
New Phosphonite Ligands with High Steric Demand and Low Basicity: Synthesis, Structural Properties and Cyclometalated Complexes of Pt(II)

 , , and
, , and 
Abstract
:1. Introduction
2. Results
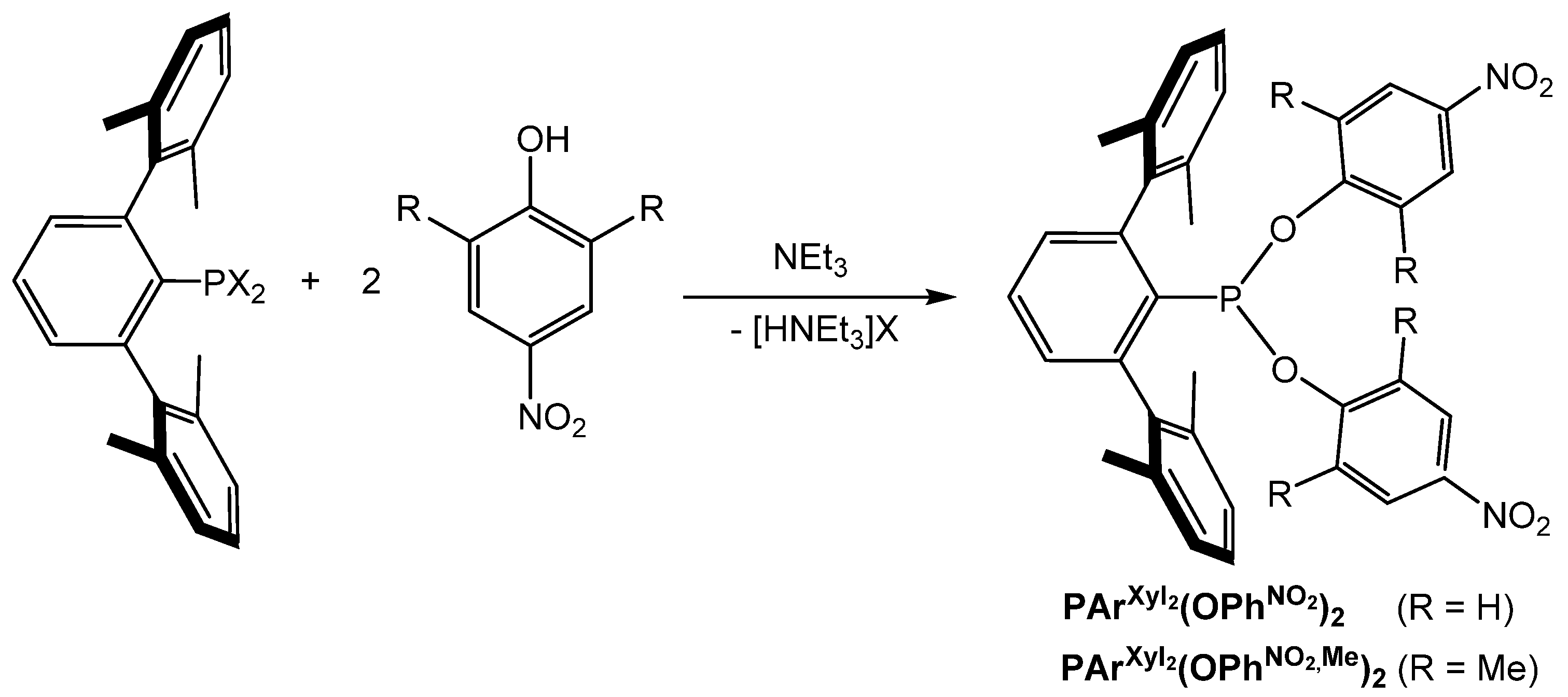
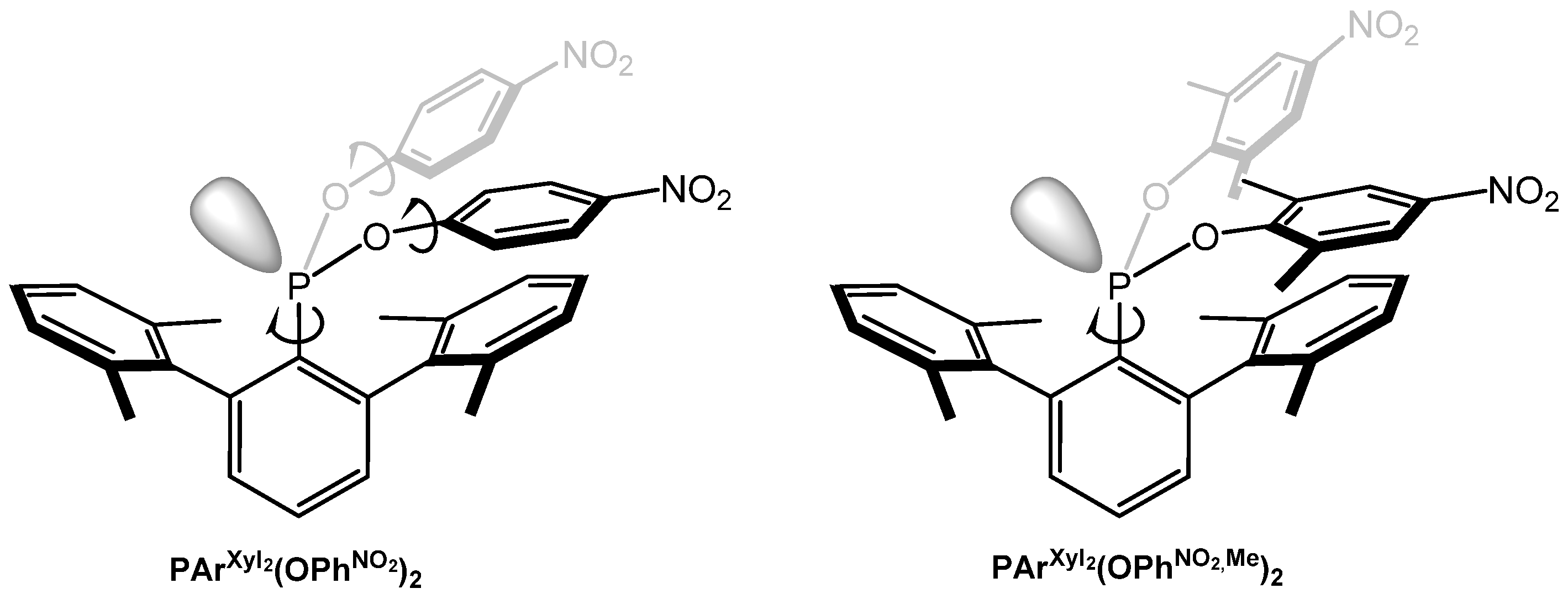
2.1. Synthesis and Properties of Terphenyl Phosphonites
- The highest TCA value among the phosphine series is 212.4°, for PArDtbp2(c-C5H9)2, in which the flanking rings of the terphenyl moiety are functionalized with two bulky tert-butyl groups at the meta positions and two cyclopentyl groups are bound to the P atom;
- The TCA values for PArXyl2R2 phosphines, which bear the same terphenyl group as the phosphonites of this work, are in the range 153.9–187.3, with the highest value for the highly steric demanding R = cyclohexyl;
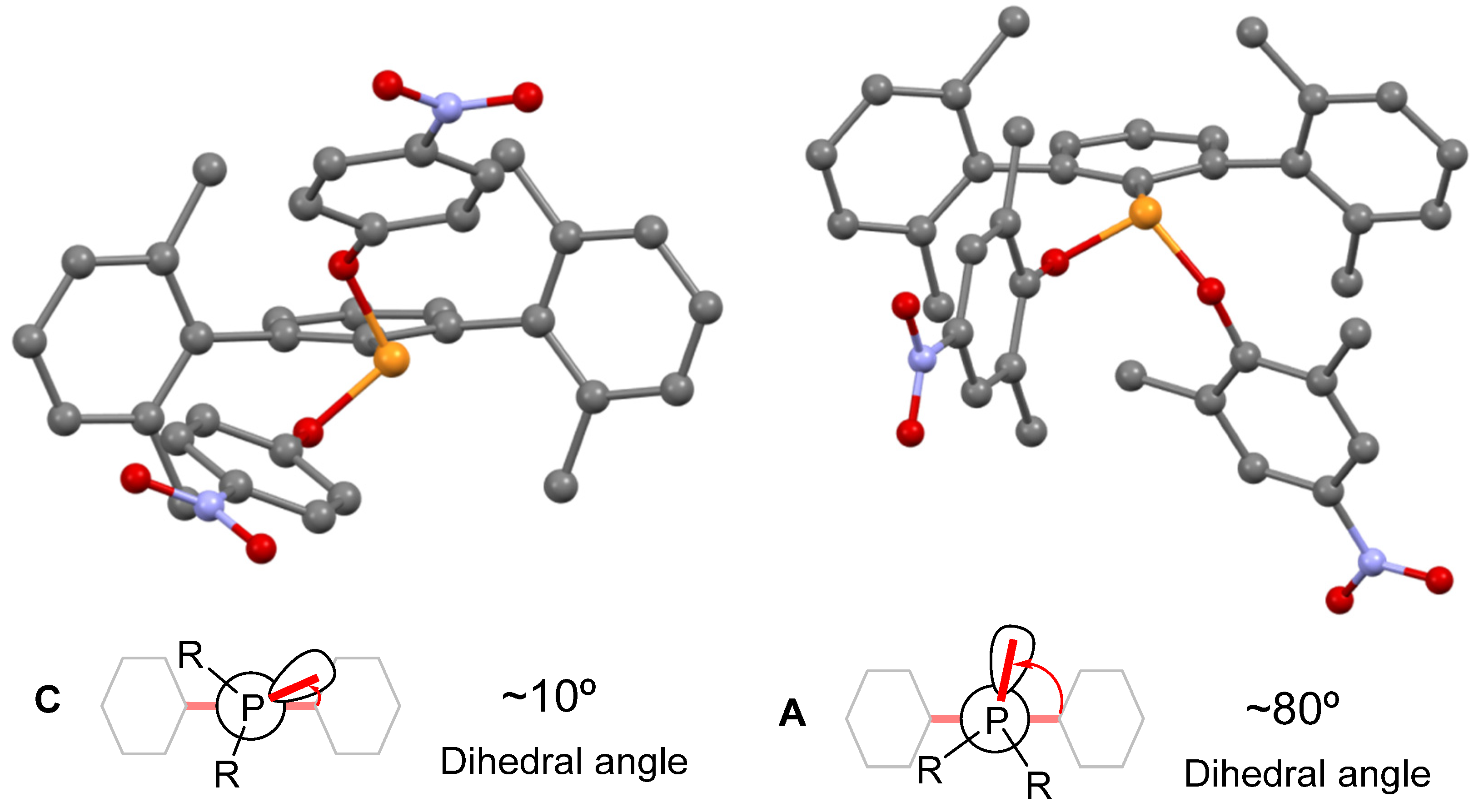
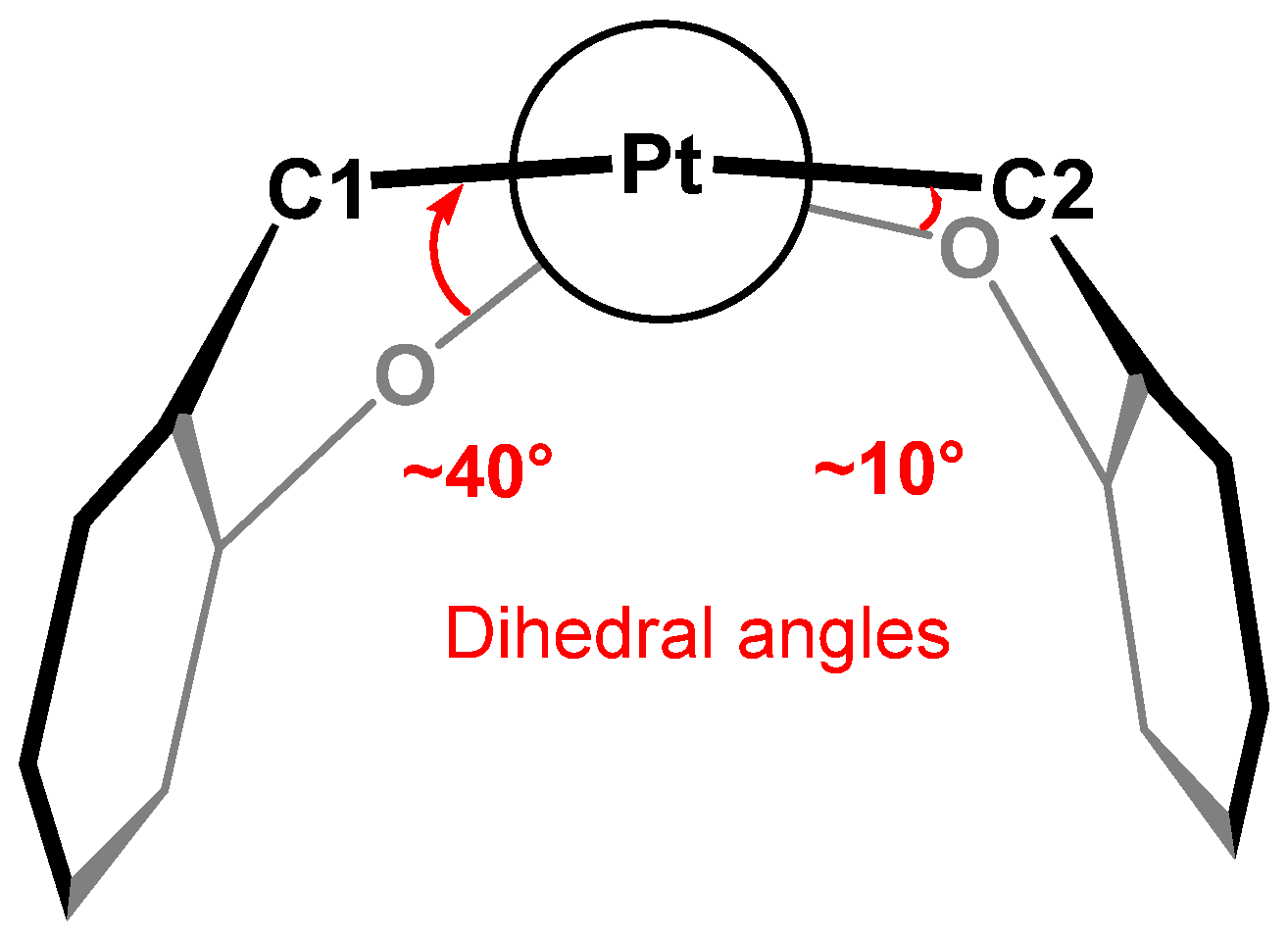
- PArXyl2(OPhNO2)2 and PArXyl2(OPhNO2,Me)2 have TCA values of 218.1 and 226.5, respectively, i.e., significantly higher than those of the related PR2ArXyl2;
- The TCA increases by ca. 60° from the dimethyl terphenyl phosphine PArXyl2Me2 to the dimethoxy phosphonite PArXyl2(OMe)2;
- Data reported for a series of PR3 and P(OR)3 ligands indicate that, in most cases, the TCA is reduced slighlty when passing from a phosphane to the related phosphite, e.g., from PPh3 (TCAtetrehedral = 165.8°) to P(OPh)3 (TCAtetrehedral = 152.9°).
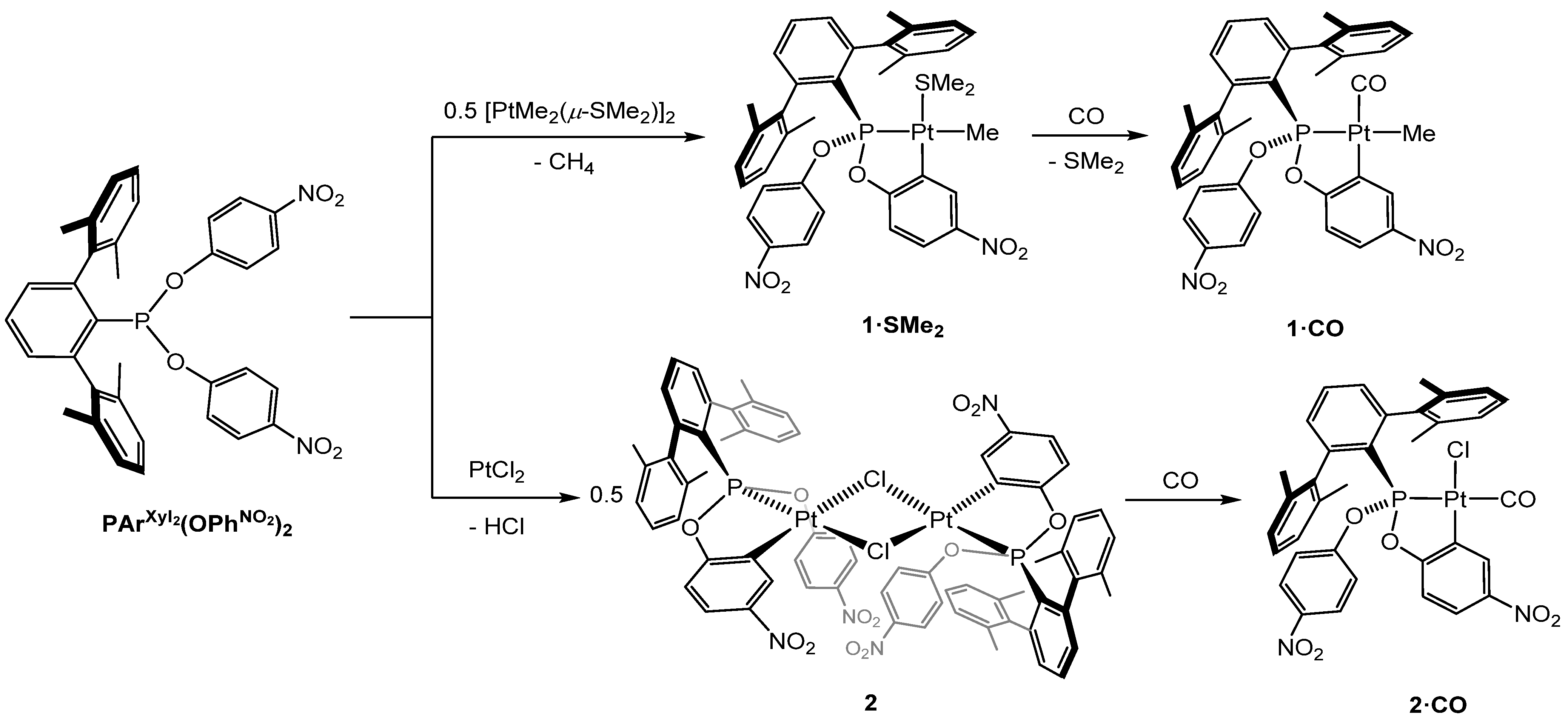
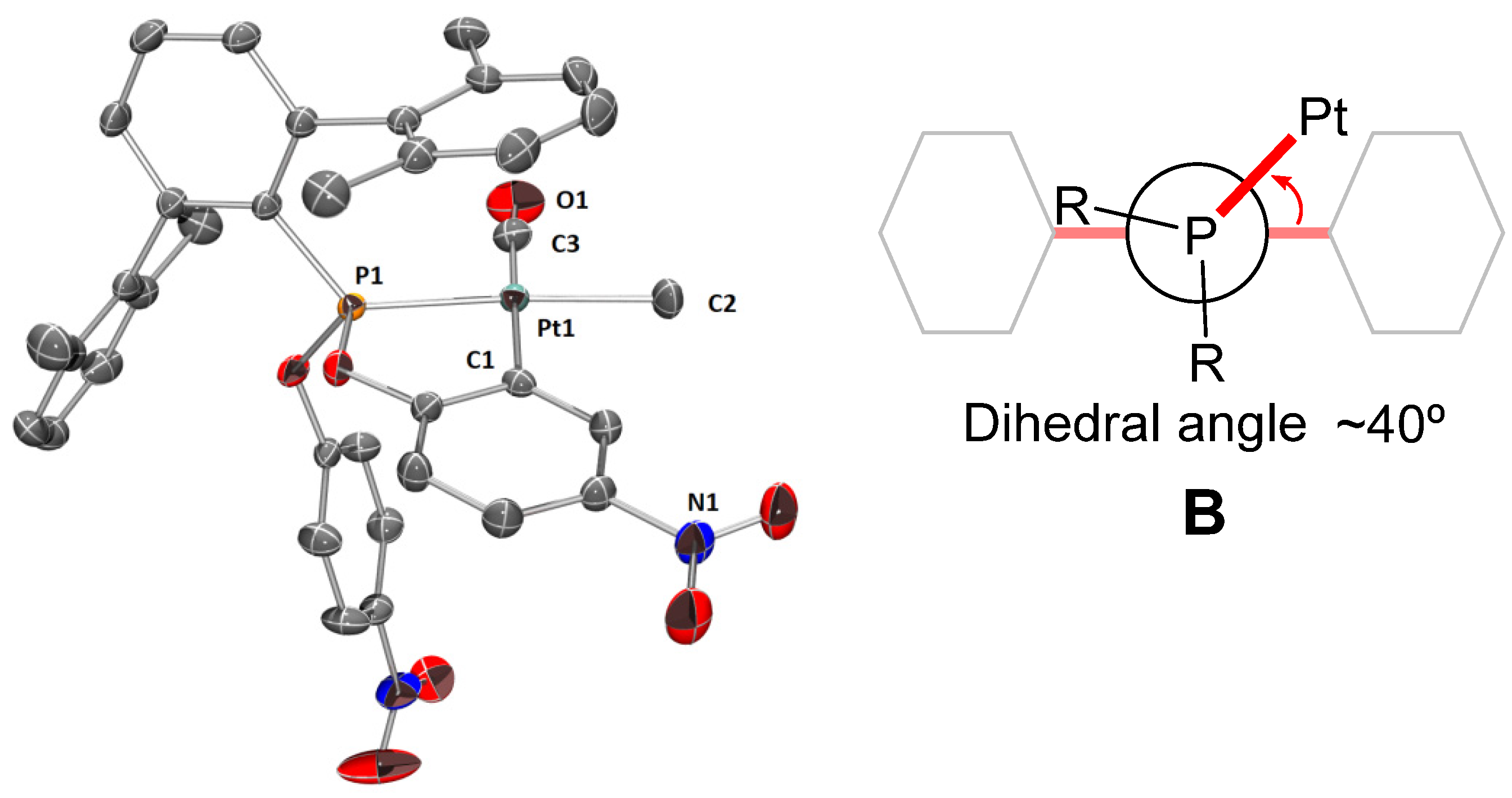
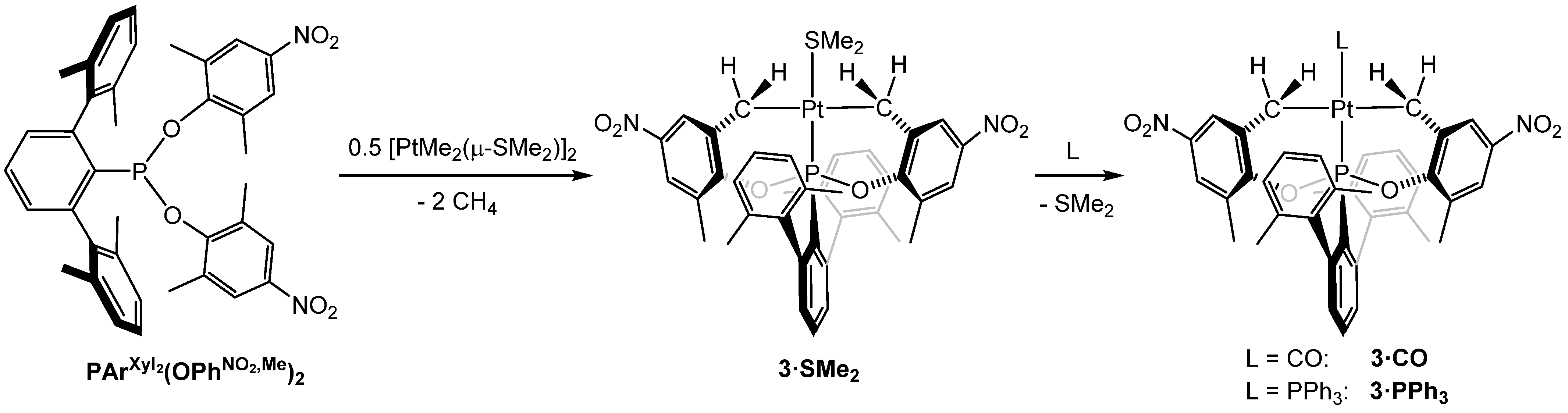
2.2. Synthesis and Characterization of Cyclometalated Complexes of Pt(II) with PArXyl2(OPhNO2)2 and PArXyl2(OPhNO2,Me)2
- The four methyl groups of the xylyl rings give rise to one singlet at 2.10 ppm and one singlet at 21.5 ppm in the 1H and 13C{1H} NMR spectra, respectively, in accordance with fast rotation around the P—C bond on the NMR time-scale, as in the free phosphonite ligands;
- The two metalated OPhNO2,Me fragments are equivalent, as shown by the presence of only one resonance for the CH3 groups in both the 1H- and 13C{1H} NMR spectra;
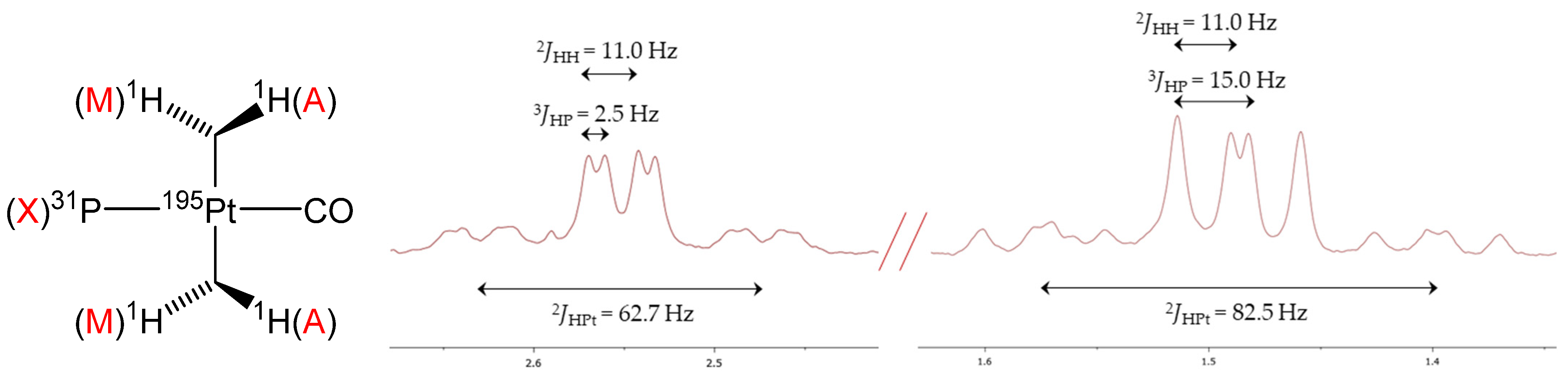
- In the 1H NMR spectrum, the four diasterotopic methylene protons are coupled with the 31P nucleus, thus constituting first-order AMX spin systems, which give rise to a doublet (2JHH = 11.5 Hz, 3JHP ≈ 0 Hz) and a triplet (2JHH ≈ 3JHP = 11.5 Hz), both with 195Pt satellites;
- The six protons of the coordinated dimethylsulfide resonate as a doublet at 2.34 ppm with 4JHP = 4.0 Hz and 3JHPt = 36.4 Hz.
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ugo, R. The coordinative reactivity of phosphine complexes of platinum(o), palladium(o) and nickel(o). Coord. Chem. Rev. 1968, 3, 319–344. [Google Scholar] [CrossRef]
- Mason, R.; Meek, D.W. The versatility of tertiary phosphines as ligands in coordination- and organometallic chemistry. Angew. Chem. Int. Ed. 1978, 90, 195–206. [Google Scholar] [CrossRef]
- Otsuka, S. Chemistry of platinum and palladium compounds of bulky phosphines. J. Organomet. Chem. 1980, 200, 191–205. [Google Scholar] [CrossRef]
- Shaw, B.L. Some steric, conformational and entropy effects of tertiary phosphine ligands. J. Organomet. Chem. 1980, 200, 307–318. [Google Scholar] [CrossRef]
- Chatt, J. Homogeneous Catalysis with Metal Phosphine Complexes; Pignolet, L.M., Ed.; Plenum Press: New York, NY, USA, 1983. [Google Scholar]
- Hartwig, J.F. Organotransition Metal Chemistry: From Bonding to Catalysis; University Science Books: Sausalito, CA, USA, 2010. [Google Scholar]
- Bader, A.; Lindner, E. Coordination chemistry and catalysis with hemilabile oxygen-phosphorus ligands. Coord. Chem. Rev. 1991, 108, 27–110. [Google Scholar] [CrossRef]
- Werner, H. The ambivalent behaviour of aryl-functionalized phosphines: Coordination, hemilability and beyond. Dalton Trans. 2003, 20, 3829–3837. [Google Scholar] [CrossRef]
- Pinault, N.; Bruce, D.W. Homogeneous catalysts based on water-soluble phosphines. Coord. Chem. Rev. 2003, 241, 1–25. [Google Scholar] [CrossRef]
- Van der Boom, M.E.; Milstein, D. Cyclometalated Phosphine-Based Pincer Complexes: Mechanistic Insight in Catalysis, Coordination, and Bond Activation. Chem. Rev. 2003, 103, 1759–1792. [Google Scholar] [CrossRef]
- Braunstein, P. Functional ligands and complexes for new structures, homogeneous catalysts and nanomaterials. J. Organomet. Chem. 2004, 689, 3953–3967. [Google Scholar] [CrossRef]
- Braunstein, P. Bonding and Organic and Inorganic Reactivity of Metal-Coordinated Phosphinoenolates and Related Functional Phosphine-Derived Anions. Chem. Rev. 2006, 106, 134–159. [Google Scholar] [CrossRef]
- Alcarazo, M. α-Cationic Phosphines: Synthesis and Applications. Chem. Eur. J. 2014, 20, 7868–7877. [Google Scholar] [CrossRef] [PubMed]
- Stuart, L.J. Phosphines as building blocks in coordination-based self-assembly. Chem. Soc. Rev. 2009, 38, 1744–1758. [Google Scholar]
- Lara, P.; Philippot, K.; Chaudret, B. Organometallic Ruthenium Nanoparticles: A Comparative Study of the Influence of the Stabilizer on their Characteristics and Reactivity. ChemCatChem 2013, 5, 28–45. [Google Scholar] [CrossRef]
- Lara, P.; Philippot, K.; Suárez, A. Phosphane-decorated Platinum Nanoparticles as Efficient Catalysts for H2 Generation from Ammonia Borane and Methanol. ChemCatChem 2019, 11, 766–771. [Google Scholar] [CrossRef]
- Bernskoetter, W.H.; Schauer, C.K.; Goldberg, K.I.; Brookhart, M. Characterization of a rhodium (I) sigma-methane complex in solution. Science 2009, 326, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Marín, M.; Moreno, J.J.; Navarro-Gilabert, C.; Álvarez, E.; Maya, C.; Peloso, R.; Nicasio, M.C.; Carmona, E. Structure and Nickel Carbonyl Complexes of Dialkylterphenyl Phosphines. Chem. Eur. J. 2018, 25, 260–272. [Google Scholar] [CrossRef]
- Marín, M.; Moreno, J.J.; Alcaide, M.M.; Álvarez, E.; López-Serrano, J.; Campos, J.; Nicasio, M.C.; Carmona, E. Evaluating stereoelectronic properties of bulky dialkylterphenyl phosphine ligands. J. Organomet. Chem. 2019, 896, 120–128. [Google Scholar] [CrossRef]
- Ortega-Moreno, L.; Peloso, R.; López-Serrano, J.; Iglesias-Sigüenza, J.; Maya, C.; Carmona, E. A Cationic Unsaturated Platinum (II) Complex that Promotes the Tautomerization of Acetylene to Vinylidene. Angew. Chem. Int. Ed. 2017, 56, 2772–2775. [Google Scholar] [CrossRef]
- Ortega-Moreno, L.; Peloso, R.; Maya, C.; Suárez, A.; Carmona, E. Platinum (0) olefin complexes of a bulky terphenylphosphine ligand. Synthetic, structural and reactivity studies. Chem. Commun. 2015, 51, 17008–17011. [Google Scholar] [CrossRef]
- Ortega-Moreno, L.; Fernández-Espada, M.; Moreno, J.J.; Navarro-Gilabert, C.; Campos, J.; Conejero, S.; López-Serrano, J.; Maya, C.; Peloso, R.; Carmona, E. Synthesis, Properties, and Some Rhodium, Iridium, and Platinum Complexes of a Series of Bulky m-Terphenylphosphine Ligands. Polyhedron 2016, 116, 170–181. [Google Scholar] [CrossRef]
- Campos, J. Dihydrogen and Acetylene Activation by a Gold (I)/Platinum (0) Transition Metal Only Frustrated Lewis Pair. J. Am. Chem. Soc. 2017, 139, 2944–2947. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, N.; Bajo, S.; Moreno, J.J.; Navarro-Gilabert, C.; Mercado, B.Q.; Campos, J. Reactivity of a gold (I)/platinum (0) frustrated Lewis pair with germanium and tin dihalides. Dalton Trans. 2019, 48, 9127–9138. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, N.; Moreno, J.J.; Pérez-Jimeénez, M.; Maya, C.; Loópez-Serrano, J.; Campos, J. Evidence for Genuine Bimetallic Frustrate Lewis Pair Activation of Dihydrogen with Gold (I)/Platinum (0) Systems. Chem. Eur. J. 2020, 26, 5982–5993. [Google Scholar] [CrossRef]
- Beltrán, Á.; Gata, I.; Maya, C.; Avó, J.; Lima, J.C.; Laia, C.A.T.; Peloso, R.; Outis, M.; Nicasio, M.C. Dinuclear Cu (I) Halides with Terphenyl Phosphines: Synthesis, Photophysical Studies, and Catalytic Applications in CuAAC Reactions. Inorg. Chem. 2020, 59, 10894–10906. [Google Scholar] [CrossRef]
- Navarro, M.; Alférez, M.G.; de Sousa, M.; Miranda-Pizarro, J.; Campos, J. Dicoordinate Au (I)–Ethylene Complexes as Hydroamination Catalysts. ACS Catal. 2022, 12, 4227–4241. [Google Scholar] [CrossRef] [PubMed]
- Rama, R.J.; Maya, C.; Nicasio, M.C. Dialkylterphenyl Phosphine-Based Palladium Precatalysts for Efficient Aryl Amination of N-Nucleophiles. Chem. Eur. J. 2020, 26, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
- Martín, M.T.; Marín, M.; Rama, R.J.; Álvarez, E.; Maya, C.; Molina, F.; Nicasio, M.C. Zero-valent ML2 complexes of group 10 metals supported by terphenyl phosphanes. Chem. Commun. 2021, 57, 3083–3086. [Google Scholar] [CrossRef]
- Deck, E.; Wagner, H.E.; Paradies, J.; Breher, F. Redox-responsive phosphonite gold complexes in hydroamination catalysis. Chem. Commun. 2019, 55, 5323–5326. [Google Scholar] [CrossRef] [PubMed]
- Rama, R.J.; Martín, M.T.; Peloso, R.; Nicasio, M.C. Low-coordinate M (0) complexes of group 10 stabilized by phosphorus (III) ligands and N-heterocyclic carbenes. Adv. Organomet. Chem. 2020, 74, 241–323. [Google Scholar]
- Peloso, R.; Pattacini, R.; Cazin, C.S.J.; Braunstein, P. Structure and Reactivity of New Iridium Complexes with Bis (Oxazoline)-Phosphonito Ligands. Inorg. Chem. 2009, 48, 11415–11424. [Google Scholar] [CrossRef]
- Volle, J.-N.; Filippini, D.; Midrier, C.; Sobecki, M.; Drag, M.; Virieux, D.; Pirat, J.-L. Revisited Synthesis of Aryl-H-phosphinates. Synthesis 2011, 15, 2490–2494. [Google Scholar] [CrossRef]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef]
- Pidcock, A.; Richards, R.E.; Venanzi, L.M. 195Pt-31P nuclear spin coupling constants and the nature of the trans-effect in platinum complexes. J. Chem. Soc. A 1966, 1707–1710. [Google Scholar] [CrossRef]
- Kühl, O. Phosphorus 31-NMR Spectroscopy: A Concise Introduction for the Synthetic Organic and Organometallic Chemist; Springer: Berlin/Heidelberg, Germany, 2008. [Google Scholar]
- Hill, G.S.; Irwin, M.J.; Levy, C.J.; Rendina, L.M.; Puddephatt, R.J.; Andersen, R.A.; Mclean, L. Platinum (II) complexes of dimethyl sulfide. Inorg. Synth. 1998, 32, 149–153. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B.01 and E.01, Gaussian, Inc.: Wallingford, CT, USA, 2010.
- Chai, J.D.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self. Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Falivene, L.; Cao, Z.; Petta, A.; Serra, L.; Poater, A.; Oliva, R.; Scarano, V.; Cavallo, L. Towards the online computer-aided design of catalytic pockets. Nat. Chem. 2019, 11, 872–879. [Google Scholar] [CrossRef]
- SambVca 2.1 A Web Application to Characterize Catalytic Pockets. Available online: https://www.molnac.unisa.it/OMtools/sambvca2.1/index.html (accessed on 31 March 2022).
- Guzei, I.A.; Wendt, M. An improved method for the computation of ligand steric effects based on solid angles. Dalton Transactions. 2006, 33, 3991–3999. [Google Scholar] [CrossRef]
- Molecular Structure Laboratory, Resources, University of Wisconsin. Available online: https://xray.chem.wisc.edu/solid-g/ (accessed on 31 March 2022).
- Diebolt, O.; Fortman, G.C.; Clavier, H.; Slawin, A.M.Z.; Escudero-Adán, E.C.; Benet-Buchholz, J.; Nolan, S.P. Steric and Electronic Parameters Characterizing Bulky and Electron-Rich Dialkylbiarylphosphines. Organometallics 2011, 30, 1668–1676. [Google Scholar] [CrossRef]
- Bruker. SAINT APEX2 2007; Bruker AXS Inc.: Madison, WI, USA, 2007. [Google Scholar]
- Sheldrick, G.M. SADABS, Programs for Scaling and Absorption Correction of Area Detector Data 1997; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Burla, M.C.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; Giacovazzo, C.; Polidori, G.; Spagna, R. SIR2002: The Program. J. Appl. Cryst. 2003, 36, 1103. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A Short History of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | %Vbur | TCA(°) | %G | Ref |
|---|---|---|---|---|
| PArXyl2(OPhNO2)2 | 59.7 | 218.1 | 57.2 | this work |
| PArXyl2(OPhNO2,Me)2 | 55.3 | 226.5 | 68.2 | this work |
| PArXyl2(OMe)2 | 49.1 | 210.8 | 50.3 | this work |
| PArXyl2Me2 | 38.6 | 153.9 | 38.8 | [18] |
| PArXyl2(c-C6H11)2 | 45.2 | 187.3 | 47.1 | [18] |
| PArDtbp2(c-C5H9)2 | 56.7 | 212.4 | 46.2 | [18] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alcaide, M.M.; Pugliesi, M.; Álvarez, E.; López-Serrano, J.; Peloso, R. New Phosphonite Ligands with High Steric Demand and Low Basicity: Synthesis, Structural Properties and Cyclometalated Complexes of Pt(II). Inorganics 2022, 10, 109. https://doi.org/10.3390/inorganics10080109
Alcaide MM, Pugliesi M, Álvarez E, López-Serrano J, Peloso R. New Phosphonite Ligands with High Steric Demand and Low Basicity: Synthesis, Structural Properties and Cyclometalated Complexes of Pt(II). Inorganics. 2022; 10(8):109. https://doi.org/10.3390/inorganics10080109
Chicago/Turabian StyleAlcaide, María M., Matteo Pugliesi, Eleuterio Álvarez, Joaquín López-Serrano, and Riccardo Peloso. 2022. "New Phosphonite Ligands with High Steric Demand and Low Basicity: Synthesis, Structural Properties and Cyclometalated Complexes of Pt(II)" Inorganics 10, no. 8: 109. https://doi.org/10.3390/inorganics10080109
APA StyleAlcaide, M. M., Pugliesi, M., Álvarez, E., López-Serrano, J., & Peloso, R. (2022). New Phosphonite Ligands with High Steric Demand and Low Basicity: Synthesis, Structural Properties and Cyclometalated Complexes of Pt(II). Inorganics, 10(8), 109. https://doi.org/10.3390/inorganics10080109