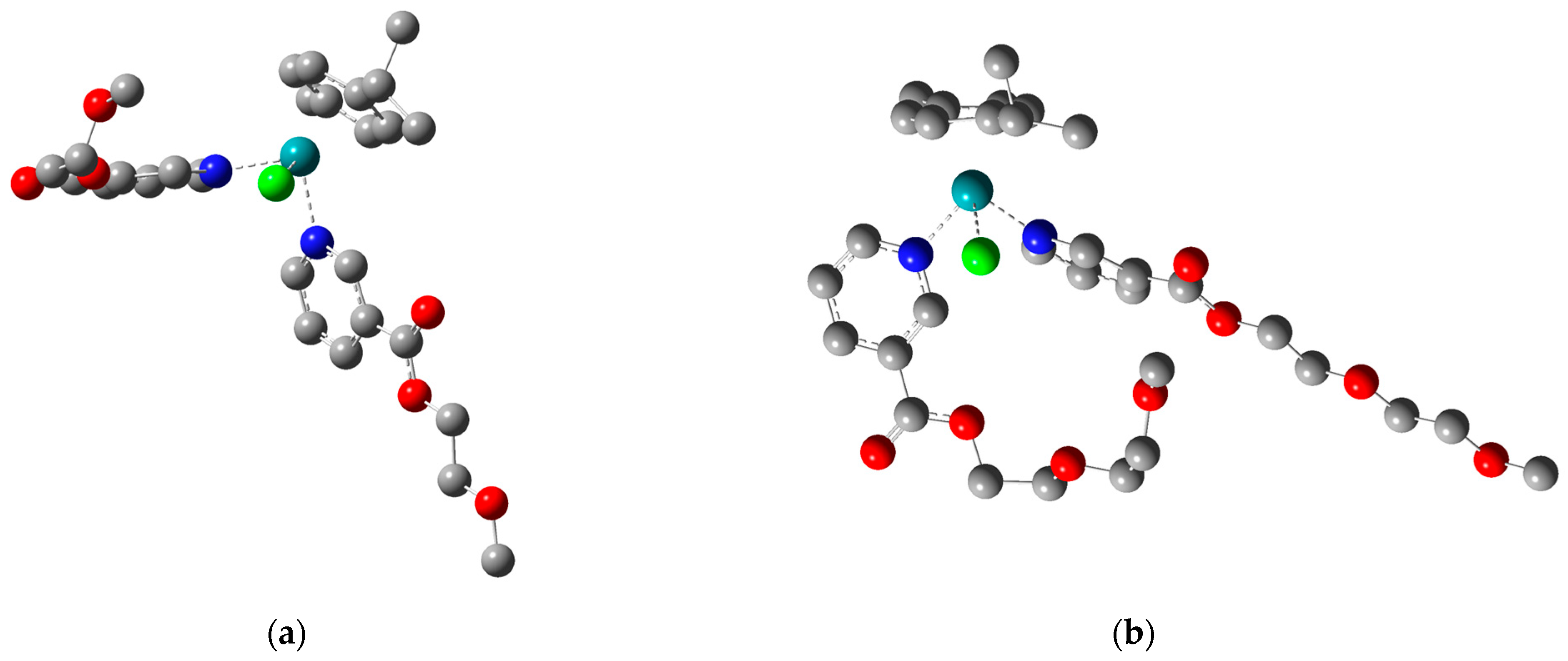
2.2. Theoretical Analysis of the Structure of Ligands L1·HCl–L3·HCl
2.3. Synthesis and Characterization of Neutral [RuCl2(η6-p-cym)(L-κN)] (L = L2: 2; L3: 3) Complexes
2.4. Theoretical Analysis of Neutral [RuCl2(η6-p-cym)(L-κN)] (L = L1: 1; L2: 2; L3: 3) Complexes
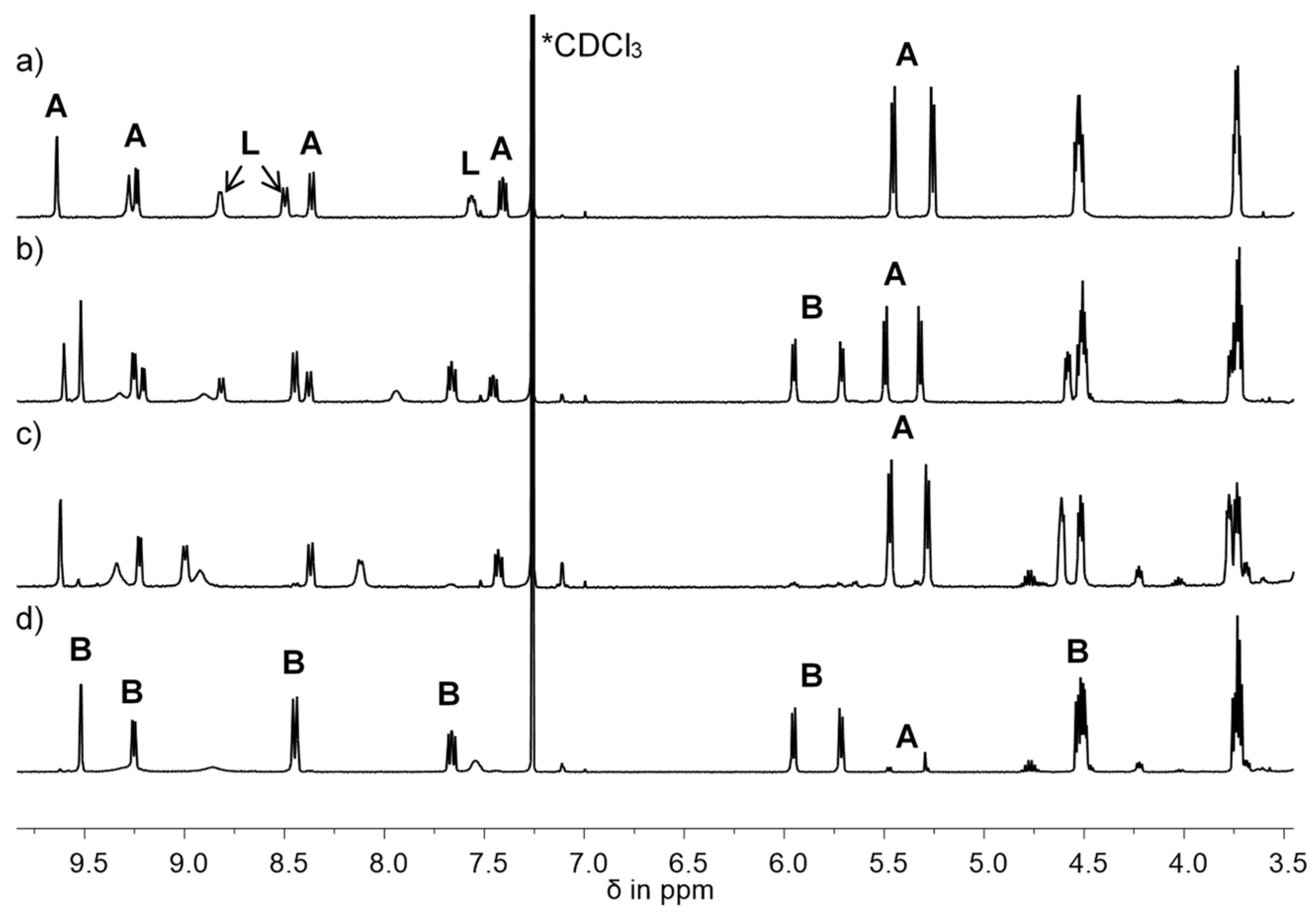
2.5. Synthesis and Structural Characterization of Cationic [RuCl(η6-p-cym)(L-κN)2][PF6] (L = L1: 4; L2: 5) Complexes
2.6. Theoretical Analysis of Cationic [RuCl(η6-p-cym)(L-κN)2][PF6] (L = L1: 4; L2: 5) Complexes
3. Materials and Methods
3.1. Purchased Chemicals
3.2. Preparative Technique and Instrumental Methods
3.3. Preparation of Ligands
- L2·HCl (2-(2-Methoxyethoxy)ethyl nicotinate hydrochloride): Viscous, hygroscopic oil; soluble in water, methanol, ethanol, and iso-propanol; insoluble in diethyl ether. Yield: 95%. 1H-NMR (400 MHz, D2O): δ 3.34 (s, 3H, OCH3), 3.66, 3.79 (m, 4H, CH3OCH2CH2), 3.97 (m, 2H, OOCCH2CH2), 4.66 (m, 2H, COOCH2), 8.28 (ddd, 3JH5,H6 = 8.2 Hz, 3JH5,H4 = 5.9 Hz, 4JH5,H2 = 0.8 Hz, 1H, H5), 9.06 (d(b), 3JH,H = 5.9 Hz, 1H, H4), 9.18 (d(b), 3JH,H = 8.2 Hz, 1H, H6), 9.44 (m, 1H, H2). 13C-NMR (100 MHz, D2O): δ 58.0 (CH3O), 65.7 (CH2OOC), 68.2, 69.5, 70.9 (CH2O), 127.7 (C5), 129.7 (C3), 142.7 (C2), 144.5 (C4), 147.1 (C6), 162.9 (COO). ESI-HRMS (CH3OH), positive mode: Calcd for [C11H16NO4]+ 226.10738, m/z 226.10733 [M + H]+. IR: ν (cm−1) 3061(w), 2876(w), 2400(b), 2090(w), 1968(w), 1728(s), 1633(w), 1606(w), 1544(w), 1461(m), 1359(w), 1286(s), 1198(w), 1103(s), 1019(m), 928(w), 837(w), 740(s), 690(m), 677(m), 619(s), 520(w), 469(w).
- L3·HCl (2-[2-(2-Methoxyethoxy)ethoxy]ethyl nicotinate hydrochloride): Viscous, hygroscopic oil; soluble in water, methanol, ethanol, iso-propanol, chloroform, and dichloromethane; insoluble in diethyl ether. Yield: 92%. 1H-NMR (400 MHz, D2O): δ 3.33 (s, 3H, OCH3), 3.58, 3.66, 3.69, 3.78 (m, 8H, OCH2CH2O), 3.95 (m, 2H, OOCCH2CH2), 4.63 (m, 2H, COOCH2), 8.27 (dd, 3JH5,H6 = 8.2 Hz, 3JH5,H4 = 6.0 Hz, 1H, H5), 9.05 (d(b), 3JH,H = 5.9 Hz, 1H, H4), 9.17 (d(b), 3JH,H = 8.2 Hz, 1H, H6), 9.43 (s(b), 1H, H2). 13C-NMR (100 MHz, D2O): δ 58.0 (CH3O), 65.7 (CH2OOC), 68.2, 69.4, 69.4, 69.7, 70.9 (CH2O), 127.7 (C5), 129.7 (C3), 142.7 (C2), 144.5 (C4), 147.1 (C6), 162.9 (COO). ESI-HRMS (CH3OH), positive mode: Calcd for [C13H20NO5]+ 270.13360, m/z 270.13355 [M + H]+. IR: ν (cm−1) 3065(w), 2876(w), 2400(b), 2092(w), 1970(w), 1730(s), 1633(w), 1606(w), 1540(w), 1461(m), 1355(w), 1286(s), 1198(w), 1100(s), 1016(m), 935(w), 837(m), 740(s), 695(m), 677(m), 619(s), 520(w), 479(w).
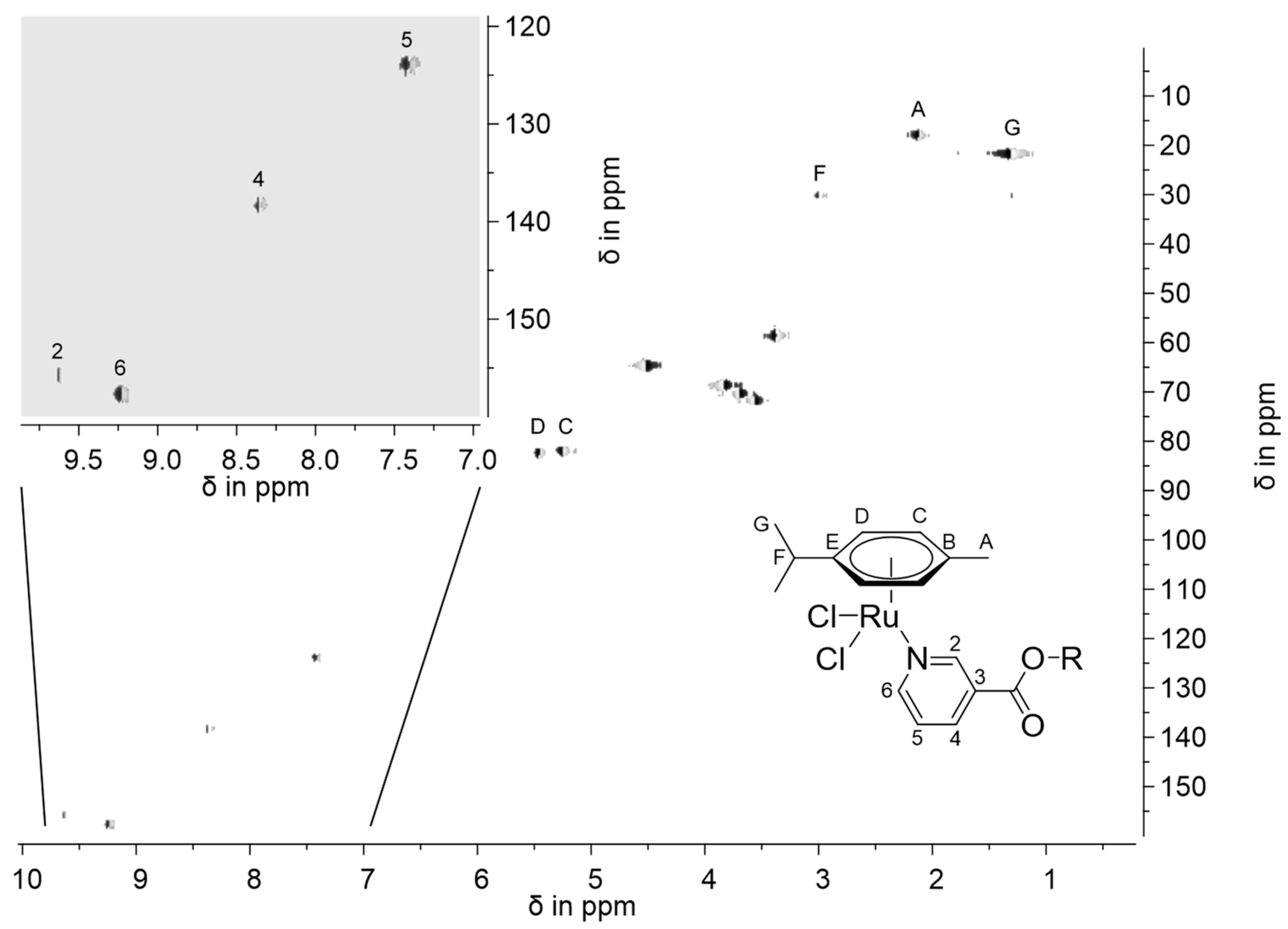
3.4. Preparation of [RuCl2(η6-p-cym)(L-κN)] Complexes (L = L2: 2; L3: 3)
- 2: Orange powder; soluble in chloroform, dichloromethane, dimethylformamide, acetone, and acetonitrile; moderately soluble in methanol, ethanol, iso-propanol, and tetrahydrofuran; insoluble in diethyl ether and toluene. Yield: 96%. EA: Anal. Found: C, 47.03; H, 5.19; N, 2.62. Calcd for C21H29Cl2NO4Ru (531.43): C, 47.46; H, 5.50; N, 2.64. 1H-NMR (400 MHz, CDCl3): δ 1.32 (d, 3JH,H = 7.2 Hz, 6H, CH(CH3)2), 2.13 (s, 3H, CCH3), 2.99 (sept, 3JH,H = 6.9 Hz, 1H, CH(CH3)2), 3.38 (s, 3H, OCH3), 3.57, 3.70 (m, 4H, CH3OCH2CH2), 3.84 (m, 2H, OOCCH2CH2), 4.53 (m, 2H, COOCH2), 5.25 (d, 3JH,H = 6.0 Hz, 2H, CHCCH3), 5.46 (d, 3JH,H = 5.9 Hz, 2H, CHCHCCH3), 7.41 (dd, 3JH5,H4 = 7.8 Hz, 3JH5,H6 = 5.7 Hz, 1H, H5), 8.35 (d, 3JH,H = 7.9 Hz, 1H, H4), 9.23 (d, 3JH,H = 5.6 Hz, 1H, H6), 9.63 (s, 1H, H2). 13C-NMR (100 MHz, CDCl3): δ 18.4 (CCH3), 22.4 (C(CH3)2), 30.8 (C(CH3)2), 59.2 (CH3O), 65.3 (CH2OOC), 69.0, 70.8, 72.1 (CH2O), 82.5, 82.8 (CH), 97.4, 103.9 (CCH), 124.2 (C5), 127.4 (C3), 138.7 (C4), 156.3 (C2), 158.0 (C6), 163.6 (COO). ESI-HRMS (CH3OH), positive mode: Calcd for [C21H29ClNO496Ru]+ 490.08556, m/z 490.08585 [M-Cl]+. IR: ν (cm−1) 3088(w), 3026(w), 2968(w), 2880(w), 1724(s), 1604(w), 1469(w), 1451(w), 1428(w), 1381(w), 1353(w), 1319(w), 1286(s), 1250(m), 1202(w), 1144(m), 1111(s), 1053(m), 1016(m), 944(m), 881(m), 845(m), 802(w), 751(s), 695(m), 670(w), 528(w), 500(w), 470(w), 455(w), 371(w), 287(s), 269(s), 229(s).
- 3: Orange powder; soluble in chloroform, dichloromethane, dimethylformamide, acetone, and acetonitrile; moderately soluble in methanol, ethanol, iso-propanol, and tetrahydrofuran; insoluble in diethyl ether and toluene. Yield: 90%. EA: Anal. Found: C, 47.85; H, 5.41; N, 2.40. Calcd for C23H33Cl2NO5Ru (575.49): C, 48.00; H, 5.78; N, 2.43. 1H-NMR (400 MHz, CDCl3): δ 1.32 (d, 3JH,H = 6.9 Hz, 6H, CH(CH3)2), 2.13 (s, 3H, CCH3), 2.99 (sept, 3JH,H = 6.9 Hz, 1H, CH(CH3)2), 3.36 (s, 3H, OCH3), 3.54, 3.64, 3.69, 3.71 (m, 8H, CH3OCH2CH2), 3.84 (m, 2H, OOCCH2CH2), 4.52 (m, 2H, COOCH2), 5.26 (d, 3JH,H = 6.0 Hz, 2H, CHCCH3), 5.46 (d, 3JH,H = 6.0 Hz, 2H, CHCHCCH3), 7.41 (dd, 3JH5,H4 = 7.8 Hz, 3JH5,H6 = 5.8 Hz, 1H, H5), 8.35 (d, 3JH,H = 7.9 Hz, 1H, H4), 9.23 (d, 3JH,H = 5.6 Hz, 1H, H6), 9.62 (s, 1H, H2). 13C-NMR (100 MHz, CDCl3): δ 18.4 (CCH3), 22.4 (C(CH3)2), 30.8 (C(CH3)2), 59.2 (CH3O), 65.3 (CH2OOC), 69.0, 70.7, 70.8, 70.9, 72.1 (CH2O), 82.5, 82.8 (CH), 97.4, 103.9 (CCH), 124.2 (C5), 127.5 (C3), 138.7 (C4), 156.3 (C2), 158.0 (C6), 163.6 (COO). ESI-HRMS (CH3OH), positive mode: Calcd for [C23H33ClNO596Ru]+ 534.11178, m/z 534.11190 [M-Cl]+. IR: ν (cm−1) 3065(w), 2964(w), 2877(w), 1722(s), 1600(w), 1472(w), 1451(w), 1428(w), 1380(w), 1282(s), 1198(m), 1105(s), 1053(m), 1028(m), 947(w), 867(m), 845(m), 798(w), 751(s), 693(m), 455(w), 371(w), 284(s), 232(s).
3.5. Preparation of [RuCl(η6-p-cym)(L-κN)2][PF6] (L = L1: 4; L2: 5) Complexes
- 4: Orange, highly viscous oil; soluble in chloroform and dichloromethane; moderately soluble in methanol, ethanol, and iso-propanol; insoluble in n-pentane. Yield: 19%. 1H-NMR (400 MHz, CDCl3): δ 1.16 (d, 3JH,H = 6.9 Hz, 6H, CH(CH3)2), 1.77 (s, 3H, CCH3), 2.59 (sept, 3JH,H = 6.9 Hz, 1H, CH(CH3)2), 3.38 (s, 6H, OCH3), 3.70 (t, 3JH,H = 4.7 Hz, 4H, OCH2), 4.48 (m, 4H, COOCH2), 5.69 (d, 3JH,H = 6.1 Hz, 2H, CHCCH3), 5.94 (d, 3JH,H = 6.1 Hz, 2H, CHCHCCH3), 7.65 (dd, 3JH5,H4 = 7.8 Hz, 3JH5,H6 = 5.8 Hz, 2H, H5), 8.43 (d, 3JH,H = 8.0 Hz, 2H, H4), 9.21 (d, 3JH,H = 5.6 Hz, 2H, H6), 9.47 (s, 2H, H2). 13C-NMR (100 MHz, CDCl3): δ 18.0 (CCH3), 22.3 (C(CH3)2), 31.0 (C(CH3)2), 59.1 (CH3O), 65.3 (CH2OOC), 70.2 (CH2O), 82.5, 88.5 (CH), 102.1, 103.4 (CCH), 126.7 (C5), 128.6 (C3), 140.2 (C4), 154.4 (C2), 158.5 (C6), 163.2 (COO). 31P-NMR (162 MHz, CDCl3): δ –144.2 (sept, JP,F = 713 Hz, PF6) IR: ν (cm−1) 3080(w), 2968(w), 2887(w), 1726(s), 1606(w), 1433(w), 1369(w), 1290(s), 1197(w), 1118(m), 1055(w), 1026(w), 837(s), 746(m), 694(w), 557(m), 293(w).
- 5: Orange, highly viscous oil; soluble in chloroform and dichloromethane; moderately soluble in methanol, ethanol, and iso-propanol; insoluble in n-pentane. Yield: 39%. 1H-NMR (400 MHz, CDCl3): δ 1.17 (d, 3JH,H = 6.9 Hz, 6H, CH(CH3)2), 1.78 (s, 3H, CCH3), 2.58 (sept, 3JH,H = 7.0 Hz, 1H, CH(CH3)2), 3.35 (s, 6H, OCH3), 3.56, 3.69, 3.84 (m, 12H, OCH2), 4.53 (m, 4H, COOCH2), 5.71 (d, 3JH,H = 6.1 Hz, 2H, CHCCH3), 5.95 (d, 3JH,H = 6.1 Hz, 2H, CHCHCCH3), 7.66 (dd, 3JH5,H4 = 7.9 Hz, 3JH5,H6 = 5.8 Hz, 2H, H5), 8.45 (d, 3JH,H = 8.0 Hz, 2H, H4), 9.23 (d, 3JH,H = 5.8 Hz, 2H, H6), 9.53 (s, 2H, H2). 13C-NMR (100 MHz, CDCl3): δ 17.9 (CCH3), 22.3 (C(CH3)2), 31.0 (C(CH3)2), 59.2 (CH3O), 64.7 (CH2OOC), 69.2, 70.7, 72.0 (CH2O), 82.4, 88.5 (CH), 102.2, 103.3 (CCH), 126.7 (C5), 128.6 (C3), 140.2 (C4), 154.4 (C2), 158.4 (C6), 163.2 (COO). 31P-NMR (162 MHz, CDCl3): δ –144.2 (sept, JP,F = 713 Hz, PF6).
3.6. Theoretical Methods
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Arnesano, F.; Natile, G. Mechanistic insight into the cellular uptake and processing of cisplatin 30 years after its approval by FDA. Coord. Chem. Rev. 2009, 253, 2070–2081. [Google Scholar] [CrossRef]
- Hambley, T.W. Developing new metal-based therapeutics: Challenges and opportunities. Dalt. Trans. 2007, 43, 4929–4937. [Google Scholar] [CrossRef]
- Monneret, C. Platinum anticancer drugs. From serendipity to rational design. Ann. Pharm. Françaises 2011, 69, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Komeda, S.; Casini, A. Next-Generation Anticancer Metallodrugs. Curr. Top. Med. Chem. 2012, 12, 219–235. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Ruiz, S.; Maksimović-Ivanić, D.; Mijatović, S.; Kaluđerović, G.N. On the Discovery, Biological Effects, and Use of Cisplatin and Metallocenes in Anticancer Chemotherapy. Bioinorg. Chem. Appl. 2012, 2012, 140284. [Google Scholar] [CrossRef]
- Giaccone, G. Clinical Perspectives on Platinum Resistance. Drugs 2000, 59, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Thota, S.; Rodrigues, D.A.; Crans, D.C.; Barreiro, E.J. Ru(II) Compounds: Next-Generation Anticancer Metallotherapeutics? J. Med. Chem. 2018, 61, 5805–5821. [Google Scholar] [CrossRef] [PubMed]
- Rilak Simović, A.; Masnikosa, R.; Bratsos, I.; Alessio, E. Chemistry and reactivity of ruthenium(II) complexes: DNA/protein binding mode and anticancer activity are related to the complex structure. Coord. Chem. Rev. 2019, 398, 113011. [Google Scholar] [CrossRef]
- Conti, L.; Macedi, E.; Giorgi, C.; Valtancoli, B.; Fusi, V. Combination of light and Ru(II) polypyridyl complexes: Recent advances in the development of new anticancer drugs. Coord. Chem. Rev. 2022, 469, 214656. [Google Scholar] [CrossRef]
- Garza-Ortiz, A.; Maheswari, P.U.; Siegler, M.; Spek, A.L.; Reedijk, J. Ruthenium(III) Chloride Complex with a Tridentate Bis(arylimino)pyridine Ligand: Synthesis, Spectra, X-ray Structure, 9-Ethylguanine Binding Pattern, and In Vitro Cytotoxicity. Inorg. Chem. 2008, 47, 6964–6973. [Google Scholar] [CrossRef]
- Schuecker, R.; John, R.O.; Jakupec, M.A.; Arion, V.B.; Keppler, B.K. Water-Soluble Mixed-Ligand Ruthenium(II) and Osmium(II) Arene Complexes with High Antiproliferative Activity. Organometallics 2008, 27, 6587–6595. [Google Scholar] [CrossRef]
- Peacock, A.F.A.; Sadler, P.J. Medicinal Organometallic Chemistry: Designing Metal Arene Complexes as Anticancer Agents. Chem.—Asian J. 2008, 3, 1890–1899. [Google Scholar] [CrossRef] [PubMed]
- Renfrew, A.K.; Phillips, A.D.; Egger, A.E.; Hartinger, C.G.; Bosquain, S.S.; Nazarov, A.A.; Keppler, B.K.; Gonsalvi, L.; Peruzzini, M.; Dyson, P.J. Influence of Structural Variation on the Anticancer Activity of RAPTA-Type Complexes: Ptn versus pta. Organometallics 2009, 28, 1165–1172. [Google Scholar] [CrossRef]
- Therrien, B.; Süss-Fink, G.; Govindaswamy, P.; Renfrew, A.K.; Dyson, P.J. The “Complex-in-a-Complex” Cations [(acac)2 M⊂Ru6(p-iPrC6H4Me)6(tpt)2(dhbq)3]6+: A Trojan Horse for Cancer Cells. Angew. Chem. 2008, 120, 3833–3836. [Google Scholar] [CrossRef]
- Bergamo, A.; Masi, A.; Dyson, P.J.; Sava, G. Bergamo Modulation of the metastatic progression of breast cancer with an organometallic ruthenium compound. Int. J. Oncol. 1992, 33, 1281–1289. [Google Scholar] [CrossRef]
- Lee, S.Y.; Kim, C.Y.; Nam, T.-G. Ruthenium Complexes as Anticancer Agents: A Brief History and Perspectives. Drug Des. Devel. Ther. 2020, 14, 5375–5392. [Google Scholar] [CrossRef] [PubMed]
- Allardyce, C.S.; Dyson, P.J.; Ellis, D.J.; Heath, S.L. [Ru(η6-p-cymene)Cl2(pta)] (pta=1,3,5-triaza-7-phosphatricyclo[3.3.1.1]decane): A water soluble compound that exhibits pH dependent DNA binding providing selectivity for diseased cells. Chem. Commun. 2001, 15, 1396–1397. [Google Scholar] [CrossRef]
- Therrien, B. Functionalised η6-arene ruthenium complexes. Coord. Chem. Rev. 2009, 253, 493–519. [Google Scholar] [CrossRef]
- Clarke, M.J. Ruthenium metallopharmaceuticals. Coord. Chem. Rev. 2002, 232, 69–93. [Google Scholar] [CrossRef]
- Smith, G.S.; Therrien, B. Targeted and multifunctional arene ruthenium chemotherapeutics. Dalt. Trans. 2011, 40, 10793. [Google Scholar] [CrossRef]
- Kenny, R.G.; Marmion, C.J. Toward Multi-Targeted Platinum and Ruthenium Drugs—A New Paradigm in Cancer Drug Treatment Regimens? Chem. Rev. 2019, 119, 1058–1137. [Google Scholar] [CrossRef]
- Morris, R.E.; Aird, R.E.; del Socorro Murdoch, P.; Chen, H.; Cummings, J.; Hughes, N.D.; Parsons, S.; Parkin, A.; Boyd, G.; Jodrell, D.I.; et al. Inhibition of Cancer Cell Growth by Ruthenium(II) Arene Complexes. J. Med. Chem. 2001, 44, 3616–3621. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.K.; Melchart, M.; Habtemariam, A.; Sadler, P.J. Organometallic chemistry, biology and medicine: Ruthenium arene anticancer complexes. Chem. Commun. 2005, 38, 4764–4776. [Google Scholar] [CrossRef] [PubMed]
- Aird, R.E.; Cummings, J.; Ritchie, A.A.; Muir, M.; Morris, R.E.; Chen, H.; Sadler, P.J.; Jodrell, D.I. In Vitro and In Vivo activity and cross resistance profiles of novel ruthenium (II) organometallic arene complexes in human ovarian cancer. Br. J. Cancer 2002, 86, 1652–1657. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Xu, C.; Li, T.; Lu, S.; Luo, F.; Wang, H. Novel NHC-coordinated ruthenium(II) arene complexes achieve synergistic efficacy as safe and effective anticancer therapeutics. Eur. J. Med. Chem. 2020, 203, 112605. [Google Scholar] [CrossRef] [PubMed]
- Muralisankar, M.; Chen, J.-R.; Haribabu, J.; Ke, S.-C. Effective and Selective Ru(II)-Arene Complexes Containing 4,4′-Substituted 2,2′ Bipyridine Ligands Targeting Human Urinary Bladder Cancer Cells. Int. J. Mol. Sci. 2023, 24, 11896. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Karotki, A.; Gabbiani, C.; Rugi, F.; Vašák, M.; Messori, L.; Dyson, P.J. Reactivity of an antimetastatic organometallic ruthenium compound with metallothionein-2: Relevance to the mechanism of action. Metallomics 2009, 1, 434. [Google Scholar] [CrossRef] [PubMed]
- Süss-Fink, G.; Khan, F.-A.; Juillerat-Jeanneret, L.; Dyson, P.J.; Renfrew, A.K. Synthesis and Anticancer Activity of Long-Chain Isonicotinic Ester Ligand-Containing Arene Ruthenium Complexes and Nanoparticles. J. Clust. Sci. 2010, 21, 313–324. [Google Scholar] [CrossRef]
- Eichhorn, T.; Hey-Hawkins, E.; Maksimović-Ivanić, D.; Mojić, M.; Schmidt, J.; Mijatović, S.; Schmidt, H.; Kaluderović, G.N. Binuclear dichlorido(η6-p-cymene)ruthenium(II) complexes with bis(nicotinate)- And bis(isonicotinate)-polyethylene glycol ester ligands. Appl. Organomet. Chem. 2015, 29, 20–25. [Google Scholar] [CrossRef]
- Kaluđerović, G.N.; Kommera, H.; Schwieger, S.; Paethanom, A.; Kunze, M.; Schmidt, H.; Paschke, R.; Steinborn, D. Synthesis, characterization, in vitro antitumoral investigations and interaction with plasmid pBR322 DNA of R2eddp-platinum(iv) complexes (R = Et, n-Pr). Dalt. Trans. 2009, 48, 10720–10726. [Google Scholar] [CrossRef]
- Kaluđerović, G.N.; Sabo, T.J. Synthesis and characterization of the cobalt(III) complexes with ethylenediamine-N,N′-di-3-propanoate ligand and its esters. Polyhedron 2002, 21, 2277–2282. [Google Scholar] [CrossRef]
- Haydock, D.B.; Mulholland, T.P.C. Some derivatives of NN′-biscarboxymethyl- and NN′-bis-(β-carboxyethyl)-ethylenediamine. J. Chem. Soc. C 1971, 13, 2389–2395. [Google Scholar] [CrossRef] [PubMed]
- Eichhorn, T.; Kolbe, F.; Mišić, S.; Dimić, D.; Morgan, I.; Saoud, M.; Milenković, D.; Marković, Z.; Rüffer, T.; Dimitrić Marković, J.; et al. Synthesis, Crystallographic Structure, Theoretical Analysis, Molecular Docking Studies, and Biological Activity Evaluation of Binuclear Ru(II)-1-Naphthylhydrazine Complex. Int. J. Mol. Sci. 2023, 24, 689. [Google Scholar] [CrossRef] [PubMed]
- Eichhorn, T.; Dimic, D.; Markovic, Z.; Kaludjerovic, G. Synthesis, spectroscopic characterization, and DFT analysis of dichlorido(η6-p-cymene)ruthenium(II) complexes with isonicotinate-polyethylene glycol ester ligands. J. Serbian Chem. Soc. 2023, 70. [Google Scholar] [CrossRef]
- Lalrempuia, R.; Kollipara, M.R.; Carroll, P.J. Syntheses and characterization of arene ruthenium (II) complexes containing N,N′-donor Schiff base ligands. Crystal and molecular structure of [(η6-C6Me6)Ru(C5H4N-2-CH=N-C6H4-p-NO2)]PF6. Polyhedron 2003, 22, 605–609. [Google Scholar] [CrossRef]
- Al-Noaimi, M.; AlDamen, M.A. Ruthenium complexes incorporating azoimine and α-diamine based ligands: Synthesis, crystal structure, electrochemistry and DFT calculation. Inorganica Chim. Acta 2012, 387, 45–51. [Google Scholar] [CrossRef]
- Savić, A.; Dulović, M.; Poljarević, J.M.; Misirlić-Denčić, S.; Jovanović, M.; Bogdanović, A.; Trajković, V.; Sabo, T.J.; Grgurić-Šipka, S.; Marković, I. Synthesis and in vitro Anticancer Activity of Ruthenium–Cymene Complexes with Cyclohexyl-Functionalized Ethylenediamine-N,N′-diacetate-Type Ligands. ChemMedChem 2011, 6, 1884–1891. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitale. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Dennington, R.; Todd, K.; Millam, J. GausView 2009; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Ditchfield, R. Self-consistent perturbation theory of diamagnetism. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1H | 13C | ||||
|---|---|---|---|---|---|
| H Atom | Exp. (ppm) | Calc. (ppm) | C Atom | Exp. (ppm) | Calc. (ppm) |
| OCH3 | 3.34 | 3.53 | CH3O | 58.0 | 57.9 |
| CH3OCH2CH2 | 3.79 | 3.78 | CH2OOC | 65.7 | 68.2 |
| OOCCH2CH2 | 3.97 | 4.01 | CH2O | 68.2 | 69.1 |
| COOCH2 | 4.66 | 4.61 | CH2O | 69.5 | 72.4 |
| C5–H | 8.28 | 8.44 | CH2O | 70.9 | 73.4 |
| C4–H | 9.06 | 8.88 | C5 | 127.7 | 127.0 |
| C6–H | 9.18 | 9.30 | C3 | 129.7 | 132.2 |
| C2–H | 9.44 | 10.00 | C2 | 142.7 | 141.2 |
| R | 0.997 | C4 | 144.5 | 141.4 | |
| MAE (ppm) | 0.16 | C6 | 147.1 | 148.8 | |
| COO | 162.9 | 158.6 | |||
| R | 0.999 | ||||
| MAE (ppm) | 1.0 | ||||
| 1H | 13C | ||||
|---|---|---|---|---|---|
| H Atom | Exp. (ppm) | Calc. (ppm) | C Atom | Exp. (ppm) | Calc. (ppm) |
| CH(CH3)2 | 1.32 | 2.12 | CCH3 | 18.4 | 22.0 |
| CCH3 | 2.13 | 2.44 | C(CH3)2 | 22.4 | 25.7 |
| CH(CH3)2 | 2.99 | 3.75 | C(CH3)2 | 30.8 | 40.4 |
| OCH3 | 3.38 | 3.87 | CH3O | 59.2 | 61.0 |
| CH3OCH2 | 3.57 | 3.92 | CH2OOC | 65.3 | 68.3 |
| CH3OCH2CH2 | 3.70 | 4.04 | CH2CH2OOC | 69.5 | 71.9 |
| COOCH2CH2 | 3.84 | 4.15 | CH3OCH2CH2 | 70.8 | 73.9 |
| COOCH2 | 4.53 | 4.56 | CH3OCH2 | 72.1 | 74.5 |
| CHCCH3 | 5.25 | 5.09 | CH3CCH | 82.5 | 83.7 |
| CHCHCCH3 | 5.46 | 5.58 | CH3CCHCH | 82.8 | 92.9 |
| C5–H | 7.41 | 7.23 | CCH(CH3)2 | 97.4 | 102.7 |
| C4–H | 8.35 | 8.13 | CCH3 | 103.9 | 106.1 |
| C6–H | 9.23 | 8.97 | C5 | 124.2 | 121.0 |
| C2–H | 9.63 | 9.36 | C3 | 127.4 | 126.5 |
| R | 0.997 | C4 | 138.7 | 134.9 | |
| MAE (ppm) | 0.32 | C2 | 156.3 | 151.8 | |
| C6 | 158.0 | 155.0 | |||
| COO | 163.6 | 161.7 | |||
| R | 0.997 | ||||
| MAE (ppm) | 3.6 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dimić, D.; Eichhorn, T.; Milenković, D.; Kaluđerović, G.N. Synthesis, Structural, and Quantum Chemical Analysis of Neutral and Cationic Ruthenium(II) Complexes with Nicotinate-Polyethylene Glycol Ester Ligands. Inorganics 2023, 11, 460. https://doi.org/10.3390/inorganics11120460
Dimić D, Eichhorn T, Milenković D, Kaluđerović GN. Synthesis, Structural, and Quantum Chemical Analysis of Neutral and Cationic Ruthenium(II) Complexes with Nicotinate-Polyethylene Glycol Ester Ligands. Inorganics. 2023; 11(12):460. https://doi.org/10.3390/inorganics11120460
Chicago/Turabian StyleDimić, Dušan, Thomas Eichhorn, Dejan Milenković, and Goran N. Kaluđerović. 2023. "Synthesis, Structural, and Quantum Chemical Analysis of Neutral and Cationic Ruthenium(II) Complexes with Nicotinate-Polyethylene Glycol Ester Ligands" Inorganics 11, no. 12: 460. https://doi.org/10.3390/inorganics11120460
APA StyleDimić, D., Eichhorn, T., Milenković, D., & Kaluđerović, G. N. (2023). Synthesis, Structural, and Quantum Chemical Analysis of Neutral and Cationic Ruthenium(II) Complexes with Nicotinate-Polyethylene Glycol Ester Ligands. Inorganics, 11(12), 460. https://doi.org/10.3390/inorganics11120460