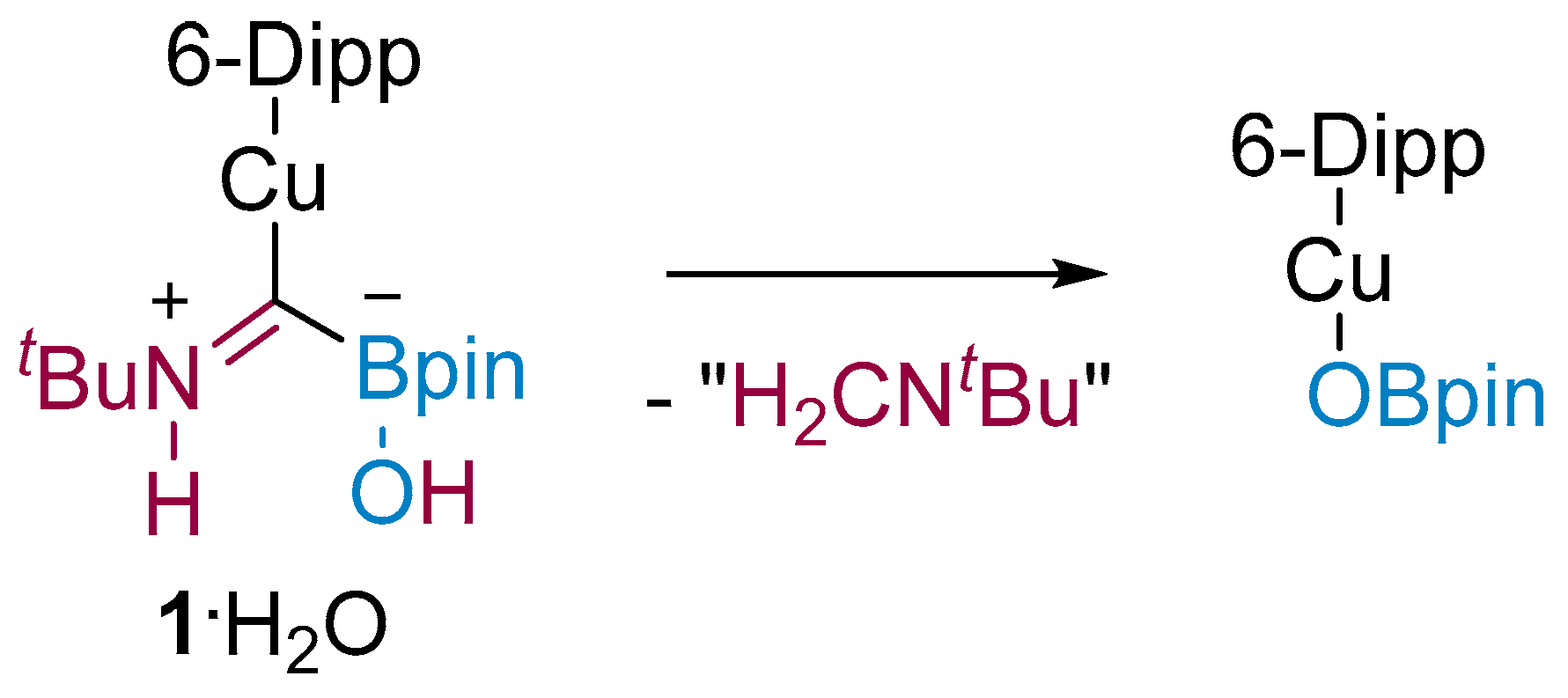1. Introduction
The “Frustrated Lewis Pair” (FLP) concept has contributed to a revolution in small molecule activation with transition metal-free systems, as well as in diverse other fields, including organic synthesis [
1,
2]. Though the original concept relied upon the combination of strong Lewis acid (LA) and base fragments (LB) that could not form a classical Lewis adduct, there is an increasing recognition that almost any set of appropriately oriented lone pairs and vacant orbitals can be considered within the FLP conceptual framework [
3].
The major attraction of FLPs is their ability to activate molecules in a transition metal-mimetic fashion. For example, Stephan’s first report of an FLP was noted for the reversible binding of dihydrogen [
4] that could be exploited in hydrogenation [
5]. Since this initial report, a plethora of other E-H bonds have been activated across FLPs. Of particular relevance to this work is the heterolytic activation of water to generate a Lewis acid-hydroxide fragment and a protonated Lewis base [
6]. A similar reaction has also been observed for some C-H acidic fragments, most notably acetylenes which generate a Lewis acid-acetylide and protonated Lewis base pair [
7]. Carbon dioxide and analogous heterocumulenes are also activated by FLPs, generally generating a LB-C(O)O-LA moiety [
8,
9].
One structural motif which has seen some interest for its FLP-type activity is the carbiminoborane scaffold which comprises a R
2B-C(R)=NR fragment where the 3-coordinate boron centre is Lewis acidic, and the 2-coordinate imine nitrogen, which retains a lone pair, constitutes the Lewis base. Figueroa and co-workers reported that 1,1-hydroboration of an isonitrile generated a carbiminoborane, Cy
2BC(H)=NAr (Ar = 2,6-(2,6-(
iPr)
2C
6H
3)
2C
6H
3), that displayed FLP-like reactivity towards carbon dioxide, nitriles, and acetylenes (
Scheme 1) [
10,
11]. For example, the reaction with CO
2 gave rise to a 5-membered N-C-B-O-C ring that was proposed to form via a 1,2-alkyl transfer of the cyclohexyl group on boron from Cy
2BC(H)=NAr·CO
2. In contrast, E-H bonds in water and acetylene were cleaved in a heterolytic fashion to give CyB(E)C(Cy)(H)=N(H)Ar (E = CC
tBu, OH) once again via a FLP-activation/cyclohexyl migration mechanism. Erker and co-workers reported that a mixture of an isonitrile and (Fmes)BH
2 (Fmes = 2,4,6-(F
3C)
3C
6H
2) would react with a wide range of organic unsaturates, chemistry they attributed to an iminoborane, Fmes(H)BC(H)NR (R = H
2CPh, 2,6-Me
2C
6H
3), formed in situ [
12]. For example, Fmes(H)BC(H)NCH
2Ph reacted with anisyl acetylene via a C-H cleavage FLP-type intermediate, Fmes(H)B(CCC
6H
4-4-OMe)C(H)N(H)CH
2Ph (
Scheme 1). We recently reported that the reaction of heterocumulenes with a copper(I) boryl generated isonitriles [
13]. These species reacted further with the copper(I) boryl to furnish a copper(I) boryliminomethanide, (6-Dipp)CuC(=N
tBu)Bpin, which reacted with heterocumulenes in an analogous fashion to that reported for the systems of Erker and Figueroa.
Herein we report the further exploration of this copper(I) boryliminomethanide, including our attempts at its structural characterisation, its long-term stability, and its reactivity towards a number of other substrates.
2. Results and Discussion
We began our work by resynthesising (6-Dipp)CuC(=N
tBu)Bpin (compound
1) via the reaction of tert-butyl isocyanide with isolated (6-Dipp)CuBpin. Single crystals of this compound were grown by vapour diffusion of hexane into a toluene solution of
1. Unfortunately, despite repeated data collections, a satisfactory structural refinement beyond confirmation of connectivity (
Figure 1a) could not be obtained for this compound. Thus, the metric data for
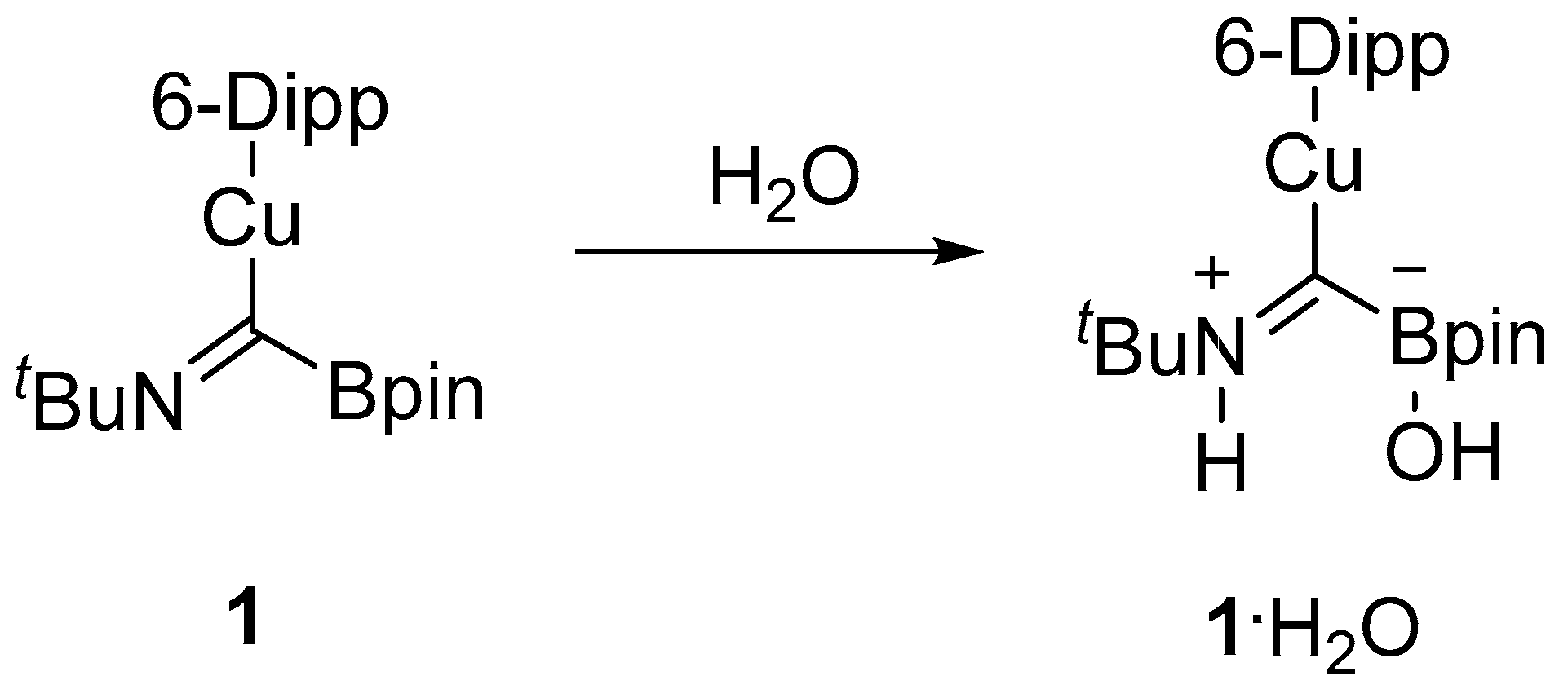
1, provided below, will not be discussed or interpreted in any detail. On one occasion, adventitious moisture resulted in hydrolysis of
1, and the crystal structure of the resulting compound was obtained to show (6-Dipp)CuC(B(OH)(pin)=N(H)
tBu (
1·H
2O) (
Figure 1b).
These results provide strong supporting evidence for the structure we previously proposed for the boryliminomethanide. In
1·H
2O, the O-H bond of water has been cleaved with the transfer of a proton to the imine N atom and a hydroxy group to the B atom (
Scheme 2). Similar reactivity was observed by Figueroa and co-workers for Cy
2BC(H)=NAr and (9-BBN)C(H)=Nar [
10,
11] and is typical for FLPs [
6]. This reaction demonstrates how
1, with a Lewis acidic boron centre in proximity to a Lewis basic nitrogen centre, can act as a reactive B/N FLP-type system in E-H cleavage reactions consistent with its previously reported reactivity towards heterocumulenes.
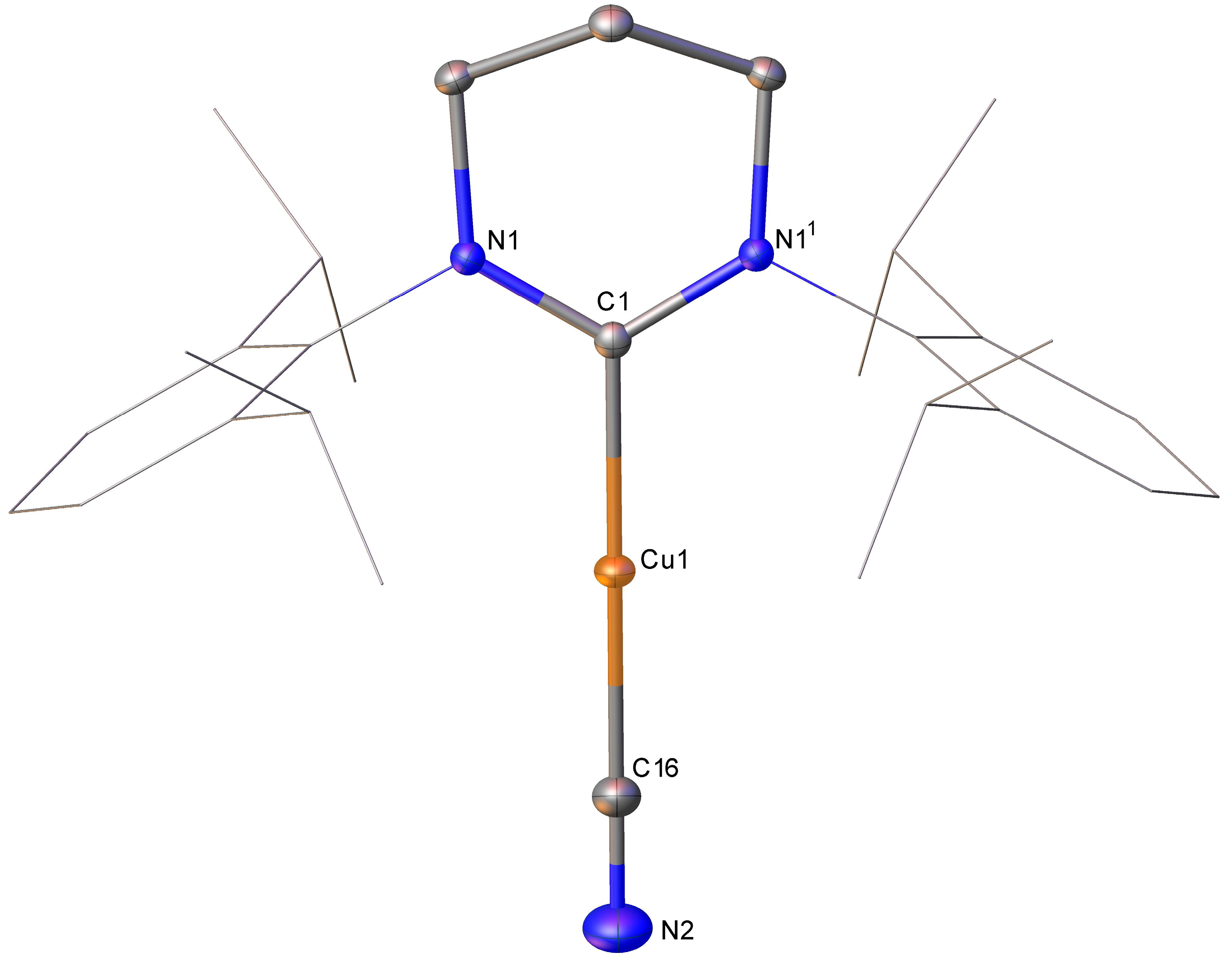
Compound
1·H
2O comprises a 2-coordinate copper atom in a near-linear coordination environment (C1-Cu1-C29 173.62(5)°) with C
carbene-Cu bond lengths (C1-Cu1 1.9331(12) Å) and N-C
carbene-N parameters commensurate (N1-C1 1.3431(15); N2-C1 1.3424(16) Å, N1-C1-N2 116.77(11) °) with other (6-Dipp)CuX systems. The boratoimidiniummethanide fragment is relatively unusual, with limited literature examples to compare to. Nevertheless, in the context of the FLP-esque activity of
1, its Cu-C(Bpin)=N
tBu core can be compared to the previously reported isocyanate activation product
1·PhNCO. The copper–carbon bond in this fragment is only marginally shorter in
1·H
2O (Cu1-C29 1.9141(12) vs. 1.925(2) Å), and the C-B bond length is very similar (C29-B1 1.6481(18) vs. 1.656(3) Å). The C-N bond lengths are similar, although the bond is slightly shorter in
1·H
2O (1.2937(17) vs. 1.314(3) Å). These data allow attribution of both systems as largely retaining a C=N double bond, but the slightly shorter bond in
1·H
2O is unsurprising in light of the more conjugated system in
1·PhNCO [
13]. We previously reported that the
11B NMR spectrum of
1 contained a broad resonance at 28.3 ppm (FWHM = 610 Hz) [
13], whereas a similar analysis of
1·H
2O provided a sharp peak at 5.3 ppm (FWHM = 36 Hz).
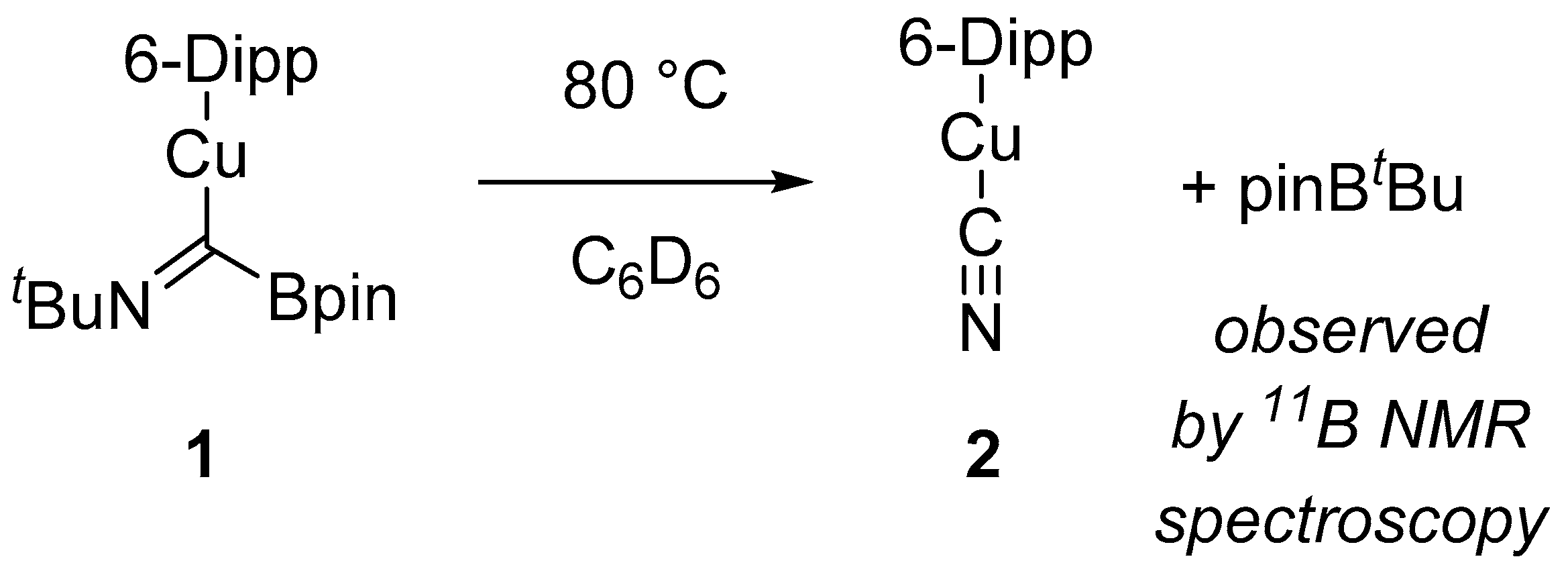
During an attempt to recrystallise a toluene solution of
1 in a boiling water bath, we noted the onset of what we suspected to be decomposition due to changes in the colour of the solution and the formation of a precipitate. To characterise the products of this decomposition, we heated a C
6D
6 solution of
1, generated in situ, to 80 °C overnight to produce a colourless solution and a crystalline material. The
11B NMR spectrum of this reaction mixture contained a resonance at 35.0 ppm that was attributed to pinB
tBu based on comparison to literature data [
14]. The crystalline material was found to be suitable for single crystal X-ray crystallography, which showed it to be (6-Dipp)CuCN (compound
2,
Figure 2).
This reactivity can be summarised as a 1,3-
tert-butyl transfer from the imine nitrogen to the pinB- fragment, as shown in
Scheme 3. Moreover, we found that samples of
1 left over the course of months at ambient temperature would undergo this reaction and were, thus, not stable long term.
Compound
2 comprises a 2-coordinate copper atom bonded to both the 6-Dipp and cyanide ligands. It is essentially linear at copper (C1-Cu1-C16 177.49(9)°), as was previously observed by Nolan and co-workers for (IPr)CuCN (177.5(3)°) [
15]. Compared to (IPr)CuCN, the copper-cyanide fragment of
2 also has a similar C-N distance (C16-N2 1.110(3) vs. 1.139(6) Å) and Cu-C-N bond angle (Cu1-C16-N2 179.9(2) vs. 177.9(7)°). The steric demand of the 6-Dipp ligand, however, provides for slightly longer C
carbene-Cu (C1-Cu1 1.9187(17) vs. 1.900(4) Å) and Cu-C
cyano (Cu1-C16 1.905(2) vs. 1.862(5) Å) distances. Attempts to observe a
13C NMR signal for the cyanide carbon were unsuccessful. Nolan and co-workers observed a resonance at 142.4 ppm to this nucleus [
15], but similar resonances in the
13C NMR spectrum of
3 (146.7, 142.4 ppm) were assigned to substituted aromatic carbons based on
3’s
1H-
13C HMBC spectrum, which showed that these carbon nuclei coupled to the iso-propyl methine and other aromatic hydrogens on the ligand.
Seeking to further extend the FLP-esque reactivity of 1, we investigated its reaction with a number of other molecules. The addition of an atmosphere of H2 to a C6D6 solution of 1 provided no evidence of reaction, and a 1H NMR spectrum containing both free H2 and compound 1, and heating of the reaction only led to the onset of decomposition. The addition of excess tert-butyl isocyanide was similar, providing no evidence of reactivity, unlike the systems described by Erker. A reaction was observed, however, with an acetylene.
The reaction of C-H acidic substrates with FLPs often gives access to C-H cleavage products analogous to the O-H cleavage product,
1·H
2O. The reaction of compound
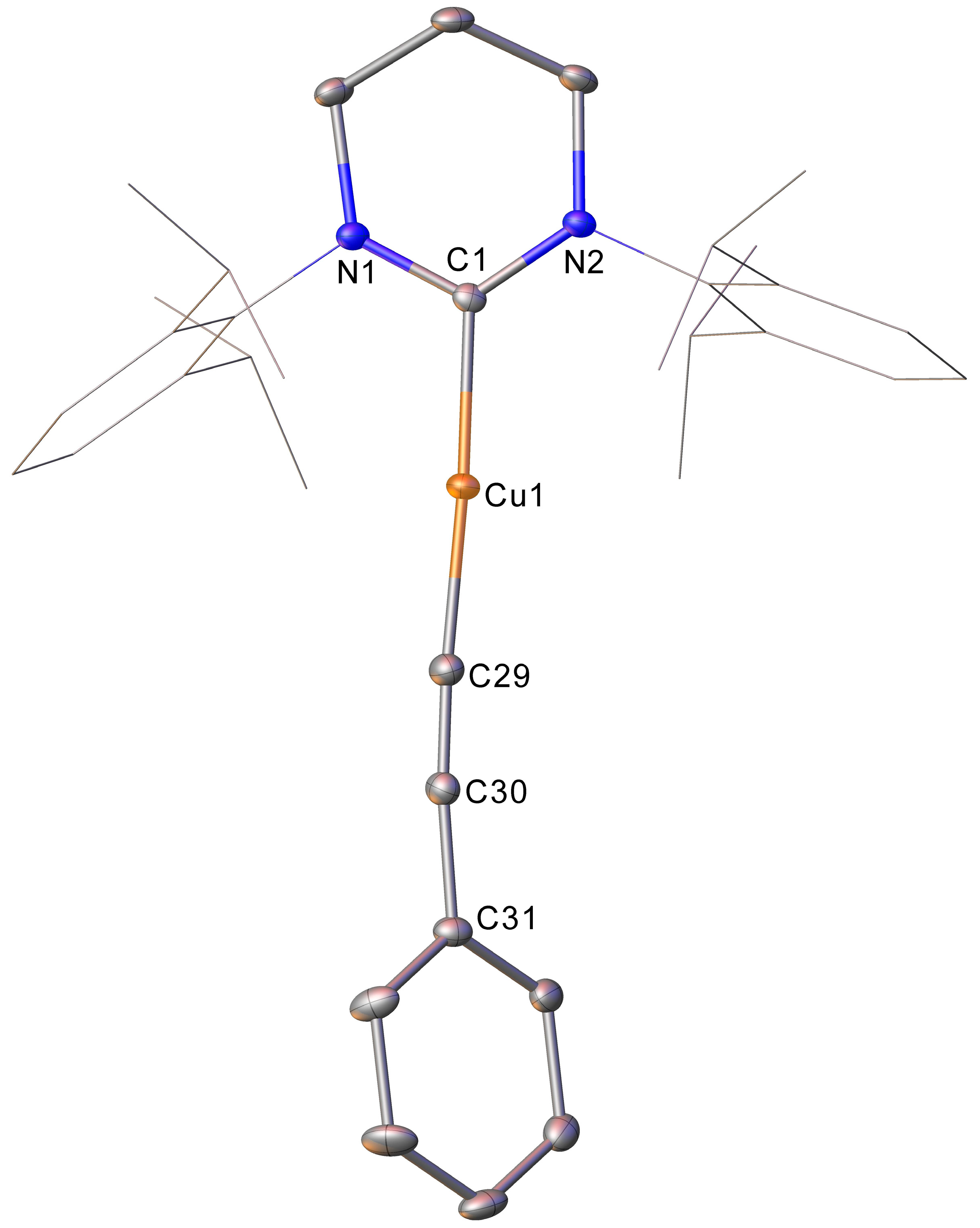
1 with a monosubstituted alkyne was thus also investigated. The addition of one equivalent of phenyl acetylene to
1 provided a
1H NMR spectrum consistent with a reaction having occurred, with the complete attenuation of the resonance associated with the acetylene C-H and a 6-Dipp region consistent with the formation of one major new ligand-containing compound. Material deposited from this reaction was subjected to single crystal X-ray crystallography, which showed it to be (6-Dipp)CuC≡CPh, compound
3 (
Figure 3).
Compound
3 is thus the result not of C-H activation across the FLP fragment but of the Cu-C bond in
1 acting as a Brønsted base towards the acidic C-H bond of the acetylene. This reaction should occur with concomitant formation of HC(=N
tBu)Bpin, evidence for which was provided by subjecting the reaction mixture to mass spectrometry (
Scheme 4). An ion with an m/z of 86.0962 was observed in the positive mode spectrum and assigned to the [M+H]
+ ion of H
2C=N
tBu. This ion was proposed to form through the hydrolysis of the C-B bond of HC(=N
tBu)Bpin by residual moisture in the solvent used to make up the sample.
Compound
3 is once again an example of a near linear (C1-Cu1-C29 176.50(10)°) copper(I) complex. The related NHC compound (IPr)CuCCPh has been reported by both Kleeberg and co-workers [
16] and the Jones group [
17]. The metric parameters of these systems are largely identical, and those reported by Jones, which were also collected at 150 K, will be discussed here. Once again, the ring-expansion to 6-Dipp results in an elongation of the C
carbene-Cu (C1-Cu1 1.919(2) vs. 1.890(4) Å) but not the Cu-C
acetylene (Cu1-C29 1.867(2) vs. 1.861(4) Å) bond lengths. The C≡C triple bond is also essentially unchanged (C29-C30 1.208(3) vs. 1.209(5) Å), and both systems display a similar distortion of the Cu-C≡C-Ph fragment away from linearity (Cu1-C29-C30 176.6(2) vs. 169.3(4)°; C29-C30-C31 174.0(5) vs. 177.4(4)°), although this is less pronounced in
3. This latter observation may reflect the effect of the marginally longer C
carbene-Cu bond, or the electronic variation between the 6-Dipp and IPr ligands. Although the
1H NMR spectrum of
3 is unremarkable, comprising the expected resonances for the 6-Dipp ligand and a phenyl group, the
13C NMR spectrum was more useful. Resonances at 122.6 and 105.3 were attributed to the alkyne carbon atoms, specifically as the carbon bonded to copper and to the phenyl substituent, respectively. Further evidence for the presence of an intact alkyne fragment in
3 could be found in the IR spectrum, which showed a sharp peak at 2086.92 cm
−1 that was assigned to a C≡C bond stretch.
The difference in the reactivity of compound
1 towards water and phenyl acetylene is notable, especially in the light of the much lower pKa of O-H bonds compared to C
sp-H bonds. We tentatively attribute this deviation to the effect of the lone pairs on the oxygen atom of water, which are capable of forming an adduct with the Bpin fragment of compound
1, thus predisposing it to the FLP-type reactivity observed to yield
1·H
2O. In contrast, the absence of such a hard donor group on phenyl acetylene precludes such a coordination, and thus, the reaction occurs at the Cu-C bond to yield the acetylide,
3. Further evidence in favour of this hypothesis comes from repeated attempts to rationally synthesise
1·H
2O, which yielded material contaminated with (6-Dipp)CuOBpin. The formation of (6-Dipp)CuOBpin can be considered as the result of the Cu-C bond in
1·H
2O deprotonating its {pinBOH} fragment and extruding a fragment of the formula H
2CN
tBu, although the precise mechanism by which this occurs is unclear (
Scheme 5). We interpreted this to indicate that
1·H
2O is the kinetic product of the activity of
1 towards water, a protic substrate with active lone pairs, but such a pathway is inhibited in the case of acetylenes, owing to the weak interaction between the C-C π-bond and vacant p-orbital on boron, thus resulting only in direct deprotonation to give
3.
In conclusion, compound 1, (6-Dipp)CuC(=NtBu)Bpin, shows less reliable FLP activity than we initially hoped. Despite its previously reported activation of isocyanates when generated and used quickly, compound 1 is unstable in our hands. Thermolysis and long-term storage both led to decomposition to generate compound 2, (6-Dipp)CuCN, the first structurally characterised ring-expanded NHC-copper(I) cyanide complex. Furthermore, no reaction towards H2 was observed. Finally, compound 1 showed divergent reactivity towards different protic reagents. The serendipitous addition of water provided O-H activation across the boryliminomethanide fragment to generate an iminium borate, 1·H2O, in a fashion reminiscent of an FLP. In contrast, the addition of a monosubstituted acetylene provided the protonation of the boryliminomethanide to generate the corresponding boryliminomethane and (6-Dipp)CuCCPh, 3, the first ring-expanded NHC-copper(I) acetylide to be structurally characterised. We tentatively attribute this deviation in reactivity to the presence of a lone pair on the oxygen atom of water that facilitates O-H cleavage across the B/N fragment via the initial formation of an adduct with the boron atom. Thus, we were able to map the range of decomposition and bond activation pathways available to compound 1 which comprise E-H activation across the boryliminomethanide, 1,3-tert-butyl transfer, and Brønsted basicity of the boryliminomethanide.
3. Materials and Methods
General Considerations and Starting Materials
All reactions dealing with air- and moisture-sensitive compounds were carried out under argon atmosphere using standard Schlenk line and glovebox techniques. NMR experiments using air sensitive compounds were conducted in J. Young’s tap NMR tubes prepared and sealed in a glovebox under argon. Toluene and hexane were purified using an MBraun Solvent Purification System and stored over 4 Å molecular sieves. C
6D
6 and
d8-THF were dried over a potassium mirror prior to vacuum transfer into a sealed ampoule and stored in a glove box under argon. All NMR data, unless otherwise stated, were acquired at 298 K on an Agilent ProPulse instrument for
1H (500 MHz),
13C{
1H} (126 MHz) and
11B (160 MHz).
1H and
13C NMR spectra were referenced using residual solvent resonances. X-ray data were collected on a New Xcalibur EosS2 diffractometer with Mo source (λ = 0.71073). or on a RIGAKU SuperNova with Cu source (λ = 1.54184). Using Olex2 [
18] the structures were solved with the SHELXT [
19] and refined with the ShelXL [
19] refinement package using Least Squares minimisation. Mass spectrometry was performed using a Bruker MicrOTOF Electrospray Time-Of-Flight Mass Spectrometer coupled to an Agilent High Performance Liquid Chromatography unit. Elemental analyses were performed by Elemental Microanalysis Ltd., Okehampton, Devon, U.K. IR data was collected with a Bruker ALPHA II spectrometer with ATR accessory inside an inert atmosphere glovebox. Tert-Butyl isocyanide was purchased from VWR and was used as received. Bis(pinacolato)diboron and phenyl acetylene were purchased from Fluorochem and used as received. (6-Dipp)CuO
tBu and (6-Dipp)CuBpin were prepared according to literature procedures [
13,
20].
Synthesis of compound 1·H2O, (6-Dipp)CuC(=N(H)tBu)B(OH)pin
In a vial in a glovebox, tBuNC (11.4 µL, 0.101 mmol) was added to a solution of (6-Dipp)CuBpin (30 mg, 0.050 mmol) in C6H6 (0.5 mL), forming a yellow solution. Volatiles were removed in vacuo, precipitating a yellow powder. The solid was re-dissolved in C6D6 (0.5 mL) and added to a solution of H2O (0.9 µL, 0.050 mmol) in C6D6 (0.5 mL). A pale brown solution formed, which was transferred to J. Young’s tap NMR tube. 1H, 11B, and 13C NMR spectra were taken, confirming the formation of compound 1·H2O, (6-Dipp)CuC(B(OH)(pin)=N(H)tBu. Colourless crystalline material precipitated out of solution overnight, which was confirmed to be (6-Dipp)CuOBpin by 1H and 11B NMR spectroscopy, yield 11.8 mg (38%).
1H NMR (500 MHz, C6D6) δ 10.59 (s, 1H, N-H), 7.24–7.20 (m, 2H, para-H), 7.12 (d, J = 7.8 Hz, 4H, meta-H), 3.08–3.01 (m, 4H, CH(CH3)2), 2.80 (t, J = 5.3 Hz, 4H, NCH2), 1.57 (s, 6H, OC(CH3)2), 1.56–1.52 (m, 14H, CH(CH3)2 and NCH2CH2), 1.20 (d, J = 7.0 Hz, 12H, CH(CH3)2), 1.11 (s, 6H, OC(CH3)2), 0.63 (s, 9H, NC(CH3)3), −0.89 (s, 1H, OH). 13C NMR (126 MHz, C6D6) δ 203.8 (CuC), 145.7 (Ar-C), 145.6 (Ar-C), 141.9 (Ar-C), 129.7 (Ar-C), 125.3 (Ar-C), 78.0 (OC(CH3)2), 54.1 (NC(CH3)3), 46.8 (NCH2), 29.5 (NC(CH3)3), 29.0 (CH(CH3)2), 27.1 (OC(CH3)2), 26.7 (OC(CH3)2), 25.2 (CH(CH3)2), 25.1 (CH(CH3)2), 20.3 (NCH2CH2). 11B NMR (160 MHz, C6D6) δ 5.3 (FWHM = 36 Hz). MS (ESI) [M+H] C39H64BCuN3O3: 696.4337, found: 696.4360 (err [ppm] = 0.44).
Synthesis of compound 2, (6-Dipp)CuCN
In a vial in a glovebox, tBuNC (9.5 µL, 0.084 mmol) was added to a solution of (6-Dipp)CuBpin (25 mg, 0.042 mmol) in C6H6 (0.5 mL), forming a yellow solution. Volatiles were removed in vacuo precipitating a yellow powder. The solid was re-dissolved in C6D6 (1 mL), and the solution was placed in a sealed ampoule. The solution was heated to 80 °C overnight, forming a brown solution. Upon cooling to room temperature, a small amount of colourless material crystallised out of the solution, which was confirmed by 1H and 13C NMR spectroscopy to be compound 2, (6-Dipp)CuCN. The crystalline material was investigated by SC-XRD.
1H NMR (500 MHz, d8-THF) δ 7.37–7.35 (m, 2H, para-H), 7.26 (d, J = 7.6 Hz, 4H, meta-H), 3.58 (residual THF), 3.46 (t, J = 5.9 Hz, 4H, NCH2), 3.12 (hept, J = 6.9 Hz, 4H, CH(CH3)2), 2.45 (water impurity), 2.35 (p, J = 5.8 Hz, 2H, NCH2CH2), 1.73 (residual THF), 1.34 (d, J = 7.0 Hz, 12H, CH(CH3)2), 1.30 (d, J = 7.0 Hz, 12H, CH(CH3)2). 13C NMR (126 MHz, d8-THF) δ 201.9 (CuCcarbene), 146.7 (Ar-C), 142.4 (Ar-C), 130.3 (Ar-C), 125.6 (Ar-C), 47.3 (NCH2), 29.6 (CH(CH3)2), 24.9 (CH(CH3)2), 21.3 (NCH2CH2). MS (ESI) [M+Na] C29H40CuN3Na: 516.2416, found: 516.2423 (err [ppm] = −1.86).
Synthesis of compound 3, (6-Dipp)CuC≡CPh,
In a vial in a glovebox, a solution of (6-Dipp)CuOtBu (30 mg, 0.055 mmol) in C6H6 (0.5 mL) was added to B2pin2 (14 mg, 0.055 mmol). Volatiles were removed in vacuo, precipitating a white powder. The powder was washed with hexane (2 mL) and dried in vacuo. The remaining solid was dissolved in C6H6 (0.5 mL) and tBuNC (12.5 µL, 0.111 mmol) was added, forming a yellow solution. Volatiles were removed in vacuo, precipitating a yellow powder. The solid was re-dissolved into C6D6 (0.5 mL), and the reaction mixture was transferred to J. Young’s tap NMR tube. An equivalent of phenyl acetylene (6.1 µL, 0.055 mmol) was added, forming an orange solution. Colourless material crystallised out of solution, which was confirmed by 1H and 13C NMR spectroscopy to be compound 4, (6-Dipp)CuCCPh. Yield 22 mg (70%). The crystalline material was investigated by SC-XRD.
1H NMR (500 MHz, C6D6) δ 7.36–7.34 (m, 2H, ortho-H), 7.18–7.12 (m, 5H, Ar-H, residual solvent signal overlapped), 7.08–7.06 (m, 4H, meta-H), 6.84–6.80 (m, 2H, meta-H), 6.77–6.73 (m, 1H, para-H), 3.02 (h, J = 6.9 Hz, 4H, CH(CH3)2), 2.68 (t, J = 5.9 Hz, 4H, NCH2), 1.52 (d, J = 6.8 Hz, 12H, CH(CH3)2), 1.45 (p, J = 5.9 Hz, 2H, NCH2CH2), 1.18 (d, J = 7.0 Hz, 12H, CH(CH3)2). 13C NMR (126 MHz, C6D6) δ 203.4 (CuC), 145.6 (Ar-C), 141.6 (Ar-C), 132.3 (Ar-C), 129.7 (Ar-C), 129.6 (Ar-C), 127.6 (Ar-C), 124.9 (Ar-C), 124.5 (Ar-C), 122.6 (CuCC), 105.3 (CuCC), 46.1 (NCH2), 28.9 (CH(CH3)2), 25.3 (CH(CH3)2), 24.6 (CH(CH3)2), 20.3 (NCH2CH2). Analysis calculated for C36H45CuN2 (MW = 569.32 g/mol): Expected: C, 75.95; H, 7.97; N, 4.92%. Found: C, 74.08; H, 7.08; N, 4.75%. Despite repeated attempts, we were unable to obtain satisfactory elemental analysis for this compound. This was most likely due to the high sensitivity towards air and moisture. IR (ATR) cm−1 2957.84 (C-H stretch), 2086.92 (C≡C stretch).



 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}