
A Computational Perspective on Carbon-Carbon Bond Formation by Single Cu Atom on Pd(111) Surface for CO Electrochemical Reduction


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
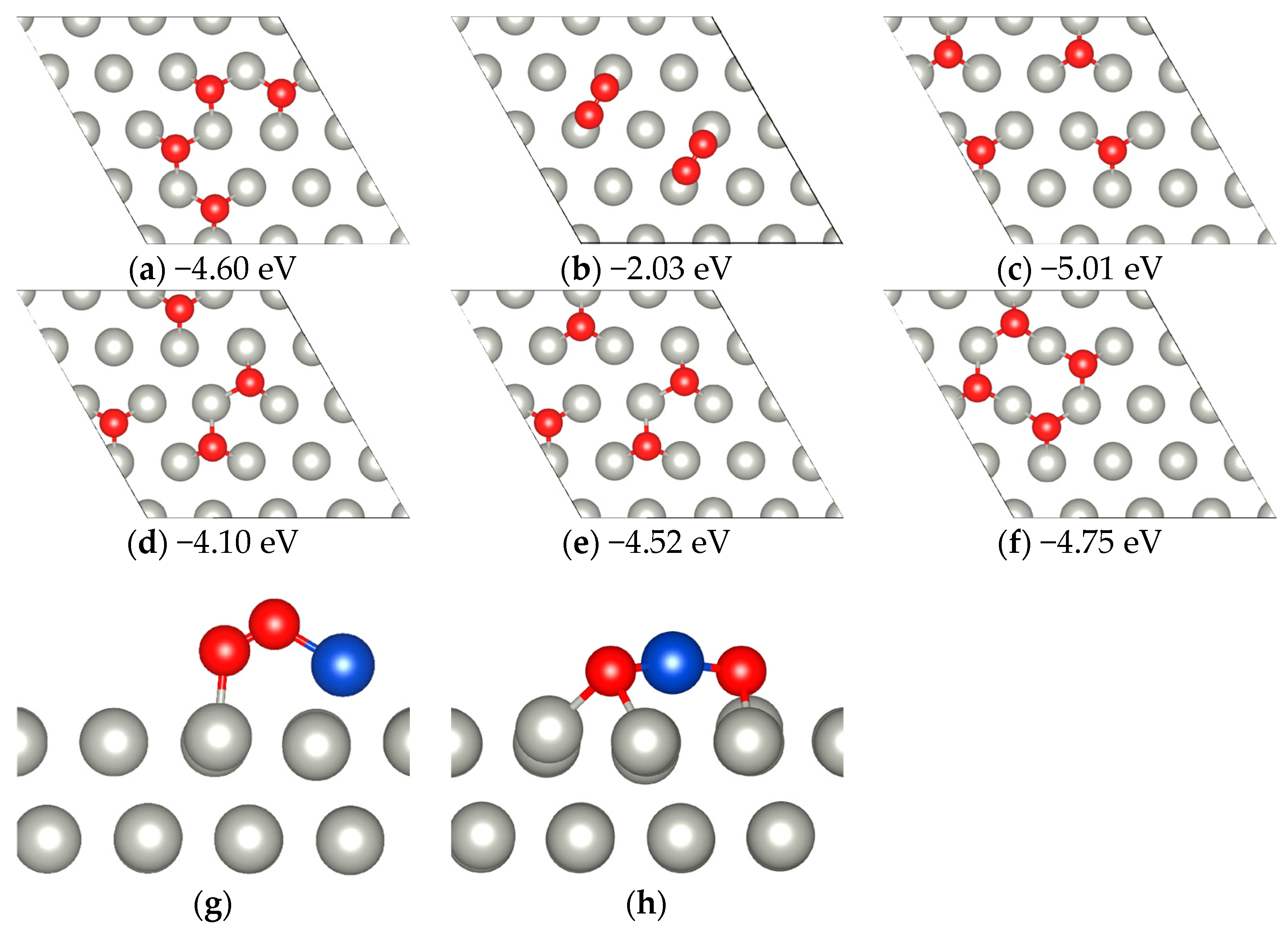
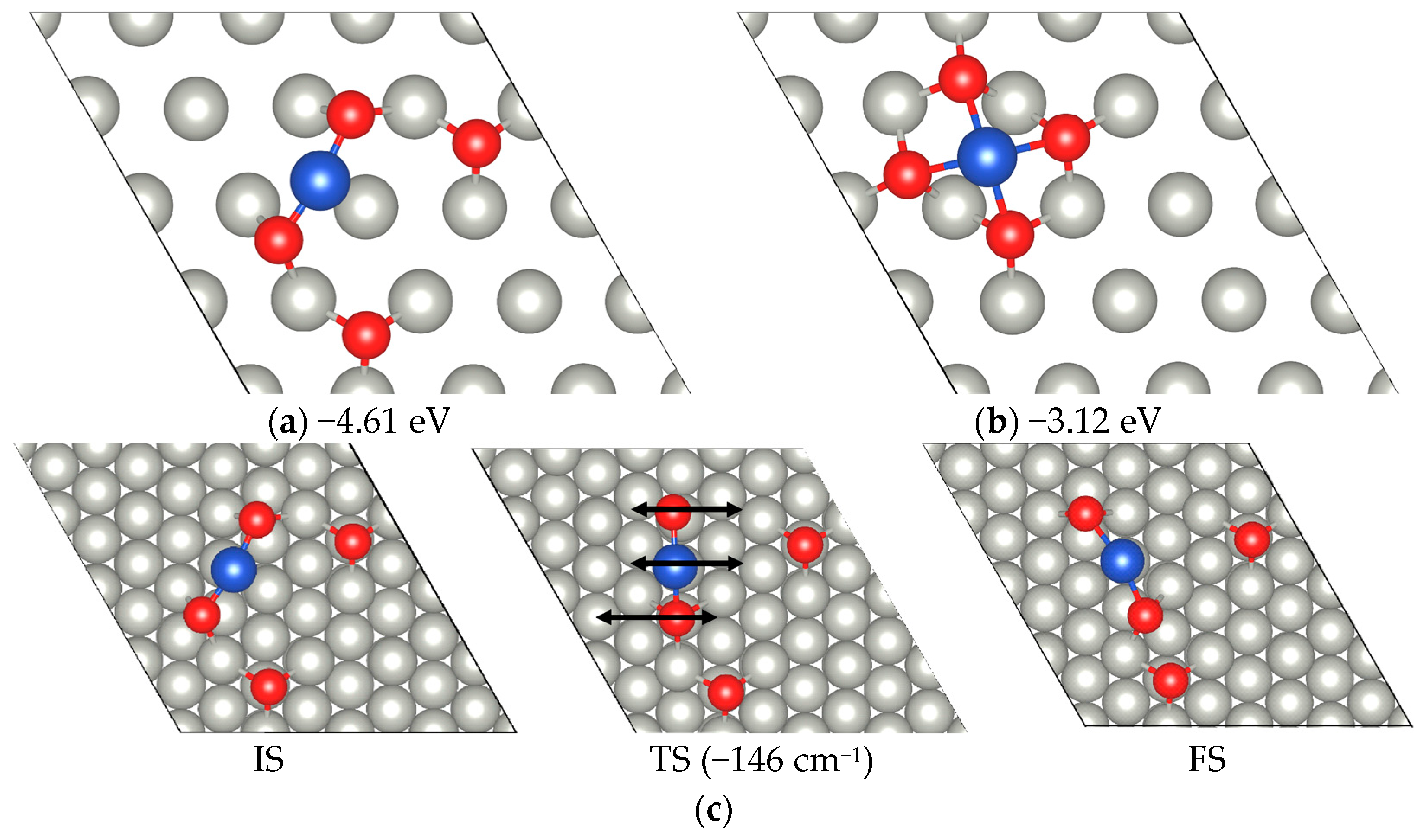
2.1. CO Dimerization Reaction on Cu-SAC
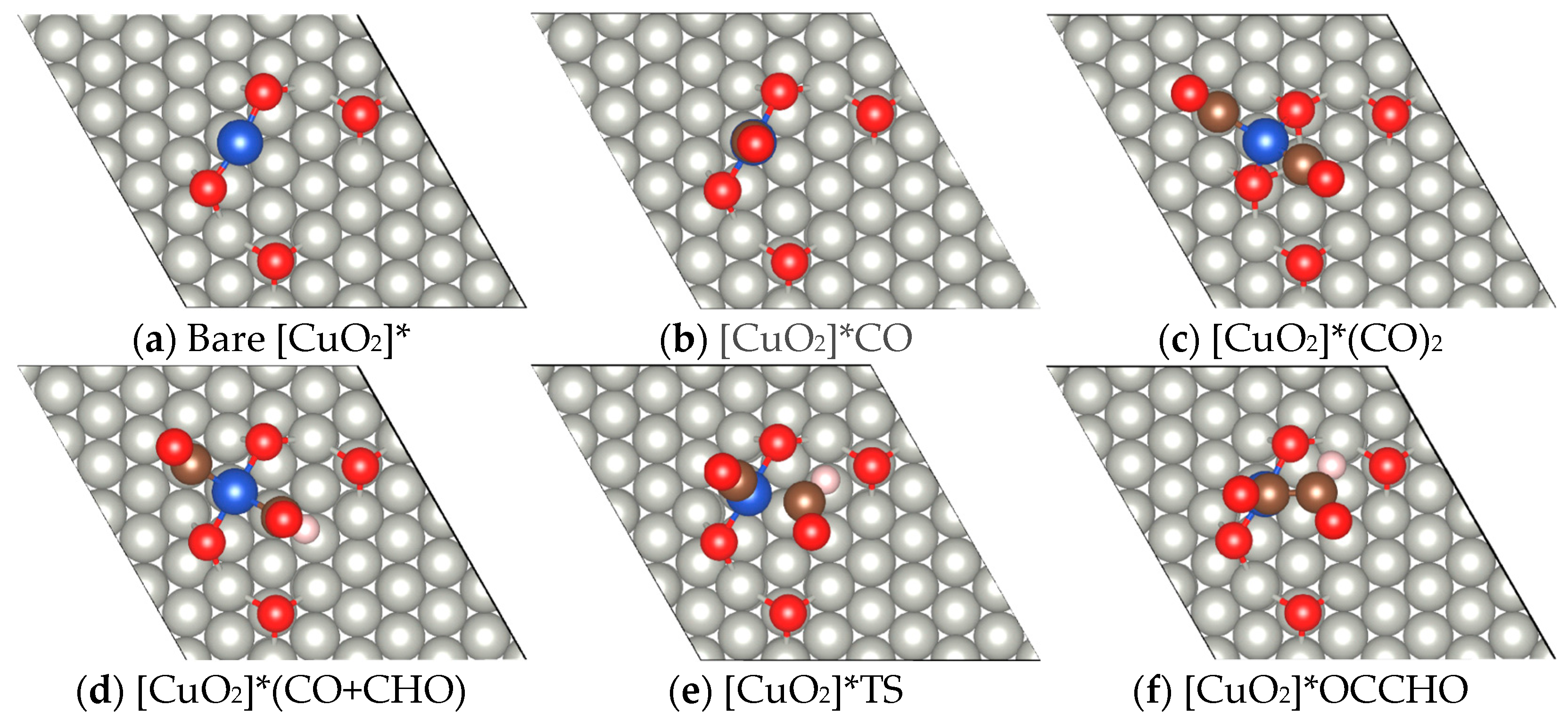
2.2. Possible Side Reactions
2.3. Kinetics Results
3. Method
3.1. First-Principle Simulations
3.2. Kinetic Modeling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nitopi, S.; Bertheussen, E.; Scott, S.B.; Liu, X.; Engstfeld, A.K.; Horch, S.; Seger, B.; Stephens, I.E.L.; Chan, K.; Hahn, C.; et al. Progress and perspectives of electrochemical CO2 reduction on copper in aqueous electrolyte. Chem. Rev. 2019, 119, 7610–7672. [Google Scholar] [CrossRef]
- Zhao, J.; Xue, S.; Barber, J.; Zhou, Y.; Meng, J.; Ke, X. An overview of Cu-based heterogeneous electrocatalysts for CO2 reduction. J. Mater. Chem. A 2020, 8, 4700–4734. [Google Scholar] [CrossRef]
- Christensen, O.; Zhao, S.; Sun, Z.; Bagger, A.; Lauritsen, J.V.; Pedersen, S.U.; Daasbjerg, K.; Rossmeisl, J. Can the CO2 Reduction Reaction Be Improved on Cu: Selectivity and Intrinsic Activity of Functionalized Cu Surfaces. ACS Catal. 2022, 12, 15737–15749. [Google Scholar] [CrossRef]
- Yoshio, H.; Katsuhei, K.; Shin, S. Production of CO and CH4 in Electrochemical Reduction of CO2 at Metal Electrodes in Aqueous Hydrogencarbonate Solution. Chem. Lett. 1985, 14, 1695–1698. [Google Scholar] [CrossRef]
- Montoya, J.H.; Shi, C.; Chan, K.; Nørskov, J.K. Theoretical Insights into a CO dimerization mechanism in CO2 electroreduction. J. Phys. Chem. Lett. 2015, 6, 2032–2037. [Google Scholar] [CrossRef]
- Wu, Z.-Z.; Zhang, X.-L.; Niu, Z.-Z.; Gao, F.-Y.; Yang, P.-P.; Chi, L.-P.; Shi, L.; Wei, W.-S.; Liu, R.; Chen, Z.; et al. Identification of Cu(100)/Cu(111) Interfaces as Superior Active Sites for CO Dimerization During CO2 Electroreduction. J. Am. Chem. Soc. 2022, 144, 259–269. [Google Scholar] [CrossRef]
- Dickinson, H.L.A.; Symes, M.D. Recent progress in CO2 reduction using bimetallic electrodes containing copper. Electrochem. Commun. 2022, 135, 107212. [Google Scholar] [CrossRef]
- Okatenko, V.; Loiudice, A.; Newton, M.A.; Stoian, D.C.; Blokhina, A.; Chen, A.N.; Rossi, K.; Buonsanti, R. Alloying as a Strategy to Boost the Stability of Copper Nanocatalysts during the Electrochemical CO2 Reduction Reaction. J. Am. Chem. Soc. 2023, 145, 5370–5383. [Google Scholar] [CrossRef]
- Robens, E.; Hecker, B.; Kungl, H.; Tempel, H.; Eichel, R.-A. Bimetallic Copper–Silver Catalysts for the Electrochemical Reduction of CO2 to Ethanol. ACS Appl. Energy Mater. 2023, 6, 7571–7577. [Google Scholar] [CrossRef]
- Xu, A.; Hung, S.-F.; Cao, A.; Wang, Z.; Karmodak, N.; Huang, J.E.; Yan, Y.; Sedighian Rasouli, A.; Ozden, A.; Wu, F.-Y.; et al. Copper/alkaline earth metal oxide interfaces for electrochemical CO2-to-alcohol conversion by selective hydrogenation. Nat. Catal. 2022, 5, 1081–1088. [Google Scholar] [CrossRef]
- Xiong, H.; Peterson, E.; Qi, G.; Datye, A.K. Trapping mobile Pt species by PdO in diesel oxidation catalysts: Smaller is better. Catal. Today 2016, 272, 80–86. [Google Scholar] [CrossRef]
- Wan, Q.; Wei, F.; Wang, Y.; Wang, F.; Zhou, L.; Lin, S.; Xie, D.; Guo, H. Single atom detachment from Cu clusters, and diffusion and trapping on CeO2(111): Implications in Ostwald ripening and atomic redispersion. Nanoscale 2018, 10, 17893–17901. [Google Scholar] [CrossRef] [PubMed]
- Sarfraz, S.; Garcia-Esparza, A.T.; Jedidi, A.; Cavallo, L.; Takanabe, K. Cu–Sn Bimetallic Catalyst for Selective Aqueous Electroreduction of CO2 to CO. ACS Catal. 2016, 6, 2842–2851. [Google Scholar] [CrossRef]
- Rasul, S.; Anjum, D.H.; Jedidi, A.; Minenkov, Y.; Cavallo, L.; Takanabe, K. A Highly Selective Copper–Indium Bimetallic Electrocatalyst for the Electrochemical Reduction of Aqueous CO2 to CO. Angew. Chem. Int. Ed. 2015, 54, 2146–2150. [Google Scholar] [CrossRef] [PubMed]
- Hoang, T.T.H.; Verma, S.; Ma, S.; Fister, T.T.; Timoshenko, J.; Frenkel, A.I.; Kenis, P.J.A.; Gewirth, A.A. Nanoporous Copper–Silver Alloys by Additive-Controlled Electrodeposition for the Selective Electroreduction of CO2 to Ethylene and Ethanol. J. Am. Chem. Soc. 2018, 140, 5791–5797. [Google Scholar] [CrossRef]
- Zhu, C.; Chen, A.; Mao, J.; Wu, G.; Li, S.; Dong, X.; Li, G.; Jiang, Z.; Song, Y.; Chen, W.; et al. Cu–Pd Bimetallic Gas Diffusion Electrodes for Electrochemical Reduction of CO2 to C2+ Products. Small Struct. 2023, 4, 2200328. [Google Scholar] [CrossRef]
- Takashima, T.; Suzuki, T.; Irie, H. Electrochemical carbon dioxide reduction on copper-modified palladium nanoparticles synthesized by underpotential deposition. Electrochim. Acta 2017, 229, 415–421. [Google Scholar] [CrossRef]
- Zhu, L.; Lin, Y.; Liu, K.; Cortés, E.; Li, H.; Hu, J.; Yamaguchi, A.; Liu, X.; Miyauchi, M.; Fu, J.; et al. Tuning the intermediate reaction barriers by a CuPd catalyst to improve the selectivity of CO2 electroreduction to C2 products. Chin. J. Catal. 2021, 42, 1500–1508. [Google Scholar] [CrossRef]
- Zhao, Y.; Tan, X.; Yang, W.; Jia, C.; Chen, X.; Ren, W.; Smith, S.C.; Zhao, C. Surface Reconstruction of Ultrathin Palladium Nanosheets during Electrocatalytic CO2 Reduction. Angew. Chem. Int. Ed. 2020, 59, 21493–21498. [Google Scholar] [CrossRef] [PubMed]
- Rahaman, M.; Dutta, A.; Broekmann, P. Size-Dependent Activity of Palladium Nanoparticles: Efficient Conversion of CO2 into Formate at Low Overpotentials. ChemSusChem 2017, 10, 1733–1741. [Google Scholar] [CrossRef]
- Ou, L.; Chen, S. Comparative Study of Oxygen Reduction Reaction Mechanisms on the Pd(111) and Pt(111) Surfaces in Acid Medium by DFT. J. Phys. Chem. C 2013, 117, 1342–1349. [Google Scholar] [CrossRef]
- Di, D.; Romanov, A.S.; Yang, L.; Richter, J.M.; Rivett, J.P.H.; Jones, S.; Thomas, T.H.; Abdi Jalebi, M.; Friend, R.H.; Linnolahti, M.; et al. High-performance light-emitting diodes based on carbene-metal-amides. Science 2017, 356, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Hamze, R.; Peltier, J.L.; Sylvinson, D.; Jung, M.; Cardenas, J.; Haiges, R.; Soleilhavoup, M.; Jazzar, R.; Djurovich, P.I.; Bertrand, G.; et al. Eliminating nonradiative decay in Cu(I) emitters: >99% quantum efficiency and microsecond lifetime. Science 2019, 363, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Soleilhavoup, M.; Melaimi, M.; Chu, J.; Bertrand, G. Air-Stable (CAAC)CuCl and (CAAC)CuBH4 Complexes as Catalysts for the Hydrolytic Dehydrogenation of BH3NH3. Angew. Chem. Int. Ed. 2015, 54, 6008–6011. [Google Scholar] [CrossRef]
- Weinberger, D.S.; Amin Sk, N.; Mondal, K.C.; Melaimi, M.; Bertrand, G.; Stückl, A.C.; Roesky, H.W.; Dittrich, B.; Demeshko, S.; Schwederski, B.; et al. Isolation of Neutral Mononuclear Copper Complexes Stabilized by Two Cyclic (Alkyl)(amino)carbenes. J. Am. Chem. Soc. 2014, 136, 6235–6238. [Google Scholar] [CrossRef]
- Li, J.; Wang, L.; Zhao, Z.; Li, X.; Yu, X.; Huo, P.; Jin, Q.; Liu, Z.; Bian, Z.; Huang, C. Two-Coordinate Copper(I)/NHC Complexes: Dual Emission Properties and Ultralong Room-Temperature Phosphorescence. Angew. Chem. Int. Ed. 2020, 59, 8210–8217. [Google Scholar] [CrossRef]
- Shi, S.; Collins, L.R.; Mahon, M.F.; Djurovich, P.I.; Thompson, M.E.; Whittlesey, M.K. Synthesis and characterization of phosphorescent two-coordinate copper(I) complexes bearing diamidocarbene ligands. Dalton Trans. 2017, 46, 745–752. [Google Scholar] [CrossRef]
- Jiao, Y.; Zheng, Y.; Chen, P.; Jaroniec, M.; Qiao, S.-Z. Molecular Scaffolding Strategy with Synergistic Active Centers To Facilitate Electrocatalytic CO2 Reduction to Hydrocarbon/Alcohol. J. Am. Chem. Soc. 2017, 139, 18093–18100. [Google Scholar] [CrossRef]
- Zhao, K.; Nie, X.; Wang, H.; Chen, S.; Quan, X.; Yu, H.; Choi, W.; Zhang, G.; Kim, B.; Chen, J.G. Selective electroreduction of CO2 to acetone by single copper atoms anchored on N-doped porous carbon. Nat. Commun. 2020, 11, 2455. [Google Scholar] [CrossRef]
- Liao, C.-C.; Tsai, T.-H.; Chang, C.-C.; Tsai, M.-K. The use of plate-type electric force field for the explicit simulations of electrochemical CO dimerization on Cu(111) surface. Chem. Phys. 2023, 568, 111821. [Google Scholar] [CrossRef]
- Xiao, H.; Cheng, T.; Goddard, W.A.; Sundararaman, R. Mechanistic Explanation of the pH Dependence and Onset Potentials for Hydrocarbon Products from Electrochemical Reduction of CO on Cu (111). J. Am. Chem. Soc. 2016, 138, 483–486. [Google Scholar] [CrossRef]
- Cheng, T.; Xiao, H.; Goddard, W.A. Full atomistic reaction mechanism with kinetics for CO reduction on Cu(100) from ab initio molecular dynamics free-energy calculations at 298 K. Proc. Natl. Acad. Sci. USA 2017, 114, 1795–1800. [Google Scholar] [CrossRef] [PubMed]
- Bagger, A.; Ju, W.; Varela, A.S.; Strasser, P.; Rossmeisl, J. Electrochemical CO2 Reduction: A Classification Problem. ChemPhysChem 2017, 18, 3266–3273. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metalamorphous- semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Arblaster, J.W. Crystallographic Properties of Palladium. Platin. Met. Rev. 2012, 56, 181–189. [Google Scholar] [CrossRef]
- Jónsson, H.; Mills, G.; Jacobsen, K.W. Nudged elastic band method for finding minimum energy paths of transitions. In Classical and Quantum Dynamics in Condensed Phase Simulations; World Scientific: Singapore, 1998; pp. 385–404. [Google Scholar]
- Momma, K.; Izumi, F. VESTA3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Filot, I.A.W.; van Santen, R.A.; Hensen, E.J.M. The Optimally Performing Fischer–Tropsch Catalyst. Angew. Chem. Int. Ed. 2014, 53, 12746–12750. [Google Scholar] [CrossRef] [PubMed]
- Filot, I.A.W.; Broos, R.J.P.; van Rijn, J.P.M.; van Heugten, G.J.H.A.; van Santen, R.A.; Hensen, E.J.M. First-Principles-Based Microkinetics Simulations of Synthesis Gas Conversion on a Stepped Rhodium Surface. ACS Catal. 2015, 5, 5453–5467. [Google Scholar] [CrossRef]
- Zhang, L.; Filot, I.A.W.; Su, Y.-Q.; Liu, J.-X.; Hensen, E.J.M. Transition metal doping of Pd(111) for the NO + CO reaction. J. Catal. 2018, 363, 154–163. [Google Scholar] [CrossRef]

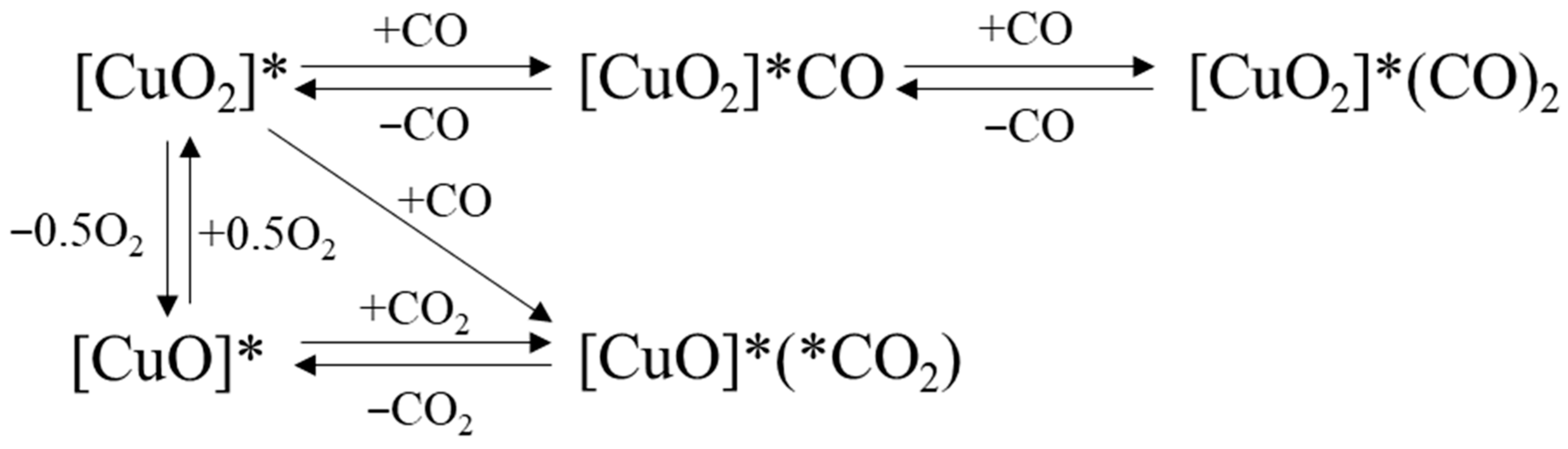


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, C.-C.; Tsai, T.-H.; Chang, C.-C.; Tsai, M.-K. A Computational Perspective on Carbon-Carbon Bond Formation by Single Cu Atom on Pd(111) Surface for CO Electrochemical Reduction. Inorganics 2023, 11, 378. https://doi.org/10.3390/inorganics11100378
Liao C-C, Tsai T-H, Chang C-C, Tsai M-K. A Computational Perspective on Carbon-Carbon Bond Formation by Single Cu Atom on Pd(111) Surface for CO Electrochemical Reduction. Inorganics. 2023; 11(10):378. https://doi.org/10.3390/inorganics11100378
Chicago/Turabian StyleLiao, Chen-Cheng, Tsung-Han Tsai, Chun-Chih Chang, and Ming-Kang Tsai. 2023. "A Computational Perspective on Carbon-Carbon Bond Formation by Single Cu Atom on Pd(111) Surface for CO Electrochemical Reduction" Inorganics 11, no. 10: 378. https://doi.org/10.3390/inorganics11100378
APA StyleLiao, C.-C., Tsai, T.-H., Chang, C.-C., & Tsai, M.-K. (2023). A Computational Perspective on Carbon-Carbon Bond Formation by Single Cu Atom on Pd(111) Surface for CO Electrochemical Reduction. Inorganics, 11(10), 378. https://doi.org/10.3390/inorganics11100378