
Latonduine-1-Amino-Hydantoin Hybrid, Triazole-Fused Latonduine Schiff Bases and Their Metal Complexes: Synthesis, X-ray and Electron Diffraction, Molecular Docking Studies and Antiproliferative Activity

 , , ,
, , ,  and
and 
Abstract
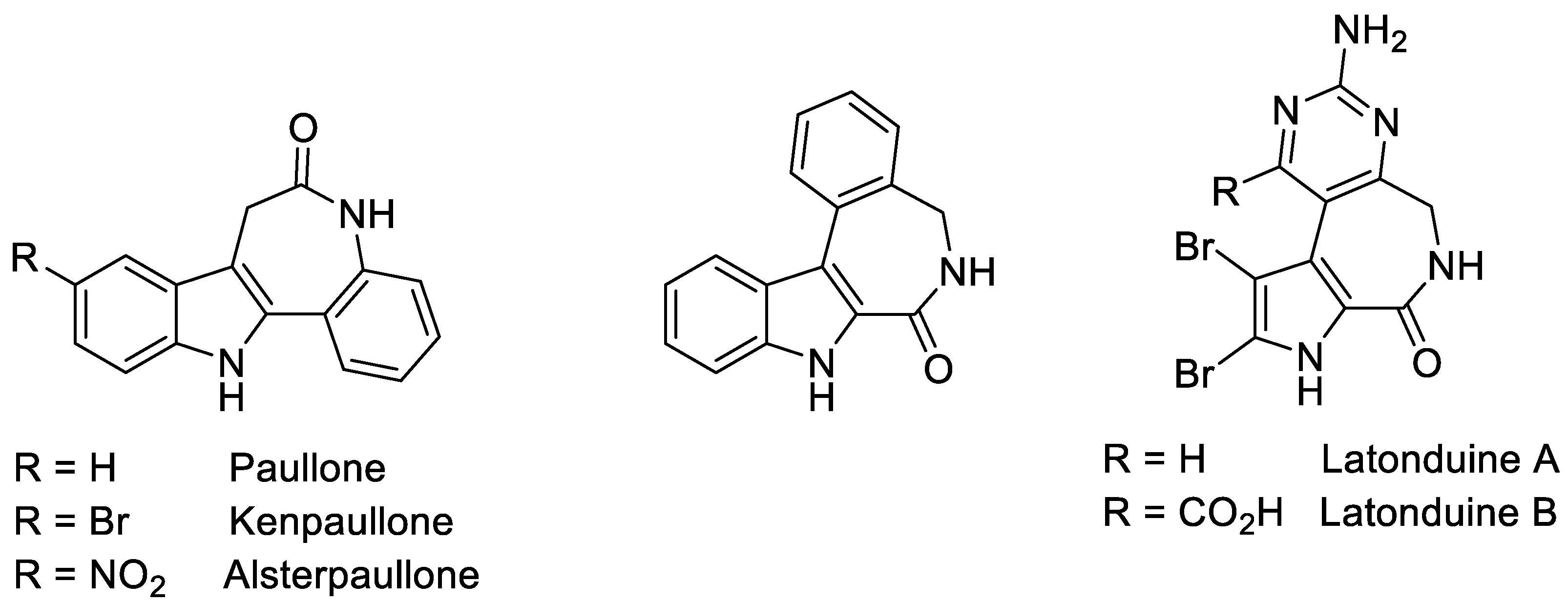
1. Introduction
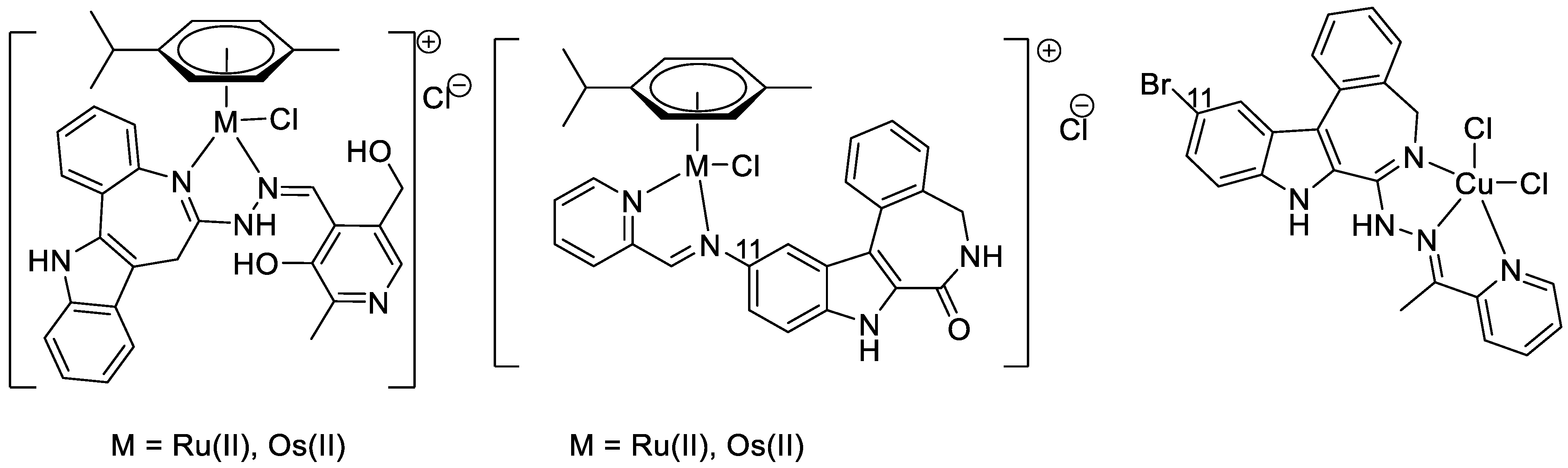
2. Results and Discussion
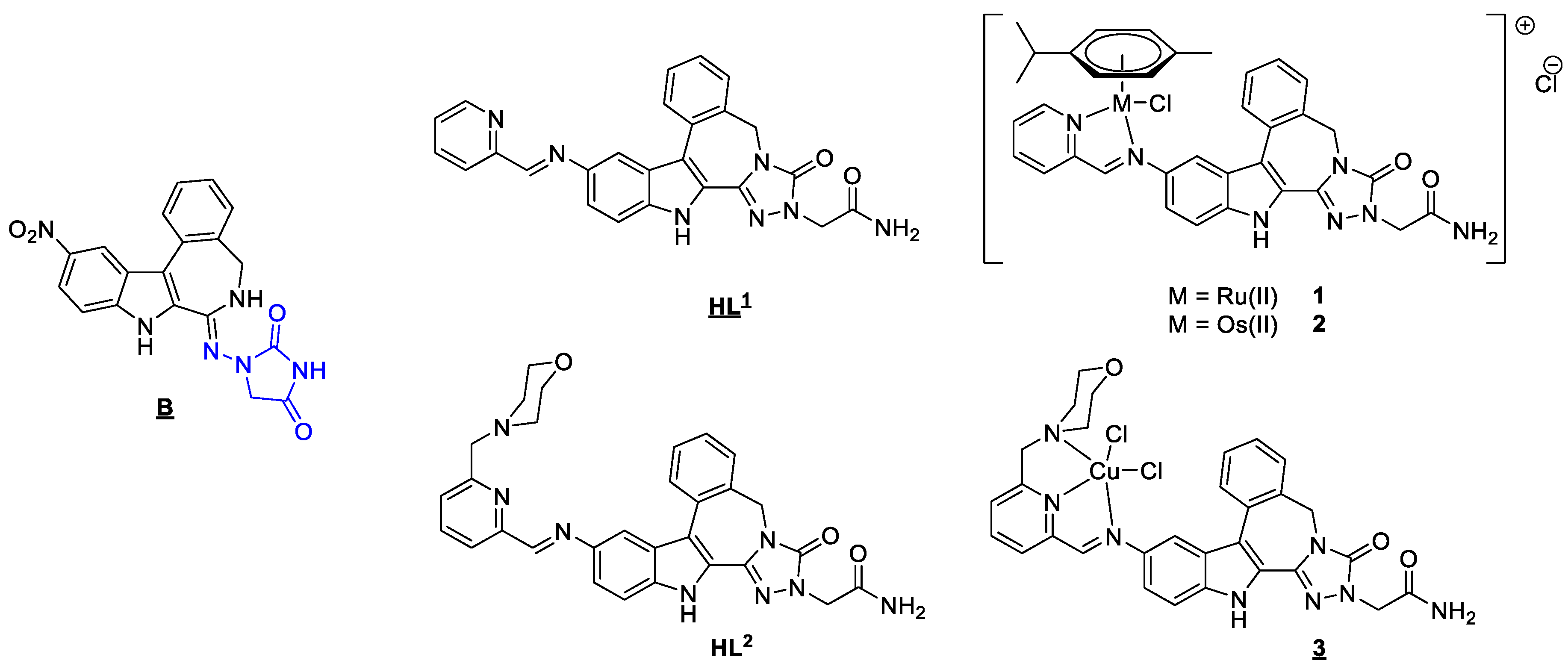
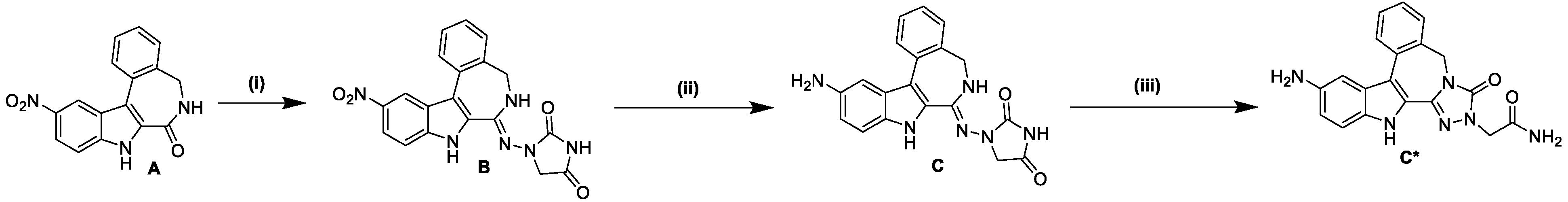
2.1. Synthesis and Spectroscopic Analysis
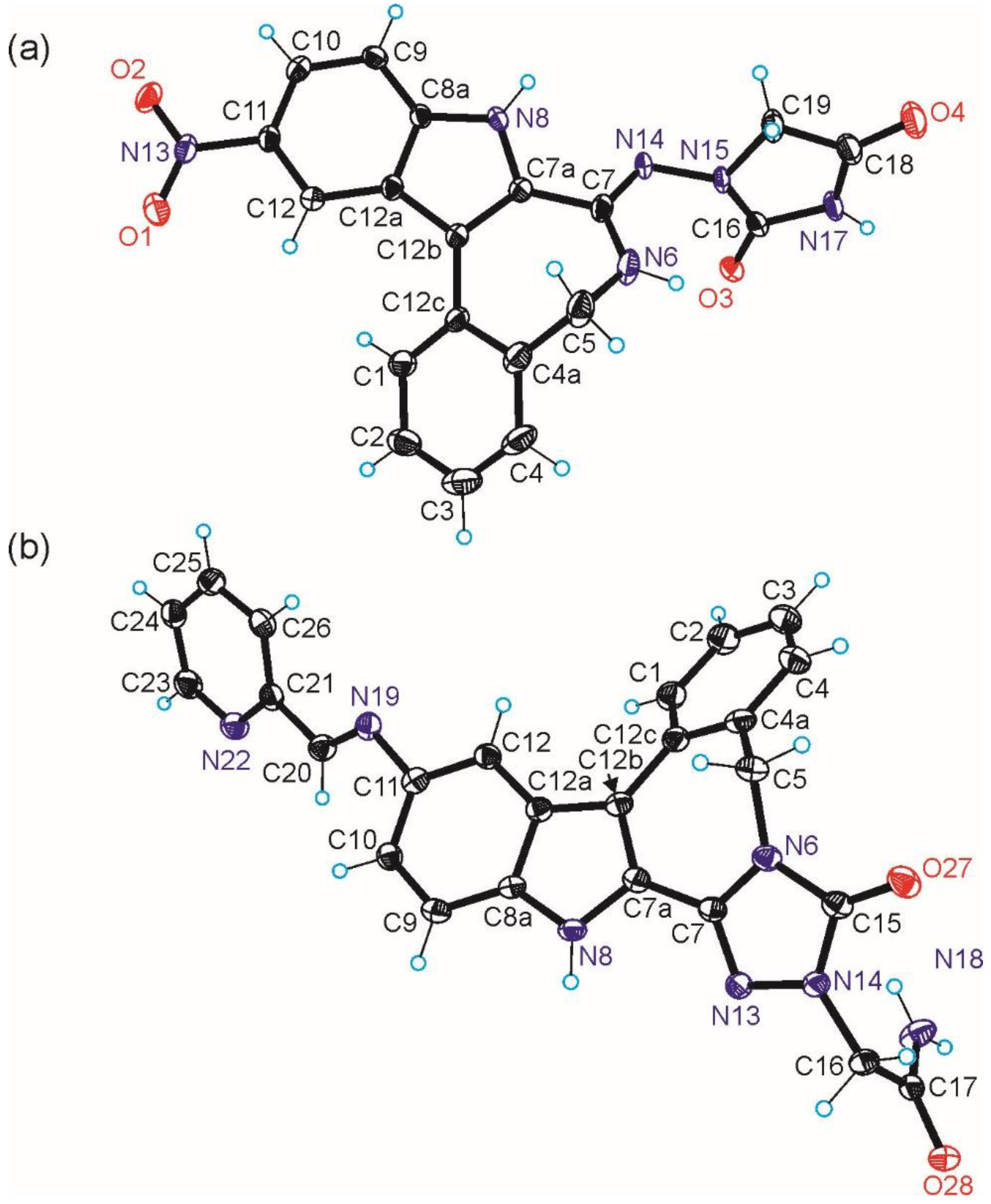
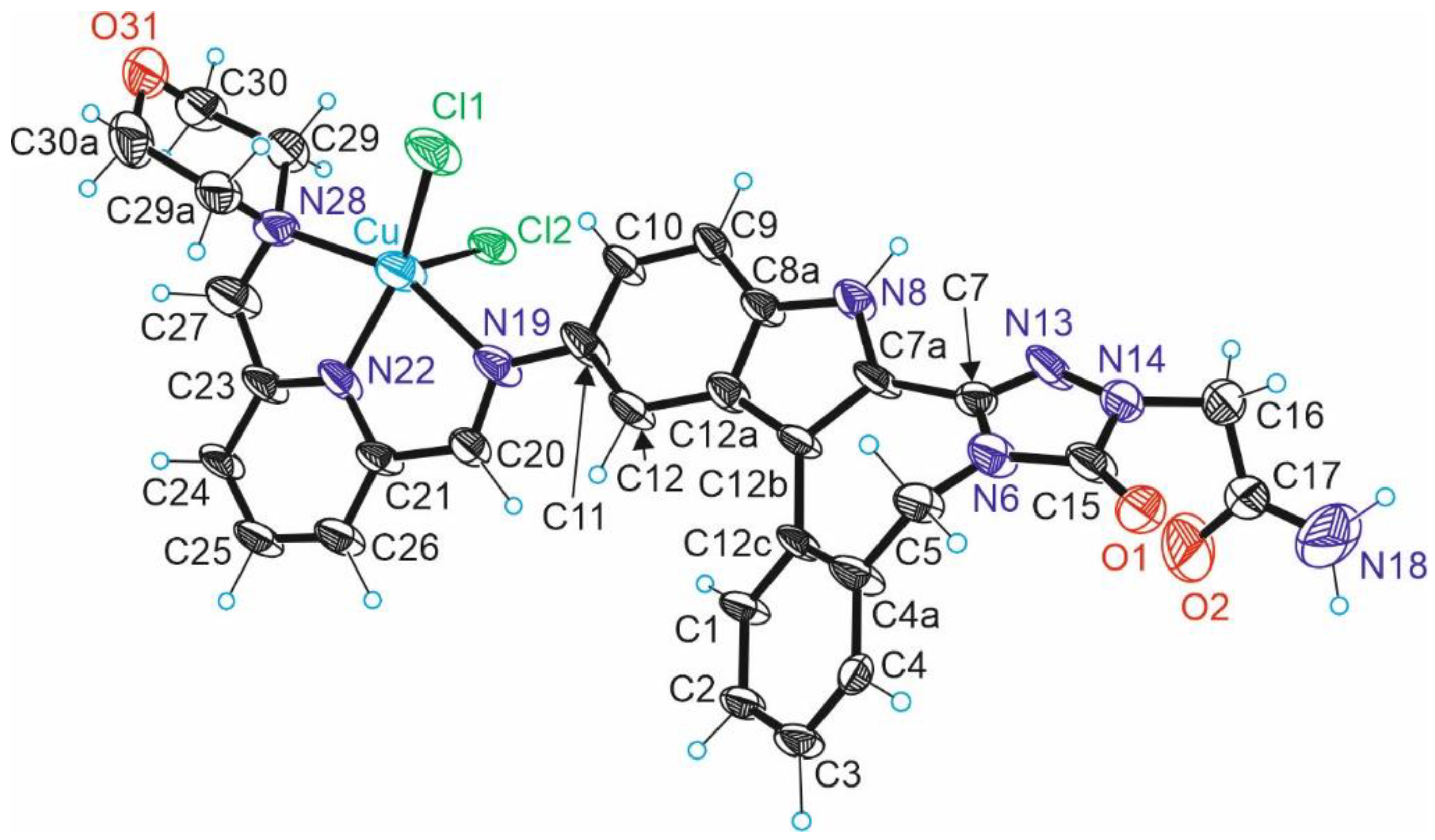
2.2. Molecular Structure Description of Species B, HL1 and 3
2.3. Stability in Solution
2.4. Antiproliferative Activity
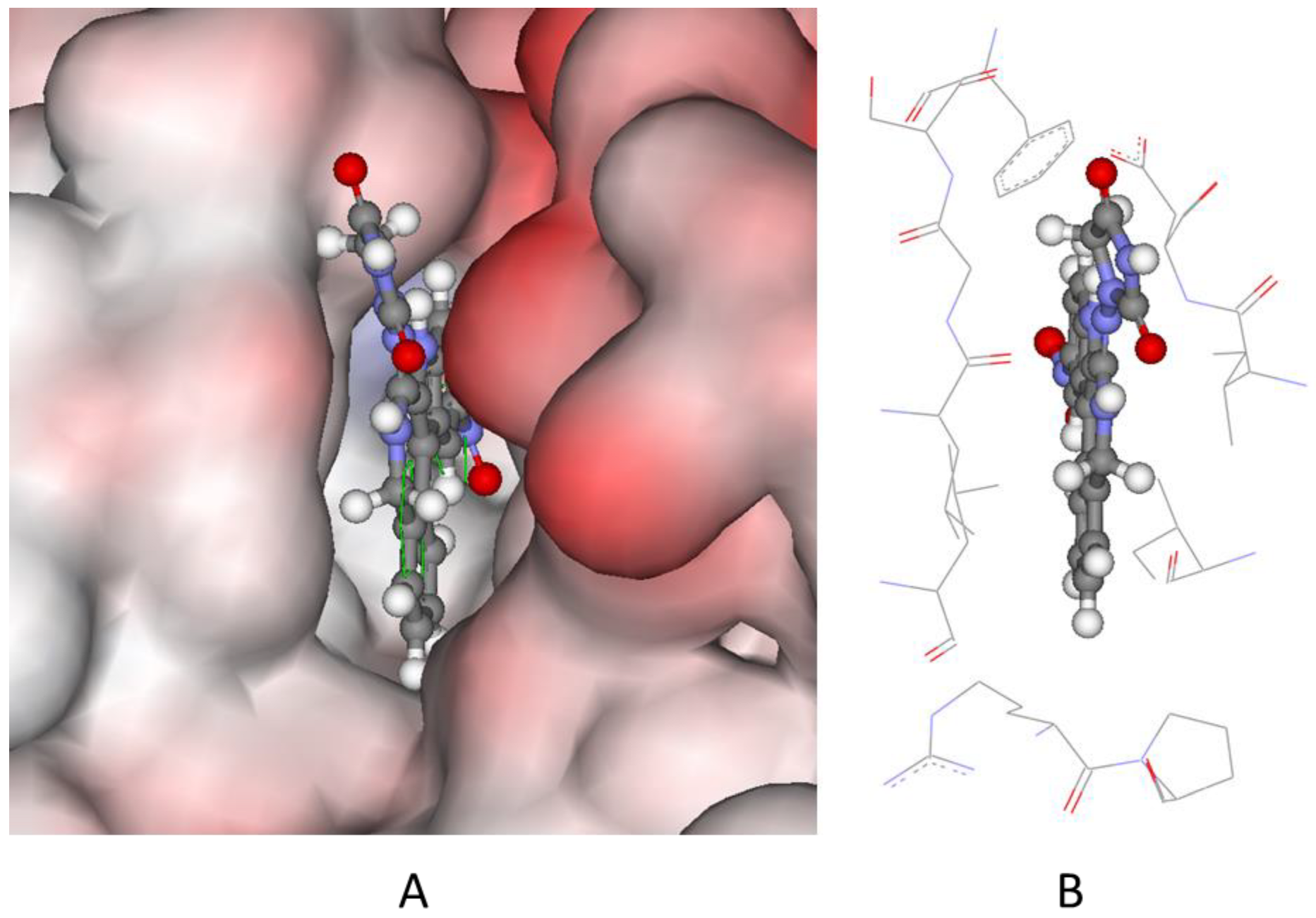
2.5. Molecular Docking of Species B to PIM-1
2.6. Chemical Space
3. Materials and Methods
3.1. Synthesis of Proligands HL1 and HL2
- 1-((11-nitro-5,8-dihydroindolo[2,3-d]benzazepine-7(6H)-ylidene)amino)imidazolidine-2,4-dione (B)
- 1-((11-amino-5,8-dihydroindolo[2,3-d]benzazepine-7(6H)-ylidene)amino)imidazolidine-2,4-dione (C)
- HL1·1.1H2O
- HL2·2H2O
3.2. Synthesis of Ru(II)/Os(II) Arene and Cu(II) Complexes
- 1·2.5H2O
- 2·2.4H2O
- 3·H2O·0.3iPrOH
3.3. Crystallographic Structure Determination
3.4. Sample Preparation and Data Collection for Complex 3 by Electron Diffraction
3.5. Cell Culture
3.6. MTT-Assay
3.7. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cancer over Time. Global Cancer Observatory. Available online: https://gco.iarc.fr/overtime/en (accessed on 4 May 2022).
- Cancer Survival. Global Cancer Observatory. Available online: https://gco.iarc.fr/survival/survmark/ (accessed on 4 May 2022).
- Cancer Tomorrow. Global Cancer Observatory. Available online: https://gco.iarc.fr/tomorrow/en (accessed on 4 May 2022).
- Primik, M.F.; Filak, L.K.; Arion, V.B. Metal-Based Indolobenzazepines and Indoloquinolines: From Moderate CDK Inhibitors to Potential Antitumor Drugs. In Advances in Organometallic Chemistry and Catalysis; Pombeiro, A.J.L., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 605–617. [Google Scholar]
- Primik, M.F.; Mühlgassner, G.; Jakupec, M.A.; Zava, O.; Dyson, P.J.; Arion, V.B.; Keppler, B.K. Highly Cytotoxic Copper(II) Complexes with Modified Paullone Ligands. Inorg. Chem. 2010, 49, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Primik, M.F.; Göschl, S.; Jakupec, M.A.; Roller, A.; Keppler, B.K.; Arion, V.B. Structure−Activity Relationships of Highly Cytotoxic Copper(II) Complexes with Modified Indolo[3,2-c]quinoline Ligands. Inorg. Chem. 2010, 49, 11084–11095. [Google Scholar] [CrossRef] [PubMed]
- Dobrov, A.; Arion, V.B.; Kandler, N.; Ginzinger, W.; Jakupec, M.A.; Rufińska, A.; Graf von Keyserlingk, N.; Galanski, M.S.; Kowol, C.; Keppler, B.K. The First Metal-Based Paullone Derivative with High Antiproliferative Activity in Vitro. Inorg. Chem. 2006, 45, 1945–1950. [Google Scholar] [CrossRef] [PubMed]
- Arion, V.B.; Dobrov, A.; Göschl, S.; Jakupec, M.A.; Keppler, B.K.; Rapta, P. Ruthenium- and Osmium-Arene-Based Paullones Bearing a TEMPO Free-Radical Unit as Potential Anticancer Drugs. Chem. Commun. 2012, 48, 8559–8561. [Google Scholar] [CrossRef] [PubMed]
- Schultz, C.; Link, A.; Leost, M.; Zaharevitz, D.W.; Gussio, R.; Sausville, E.A.; Meijer, L.; Kunick, C. Paullones, a Series of Cyclin-Dependent Kinase Inhibitors: Synthesis, Evaluation of CDK1/Cyclin B Inhibition, and in Vitro Antitumor Activity. J. Med. Chem. 1999, 42, 2909–2919. [Google Scholar] [CrossRef]
- Knockaert, M.; Wieking, K.; Schmitt, S.; Leost, M.; Grant, K.M.; Mottram, J.C.; Kunick, C.; Meijer, L. Intracellular Targets of Paullones. J. Biol. Chem. 2002, 277, 25493–25501. [Google Scholar] [CrossRef]
- Kunick, C.; Lauenroth, K.; Wieking, K.; Xie, X.; Schultz, C.; Gussio, R.; Zaharevitz, D.; Leost, M.; Meijer, L.; Weber, A.; et al. Evaluation and Comparison of 3D-QSAR CoMSIA Models for CDK1, CDK5, and GSK-3 Inhibition by Paullones. J. Med. Chem. 2004, 47, 22–36. [Google Scholar] [CrossRef]
- Kunick, C.; Zeng, Z.; Gussio, R.; Zaharevitz, D.; Leost, M.; Totzke, F.; Schächtele, C.; Kubbutat, M.H.G.; Meijer, L.; Lemcke, T. Structure-Aided Optimization of Kinase Inhibitors Derived from Alsterpaullone. ChemBioChem 2005, 6, 541–549. [Google Scholar] [CrossRef]
- Leost, M.; Schultz, C.; Link, A.; Wu, Y.-Z.; Biernat, J.; Mandelkow, E.-M.; Bibb, J.A.; Snyder, G.L.; Greengard, P.; Zaharevitz, D.W.; et al. Paullones Are Potent Inhibitors of Glycogen Synthase Kinase-3β and Cyclin-Dependent Kinase 5/P25: Paullones Inhibit GSK-3β and CDK5/P25. Eur. J. Biochem. 2000, 267, 5983–5994. [Google Scholar] [CrossRef]
- Zaharevitz, D.W.; Gussio, R.; Leost, M.; Senderowicz, A.M.; Lahusen, T.; Kunick, C.; Meijer, L.; Sausville, E.A. Discovery and Initial Characterization of the Paullones, a Novel Class of Small-Molecule Inhibitors of Cyclin-Dependent Kinases. Cancer Res. 1999, 59, 2566–2569. [Google Scholar]
- Soto, S.; Vaz, E.; Dell’Aversana, C.; Álvarez, R.; Altucci, L.; de Lera, Á.R. New Synthetic Approach to Paullones and Characterization of Their SIRT1 Inhibitory Activity. Org. Biomol. Chem. 2012, 10, 2101–2112. [Google Scholar] [CrossRef]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C.J.; Mclauchlan, H.; Klevernic, I.; Arthur, J.S.C.; Alessi, D.R.; Cohen, P. The Selectivity of Protein Kinase Inhibitors: A Further Update. Biochem. J. 2007, 408, 297–315. [Google Scholar] [CrossRef] [PubMed]
- Bain, J.; McLauchlan, H.; Elliott, M.; Cohen, P. The Specificities of Protein Kinase Inhibitors: An Update. Biochem. J. 2003, 371, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Linington, R.G.; Williams, D.E.; Tahir, A.; Van Soest, R.; Andersen, R.J. Latonduines A and B, New Alkaloids Isolated from the Marine Sponge Stylissa Carteri: Structure Elucidation, Synthesis, and Biogenetic Implications. Org. Lett. 2003, 5, 2735–2738. [Google Scholar] [CrossRef] [PubMed]
- Fouad, M.A.; Debbab, A.; Wray, V.; Müller, W.E.G.; Proksch, P. New Bioactive Alkaloids from the Marine Sponge Stylissa sp. Tetrahedron 2012, 68, 10176–10179. [Google Scholar] [CrossRef]
- Putey, A.; Joucla, L.; Picot, L.; Besson, T.; Joseph, B. Synthesis of Latonduine Derivatives via Intramolecular Heck Reaction. Tetrahedron 2007, 63, 867–879. [Google Scholar] [CrossRef]
- Wan, Y.; Li, Y.; Yan, C.; Yan, M.; Tang, Z. Indole: A Privileged Scaffold for the Design of Anti-Cancer Agents. Eur. J. Med. Chem. 2019, 183, 111691. [Google Scholar] [CrossRef]
- Brancale, A.; Silvestri, R. Indole, a Core Nucleus for Potent Inhibitors of Tubulin Polymerization. Med. Res. Rev. 2007, 27, 209–238. [Google Scholar] [CrossRef]
- Singh, A.K.; Raj, V.; Saha, S. Indole-Fused Azepines and Analogues as Anticancer Lead Molecules: Privileged Findings and Future Directions. Eur. J. Med. Chem. 2017, 142, 244–265. [Google Scholar] [CrossRef]
- Putey, A.; Popowycz, F.; Do, Q.T.; Bernard, P.; Talapatra, S.K.; Kozielski, F.; Galmarini, C.M.; Joseph, B. Indolobenzazepin-7-ones and 6-, 8-, and 9-Membered Ring Derivatives as Tubulin Polymerization Inhibitors: Synthesis and Structure-Activity Relationship Studies. J. Med. Chem. 2009, 52, 5916–5925. [Google Scholar] [CrossRef]
- Filak, L.K.; Mühlgassner, G.; Jakupec, M.A.; Heffeter, P.; Berger, W.; Arion, V.B.; Keppler, B.K. Organometallic Indolo[3,2-c]quinolines versus Indolo[3,2-d]benzazepines: Synthesis, Structural and Spectroscopic Characterization, and Biological Efficacy. J. Biol. Inorg. Chem. 2010, 15, 903–918. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, C.; Sivchenko, A.S.; Bacher, F.; Tong, K.K.H.; Guru, N.; Wilson, T.; Gonzales, J.; Rauch, H.; Kossatz, S.; Reiner, T.; et al. Inhibition of Microtubule Dynamics in Cancer Cells by Indole-Modified Latonduine Derivatives and Their Metal Complexes. Inorg. Chem. 2022, 61, 1456–1470. [Google Scholar] [CrossRef] [PubMed]
- Bacher, F.; Wittmann, C.; Nové, M.; Spengler, G.; Marć, M.A.; Enyedy, E.A.; Darvasiová, D.; Rapta, P.; Reiner, T.; Arion, V.B. Novel Latonduine Derived Proligands and Their Copper(II) Complexes Show Cytotoxicity in the Nanomolar Range in Human Colon Adenocarcinoma Cells and in Vitro Cancer Selectivity. Dalton Trans. 2019, 48, 10464–10478. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, C.; Bacher, F.; Enyedy, E.A.; Dömötör, O.; Spengler, G.; Madejski, C.; Reynisson, J.; Arion, V.B. Highly Antiproliferative Latonduine and Indolo[2,3-c]quinoline Derivatives: Complex Formation with Copper(II) Markedly Changes the Kinase Inhibitory Profile. J. Med. Chem. 2022, 65, 2238–2261. [Google Scholar] [CrossRef]
- Anand, R.; Maksimoska, J.; Pagano, N.; Wong, E.Y.; Gimotty, P.A.; Diamond, S.L.; Meggers, E.; Marmorstein, R. Toward the Development of a Potent and Selective Organoruthenium Mammalian Sterile 20 Kinase Inhibitor. J. Med. Chem. 2009, 52, 1602–1611. [Google Scholar] [CrossRef]
- Puszyńska-Tuszkanow, M.; Daszkiewicz, M.; Maciejewska, G.; Adach, A.; Cieślak-Golonka, M. Interaction of Hydantoins with Transition Metal Ions: Synthesis, Structural, Spectroscopic, Thermal and Magnetic Properties of [M(H2O)4(Phenytoinate)2] M = Ni(II), Co(II). Struct. Chem. 2010, 21, 315–321. [Google Scholar] [CrossRef]
- Rajic, Z.; Zorc, B.; Raic-Malic, S.; Ester, K.; Kralj, M.; Pavelic, K.; Balzarini, J.; De Clercq, E.; Mintas, M. Hydantoin Derivatives of L- and D-Amino Acids: Synthesis and Evaluation of Their Antiviral and Antitumoral Activity. Molecules 2006, 11, 837–848. [Google Scholar] [CrossRef]
- Levo, Y.; Trainin, N. Hydantoin Immunosuppression and Carcinogenesis. Clinincal Exp. Immunol. 1975, 19, 521–527. [Google Scholar]
- Cavazzoni, A.; Alfieri, R.R.; Carmi, C.; Zuliani, V.; Galetti, M.; Fumarola, C.; Frazzi, R.; Bonelli, M.; Bordi, F.; Lodola, A.; et al. Dual Mechanisms of Action of the 5-Benzylidene-Hydantoin UPR1024 on Lung Cancer Cell Lines. Mol. Cancer Ther. 2008, 7, 361–370. [Google Scholar] [CrossRef]
- Carmi, C.; Cavazzoni, A.; Zuliani, V.; Lodola, A.; Bordi, F.; Plazzi, P.V.; Alfieri, R.R.; Petronini, P.G.; Mor, M. 5-Benzylidene-Hydantoins as New EGFR Inhibitors with Antiproliferative Activity. Bioorg. Med. Chem. Lett. 2006, 16, 4021–4025. [Google Scholar] [CrossRef]
- Primik, M.F.; Göschl, S.; Meier, S.M.; Eberherr, N.; Jakupec, M.A.; Enyedy, É.A.; Novitchi, G.; Arion, V.B. Dicopper(II) and Dizinc(II) Complexes with Nonsymmetric Dinucleating Ligands Based on Indolo[3,2-c]quinolines: Synthesis, Structure, Cytotoxicity, and Intracellular Distribution. Inorg. Chem. 2013, 52, 10137–10146. [Google Scholar] [CrossRef] [PubMed]
- Addison, A.W.; Rao, T.N.; Reedijk, J.; van Rijn, J.; Verschoor, G.C. Synthesis, Structure, and Spectroscopic Properties of Copper(II) Compounds Containing Nitrogen–Sulphur Donor Ligands; the Crystal and Molecular Structure of Aqua[1,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]copper(II) perchlorate. J. Chem. Soc. Dalton Trans. 1984, 1349–1356. [Google Scholar] [CrossRef]
- Jungwirth, U.; Kowol, C.; Keppler, B.; Hartinger, C.; Berger, W.; Heffeter, P. Anticancer Activity of Metal Complexes: Involvement of Redox Processes. Antioxid. Redox Signal. 2011, 15, 1085–1127. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, C.; Musumeci, D.; Irace, C.; Paduano, L.; Montesarchio, D. RuIII Complexes for Anticancer Therapy: The Importance of Being Nucleolipidic. Eur. J. Org. Chem. 2017, 7, 1100–1119. [Google Scholar] [CrossRef]
- Thota, S.; Rodrigues, D.; Crans, D.; Barreiro, E.J. Ru(II) Compounds: Next-Generation Anticancer Metallotherapeutics? J. Med. Chem. 2018, 61, 5805–5821. [Google Scholar] [CrossRef] [PubMed]
- Shanbhag, V.C.; Gudekar, N.; Jasmer, K.; Papageorgiou, C.; Singh, K.; Petris, M. Copper metabolism as a unique vulnerability in cancer. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118893. [Google Scholar] [CrossRef] [PubMed]
- Denoyer, D.; Masaldan, S.; La Fontaine, S.; Cater, M. Targeting copper in cancer therapy: ’Copper That Cancer’. Metallomics 2015, 7, 1459–1476. [Google Scholar] [CrossRef] [PubMed]
- Babak, M.V.; Ahn, D. Modulation of Intracellular Copper Levels as the Mechanism of Action of Anticancer Copper Complexes: Clinical Relevance. Biomedicines 2021, 9, 852. [Google Scholar] [CrossRef]
- da Silva, D.A.; De Luca, A.; Squitti, R.; Rongioletti, M.; Rossi, L.; Machado, C.; Cerchiaro, G.J. Copper in tumors and the use of copper-based compounds in cancr treatment. J. Inorg. Biochem. 2022, 226, 111634. [Google Scholar] [CrossRef]
- Dobrov, A.; Göschl, S.; Jakupec, M.A.; Popović-Bijelić, A.; Gräslund, A.; Rapta, P.; Arion, V.B. A Highly Cytotoxic Modified Paullone Ligand Bearing a TEMPO Free-Radical Unit and Its Copper(II) Complex as Potential HR2 RNR Inhibitors. Chem. Commun. 2013, 49, 10007–10009. [Google Scholar] [CrossRef]
- Kumar, A.; Mandiyan, V.; Suzuki, Y.; Zhang, C.; Rice, J.; Tsai, J.; Artis, D.R.; Ibrahim, P.; Bremer, R. Crystal Structures of Proto-Oncogene Kinase Pim1: A Target of Aberrant Somatic Hypermutations in Diffuse Large Cell Lymphoma. J. Mol. Biol. 2005, 348, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and Validation of a Genetic Algorithm for Flexible Docking 1. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Eldridge, M.D.; Murray, C.W.; Auton, T.R.; Paolini, G.V.; Mee, R.P. Empirical Scoring Functions: I. The Development of a Fast Empirical Scoring Function to Estimate the Binding Affinity of Ligands in Receptor Complexes. J. Comput. Aided Mol. Des. 1997, 11, 425–445. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved Protein-Ligand Docking Using GOLD. Proteins Struct. Funct. Bioinform. 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical Scoring Functions for Advanced Protein−Ligand Docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef]
- Mooij, W.T.M.; Verdonk, M.L. General and Targeted Statistical Potentials for Protein-Ligand Interactions. Proteins Struct. Funct. Bioinform. 2005, 61, 272–287. [Google Scholar] [CrossRef]
- QikProp, v6.2; Schrödinger, LLC: New York, NY, USA, 2009.
- Zhu, F.; Logan, G.; Reynisson, J. Wine Compounds as a Source for HTS Screening Collections. A Feasibility Study. Mol. Inform. 2012, 31, 847–855. [Google Scholar] [CrossRef]
- Eurtivong, C.; Reynisson, J. The Development of a Weighted Index to Optimise Compound Libraries for High Throughput Screening. Mol. Inform. 2019, 38, 1800068. [Google Scholar] [CrossRef]
- Matuszek, A.M.; Reynisson, J. Defining Known Drug Space Using DFT. Mol. Inform. 2016, 35, 46–53. [Google Scholar] [CrossRef]
- Yu, B.; Reynisson, J. Bond Stability of the “Undesirable” Heteroatom–Heteroatom Molecular Moieties for High-Throughput Screening Libraries. Eur. J. Med. Chem. 2011, 46, 5833–5837. [Google Scholar] [CrossRef]
- Ohui, K.; Afanasenko, E.; Bacher, F.; Ting, R.L.X.; Zafar, A.; Blanco-Cabra, N.; Torrents, E.; Dömötör, O.; May, N.V.; Darvasiova, D.; et al. New Water-Soluble Copper(II) Complexes with Morpholine–Thiosemicarbazone Hybrids: Insights into the Anticancer and Antibacterial Mode of Action. J. Med. Chem. 2019, 62, 512–530. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.A.; Smith, A.K. Arene Ruthenium(II) Complexes Formed by Dehydrogenation of Cyclohexadienes with Ruthenium(III) Trichloride. J. Chem. Soc. Dalton Trans. 1974, 233–241. [Google Scholar] [CrossRef]
- Kiel, W.A.; Ball, R.G.; Graham, W.A.G. Carbonyl-η-hexamethylbenzene Complexes of Osmium. Carbon-Hydrogen Activation by (η-C6Me6)Os(CO)(H)2. J. Organomet. Chem. 1990, 383, 481–496. [Google Scholar] [CrossRef]
- SAINT-Plus, v8.38A; Bruker-Nonius AXS Inc.: Madison, WI, USA, 2016.
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Burnett, M.N.; Johnson, C.K. ORTEP-III: Oak Ridge Thermal Ellipsoid Plot Program for Crystal Structure Illustrations. ORNL-6895, Oak Ridge, Tennessee 37831, U.S. 1996. [Google Scholar]
- Fröjdh, E.; Wennmacher, J.; Rzepka, P.; Mozzanica, A.; Redford, S.; Schmitt, B.; van Bokhoven, J.; Gruene, T. Discrimination of Aluminum from Silicon by Electron Crystallography with the JUNGFRAU Detector. Crystals 2020, 10, 1148. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef]
- Assmann, G.M.; Wang, M.; Diederichs, K. Making a Difference in Multi-Data-Set Crystallography: Simple and Deterministic Data-Scaling/Selection Methods. Acta Crystallogr. Sect. Struct. Biol. 2020, 76, 636–652. [Google Scholar] [CrossRef]
- Karplus, P.A.; Diederichs, K. Linking Crystallographic Model and Data Quality. Science 2012, 336, 1030–1033. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Janssen, T.; Janner, A.; Looijenga-Vosb, A.; de Wolff, P.M. (Eds.) International Tables for Crystallography: Mathematical, Physical and Chemical Tables, 1st ed.; International Union of Crystallography: Chester, UK, 2006; Volume C. [Google Scholar]
- Peng, L.-M. Electron Atomic Scattering Factors and Scattering Potentials of Crystals. Micron 1999, 30, 625–648. [Google Scholar] [CrossRef]
- Gruene, T.; Hahn, H.W.; Luebben, A.V.; Meilleur, F.; Sheldrick, G.M. Refinement of Macromolecular Structures against Neutron Data with SHELXL2013. J. Appl. Crystallogr. 2014, 47, 462–466. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminf. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Smart, O.S.; Sharff, A.; Holstein, J.; Womack, T.O.; Flensburg, C.; Keller, P.; Paciorek, W.; Vonrhein, C.; Bricogne, G. Grade2. Version 1.3.0. Global Phasing Ltd.: Cambridge, UK, 2021. [Google Scholar]
- Gruene, T.; Clabbers, M.T.B.; Luebben, J.; Chin, J.M.; Reithofer, M.R.; Stowasser, F.; Alker, A.M. CELLOPT: Improved Unit-Cell Parameters for Electron Diffraction Data of Small-Molecule Crystals. J. Appl. Crystallogr. 2022, 55, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Supino, R. MTT Assays. In In Vitro Toxicity Testing Protocols; O’Hare, S., Atterwill, C.K., Eds.; Humana Press: Totowa, NJ, USA, 1995; pp. 137–149. [Google Scholar]
- Pavlović, M.; Nikolić, S.; Gligorijević, N.; Dojčinović, B.; Aranđelović, S.; Grgurić-Šipka, S.; Radulović, S. New Organoruthenium Compounds with Pyrido[2′,3′:5,6]pyrazino[2,3-f][1,10]phenanthroline: Synthesis, Characterization, Cytotoxicity, and Investigation of Mechanism of Action. J. Biol. Inorg. Chem. 2019, 24, 297–310. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, H.; Yao, X.; Li, D.; Xu, L.; Li, Y.; Tian, S.; Hou, T. Comprehensive Evaluation of Ten Docking Programs on a Diverse Set of Protein-Ligand Complexes: The Prediction Accuracy of Sampling Power and Scoring Power. Phys. Chem. Chem. Phys. 2016, 18, 12964–12975. [Google Scholar] [CrossRef]
- Bissantz, C.; Folkers, G.; Rognan, D. Protein-Based Virtual Screening of Chemical Databases. 1. Evaluation of Different Docking/Scoring Combinations. J. Med. Chem. 2000, 43, 4759–4767. [Google Scholar] [CrossRef]
- Scigress Ultra, V. F.J 2.6. Fujitsu Limited. 2016. Available online: http://www.scigress.com (accessed on 14 November 2022).
- Allinger, N.L.; Yuh, Y.H.; Lii, J.H. Molecular Mechanics. The MM3 Force Field for Hydrocarbons. 1. J. Am. Chem. Soc. 1989, 111, 8551–8566. [Google Scholar] [CrossRef]
- Lii, J.H.; Allinger, N.L. Molecular Mechanics. The MM3 Force Field for Hydrocarbons. 2. Vibrational Frequencies and Thermodynamics. J. Am. Chem. Soc. 1989, 111, 8566–8575. [Google Scholar] [CrossRef]
- Lii, J.H.; Allinger, N.L. Molecular Mechanics. The MM3 Force Field for Hydrocarbons. 3. The van Der Waals’ Potentials and Crystal Data for Aliphatic and Aromatic Hydrocarbons. J. Am. Chem. Soc. 1989, 111, 8576–8582. [Google Scholar] [CrossRef]
- Gotō, H.; Ōsawa, E. An Efficient Algorithm for Searching Low-Energy Conformers of Cyclic and Acyclic Molecules. J. Chem. Soc. Perkin Trans. 1993, 2, 187–198. [Google Scholar] [CrossRef]
- Ioakimidis, L.; Thoukydidis, L.; Mirza, A.; Naeem, S.; Reynisson, J. Benchmarking the Reliability of QikProp. Correlation between Experimental and Predicted Values. QSAR Comb. Sci. 2008, 27, 445–456. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01. 2019; Gaussian Inc.: Wallingford, CT, USA.
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-Consistent Molecular Orbital Methods 25. Supplementary Functions for Gaussian Basis Sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies. Theoret. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Wong, M.W. Vibrational Frequency Prediction Using Density Functional Theory. Chem. Phys. Lett. 1996, 256, 391–399. [Google Scholar] [CrossRef]
- Frisch, A.; Foresman, J. Exploring Chemistry with Electronic Structure Methods, 3rd ed.; Gaussian Inc.: Wallingford, CT, USA, 1996. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | A549 | LS-174 | MDA-MB-231 | MRC-5 |
|---|---|---|---|---|
| B | 157.9 ± 3.7 | 86.0 ± 0.5 | 66.7 ± 8.7 | 99.7 ± 0.3 |
| HL1 | 180.7 ± 5.8 | 84.4 ± 2.1 | 89.8 ± 1.0 | 93.5 ± 4.1 |
| HL2 | 190.2 ± 2.1 | 98.8 ± 6.6 | 81.6 ± 8.5 | 77.3 ± 3.1 |
| 1 | 41.5 ± 2.9 | 47.7 ± 6.4 | 65.4 ± 2.5 | 44.1 ± 5.7 |
| 2 | 50.0 ± 8.9 | 59.4 ± 2.9 | 74.4 ± 5.2 | 69.7 ± 3.0 |
| 3 | 37.2 ± 2.4 | 10.9 ± 0.8 | 33.7 ± 5.1 | 31.8 ± 6.6 |
| Cisplatin | 24.7 ± 3.5 | 23.6 ± 4.4 | 9.0 ± 0.5 | 14.6 ± 1.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wittmann, C.; Gruene, T.; Prado-Roller, A.; Aranđelović, S.; Reynisson, J.; Arion, V.B. Latonduine-1-Amino-Hydantoin Hybrid, Triazole-Fused Latonduine Schiff Bases and Their Metal Complexes: Synthesis, X-ray and Electron Diffraction, Molecular Docking Studies and Antiproliferative Activity. Inorganics 2023, 11, 30. https://doi.org/10.3390/inorganics11010030
Wittmann C, Gruene T, Prado-Roller A, Aranđelović S, Reynisson J, Arion VB. Latonduine-1-Amino-Hydantoin Hybrid, Triazole-Fused Latonduine Schiff Bases and Their Metal Complexes: Synthesis, X-ray and Electron Diffraction, Molecular Docking Studies and Antiproliferative Activity. Inorganics. 2023; 11(1):30. https://doi.org/10.3390/inorganics11010030
Chicago/Turabian StyleWittmann, Christopher, Tim Gruene, Alexander Prado-Roller, Sandra Aranđelović, Jóhannes Reynisson, and Vladimir B. Arion. 2023. "Latonduine-1-Amino-Hydantoin Hybrid, Triazole-Fused Latonduine Schiff Bases and Their Metal Complexes: Synthesis, X-ray and Electron Diffraction, Molecular Docking Studies and Antiproliferative Activity" Inorganics 11, no. 1: 30. https://doi.org/10.3390/inorganics11010030
APA StyleWittmann, C., Gruene, T., Prado-Roller, A., Aranđelović, S., Reynisson, J., & Arion, V. B. (2023). Latonduine-1-Amino-Hydantoin Hybrid, Triazole-Fused Latonduine Schiff Bases and Their Metal Complexes: Synthesis, X-ray and Electron Diffraction, Molecular Docking Studies and Antiproliferative Activity. Inorganics, 11(1), 30. https://doi.org/10.3390/inorganics11010030