
Rearrangement of Diferrocenyl 3,4-Thiophene Dicarboxylate

 , , , , and
, , , , and
Abstract
1. Introduction
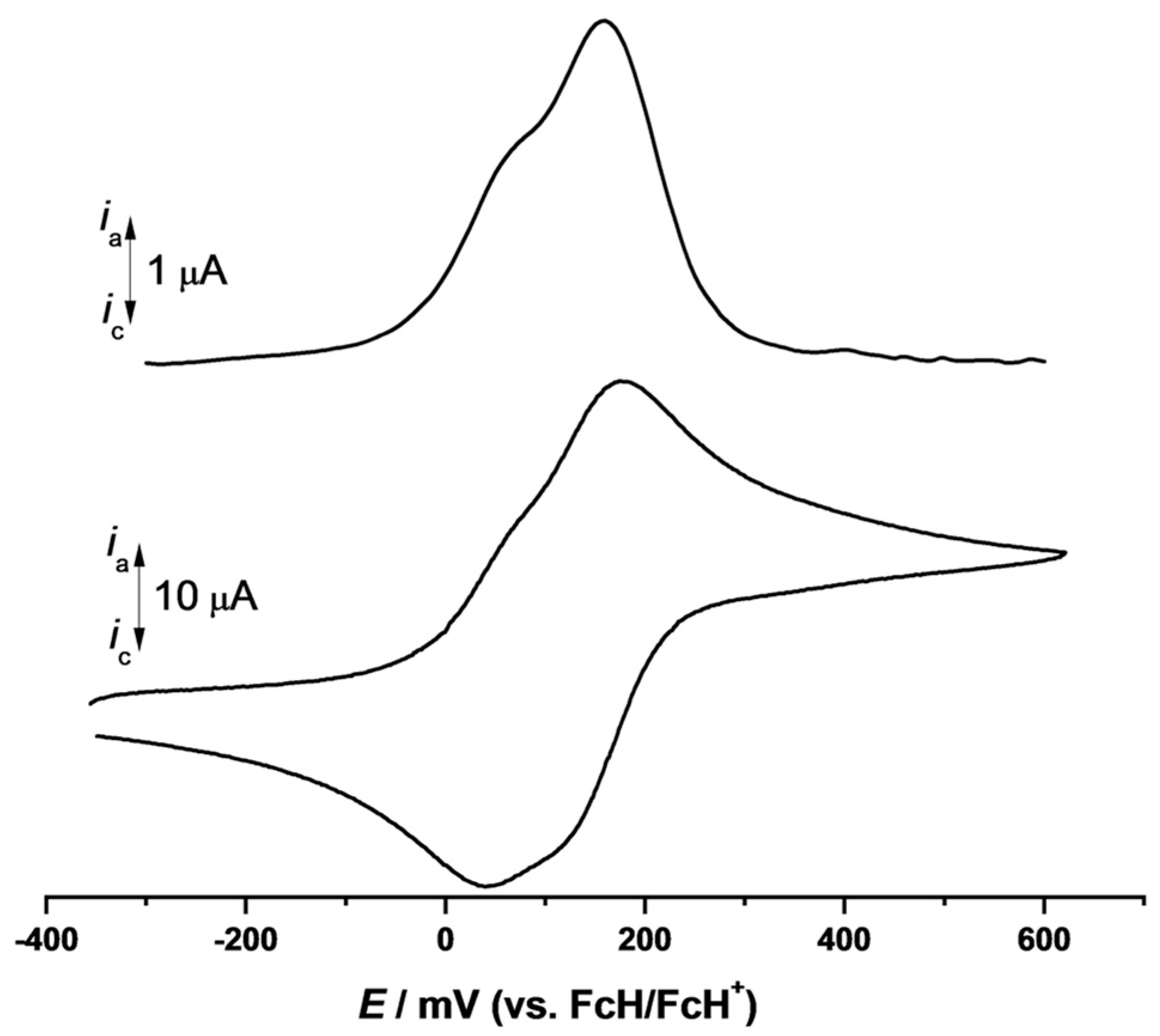
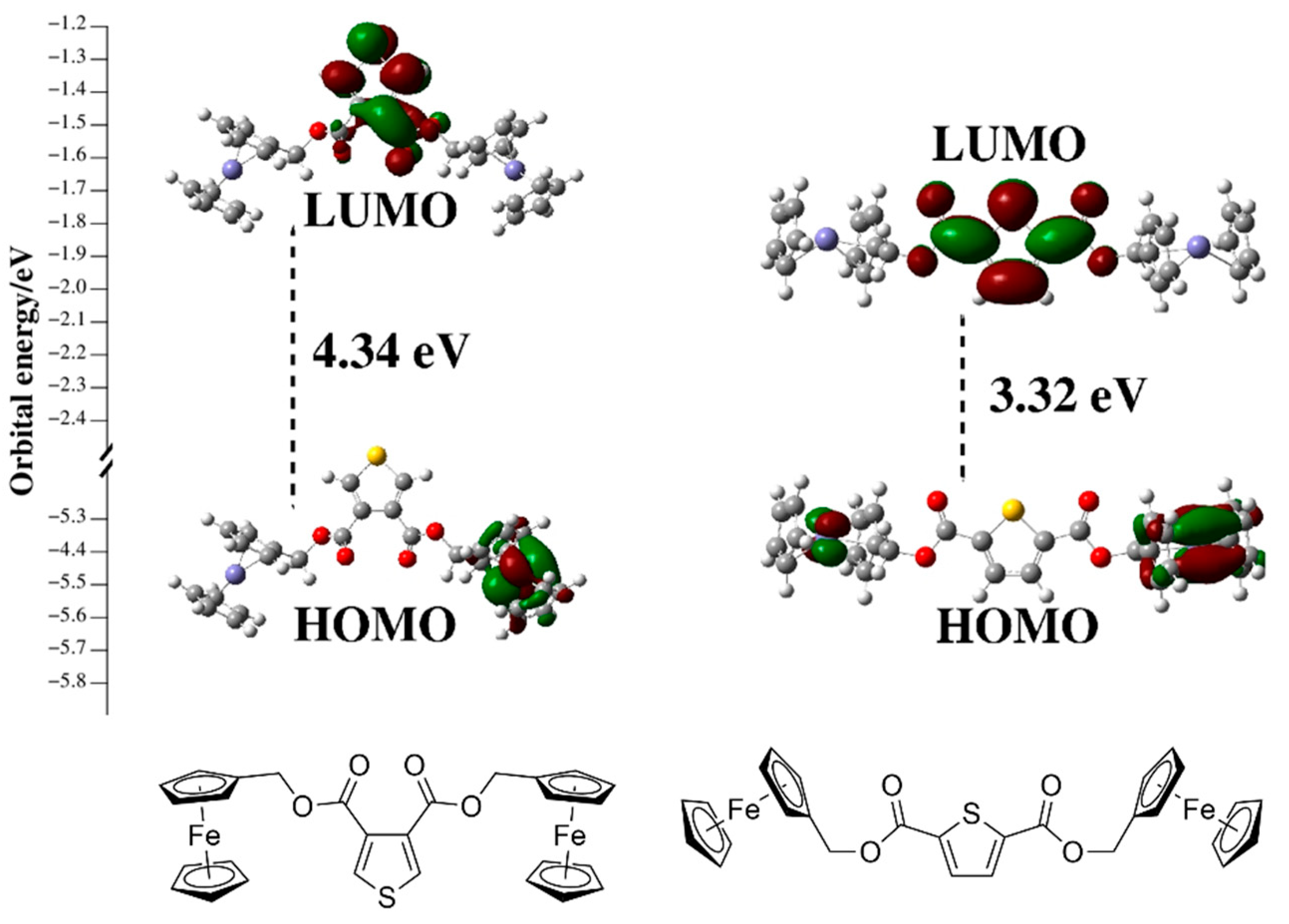
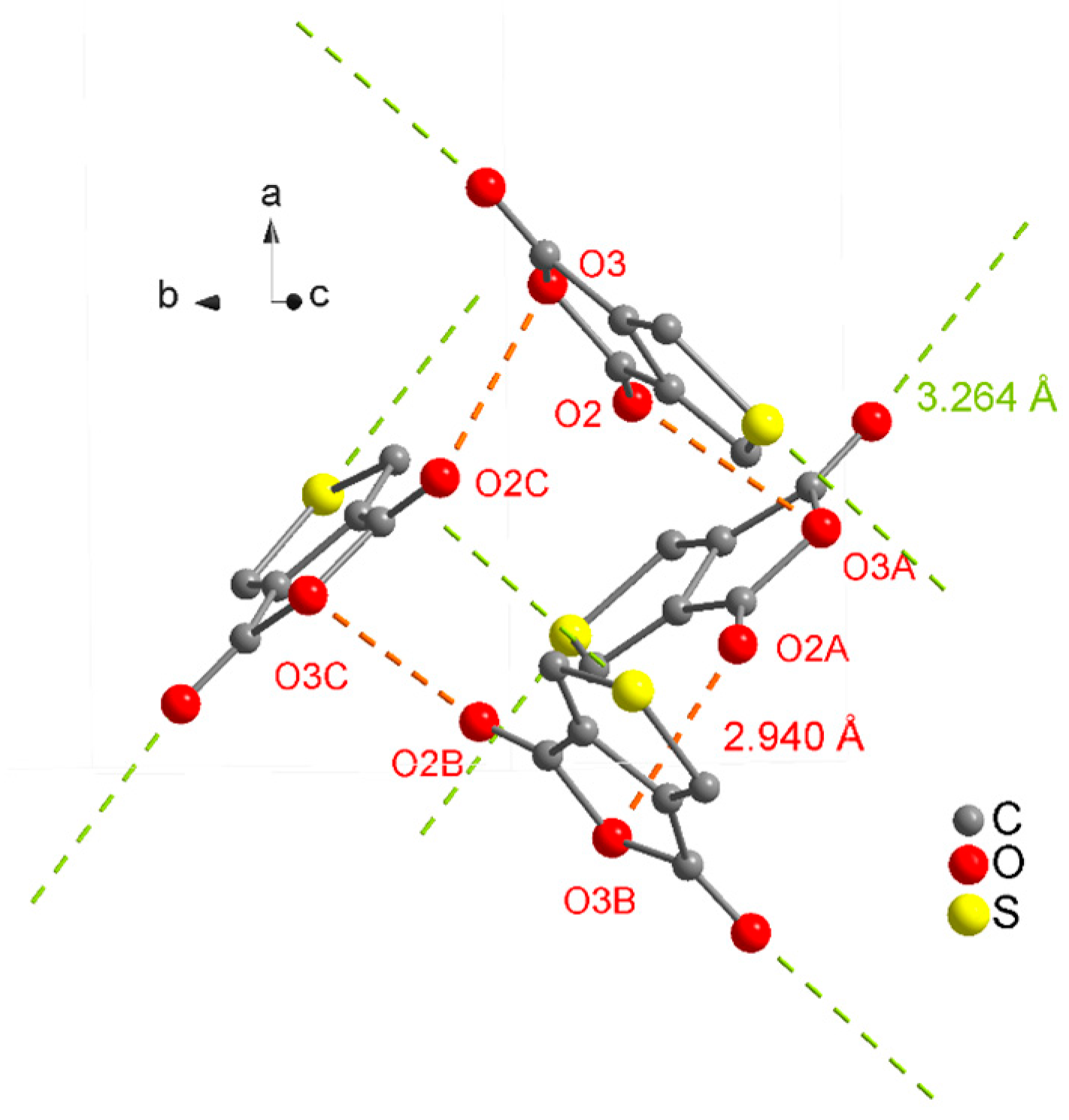
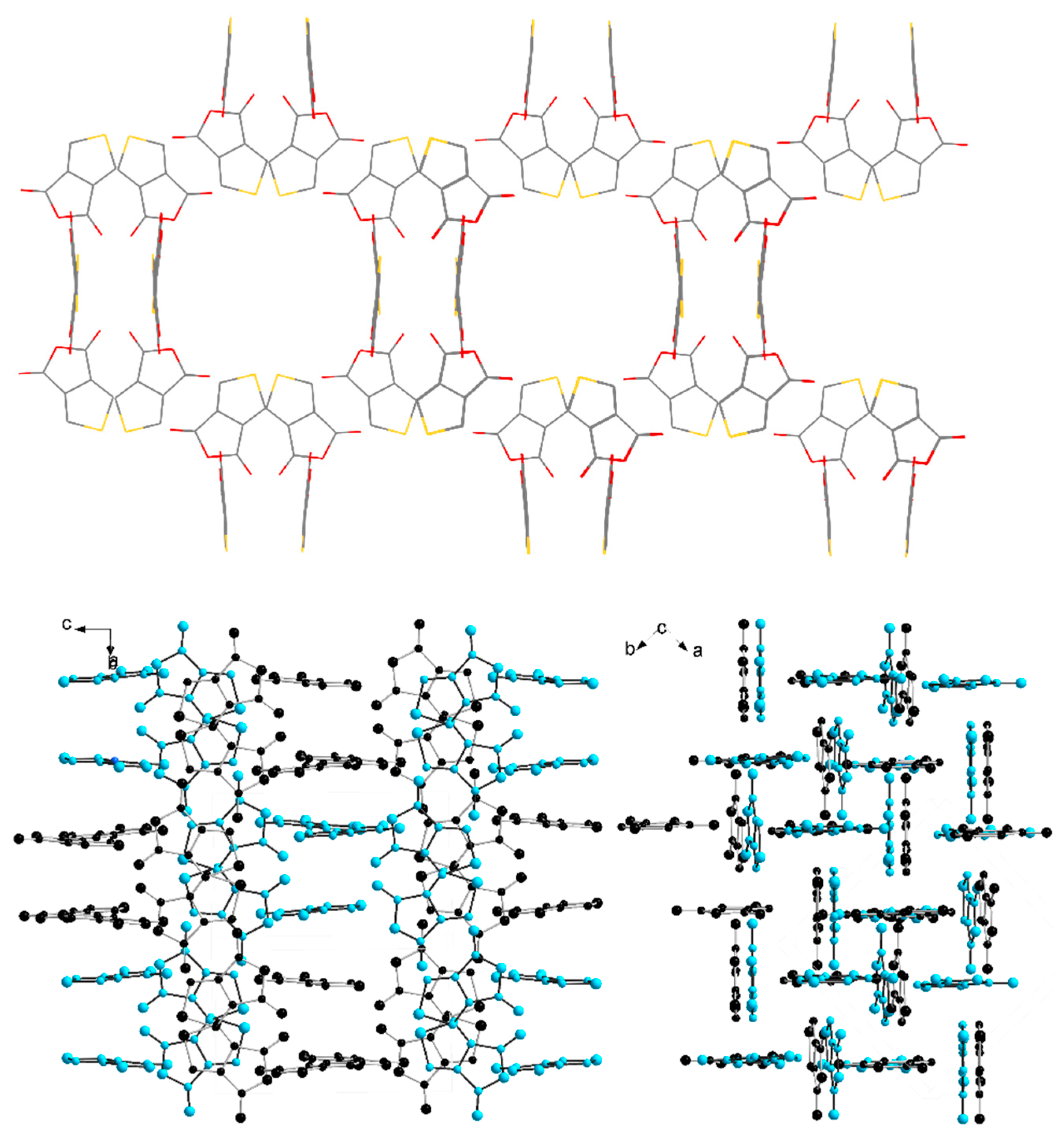
2. Results and Discussion
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Supporting Data
References
- Kealy, T.J.; Pauson, P.L. A new type of organo-iron compound. Nature 1951, 168, 1039. [Google Scholar] [CrossRef]
- Miller, S.A.; Tebboth, J.A.; Tremaine, J.F. Dicyclopentadienyliron. J. Chem. Soc. 1952, 114, 632. [Google Scholar] [CrossRef]
- Van Staveren, D.R.; Metzler-Nolte, N. Bioorganometallic chemistry of ferrocene. Chem. Rev. 2004, 104, 5931. [Google Scholar] [CrossRef] [PubMed]
- Štěpnička, P. (Ed.) Ferrocenes: Ligands, Materials and Biomolecules; Wiley & Sons: Chichester, UK, 2008. [Google Scholar]
- Tong, R.; Zhao, Y.; Wang, L.; Yu, H.; Ren, F.; Saleem, M.; Amer, W.A. Recent research progress in the synthesis and properties of burning rate catalysts based on ferrocene-containing polymers and derivatives. J. Organomet. Chem. 2014, 755, 16. [Google Scholar] [CrossRef]
- Dai, L.-X.; Hou, X.-L. (Eds.) Chiral Ferrocenes in Asymmetric Catalysis; Synthesis and Applications; Wiley-VCH: Weinheim, Germany, 2010. [Google Scholar]
- Yu, Y.; Bond, A.D.; Leonard, P.W.; Lorenz, U.J.; Timofeeva, T.V.; Vollhardt, K.P.C.; Whitener, G.D.; Yakovenko, A.A. Hexaferrocenylbenzene. Chem. Commun. 2006, 24, 2572. [Google Scholar] [CrossRef]
- Yu, Y.; Bond, A.D.; Leonard, P.W.; Vollhardt, K.P.C.; Whitener, G.D. Syntheses, structures, and reactivity of radial oligocyclopentadienyl metal complexes: Penta (ferrocenyl) cyclopentadienyl and congeners. Angew. Chem. Int. Ed. 2006, 45, 1794. [Google Scholar] [CrossRef]
- Hildebrandt, A.; Rüffer, T.; Erasmus, E.; Swarts, J.C.; Lang, H. A star-shaped supercrowded 2,3,4,5-tetraferrocenylthiophene: Synthesis, solid-state structure, and electrochemistry. Organometallics 2010, 29, 4900–4905. [Google Scholar] [CrossRef]
- Frenzel, P.; Lehrich, S.W.; Korb, M.; Hildebrandt, A.; Lang, H. Ferrocenyloxysilanes: Synthesis, characterization and electrochemical investigations. J. Organomet. Chem. 2017, 845, 98. [Google Scholar] [CrossRef]
- Packheiser, R.; Lang, H. Mixed transition–metal complexes based on multitopic 1,3,5-triethynyl-and 1-diphenylphosphino-3,5-diethynyl-benzene linking units. Inorg. Chim. Acta 2011, 366, 177–183. [Google Scholar] [CrossRef]
- Packheiser, R.; Rüffer, T.; Ecorchard, P.; Lang, H.Z. Transition-Metal Complexes Based on the 1,3,5-Triethynyl Benzene Linking Unit. Anorg. Allg. Chem. 2010, 636, 2607. [Google Scholar] [CrossRef]
- Packheiser, R.; Lang, H. A first mixed heteropentametallic transition metal complex: Synthesis and characterisation. Chem. Commun. 2007, 10, 580. [Google Scholar] [CrossRef]
- Barlow, S.; O’Hare, D. Metal−metal interactions in linked metallocenes. Chem. Rev. 1997, 97, 637. [Google Scholar] [CrossRef] [PubMed]
- Paul, F.; Lapinte, C. Organometallic molecular wires and other nanoscale-sized devices: An approach using the organoiron (dppe) Cp* Fe building block. Coord. Chem. Rev. 1998, 178–180, 431–509. [Google Scholar] [CrossRef]
- Lal, B.; Badshah, A.; Altaf, A.A.; Khan, N.; Ullah, S. Miscellaneous applications of ferrocene-based peptides/amides. Appl. Organomet. Chem. 2011, 25, 843. [Google Scholar] [CrossRef]
- Braga, S.S.; Silva, A.M.S. A new age for iron: Antitumoral ferrocenes. Organometallics 2013, 32, 5626. [Google Scholar] [CrossRef]
- Hildebrandt, A.; Lang, H. (Multi) ferrocenyl five-membered heterocycles: Excellent connecting units for electron transfer studies. Organometallics 2013, 32, 5640. [Google Scholar] [CrossRef]
- Ceccon, A.; Santi, S.; Orian, L.; Bisello, A. Electronic communication in heterobinuclear organometallic complexes through unsaturated hydrocarbon bridges. Coord. Chem. Rev. 2004, 248, 683. [Google Scholar] [CrossRef]
- Ward, M.D. Metal-metal interactions in binuclear complexes exhibiting mixed valency; molecular wires and switches. Chem. Soc. Rev. 1995, 24, 121. [Google Scholar] [CrossRef]
- Hildebrandt, A.; Lang, H. Influencing the electronic interaction in diferrocenyl-1-phenyl-1 H-pyrroles. Dalt. Trans. 2011, 40, 11831. [Google Scholar] [CrossRef]
- Li, Y.; Josowicz, M.; Tolbert, L.M. Diferrocenyl molecular wires. The role of heteroatom linkers. J. Am. Chem. Soc. 2010, 132, 10374. [Google Scholar] [CrossRef]
- Moriuchi, T.; Hirao, T. Imide-bridged diferrocene for protonation-controlled regulation of electronic communication. Tetrahedron Lett. 2007, 48, 5099. [Google Scholar] [CrossRef]
- Guldi, D.M.; Maggini, M.; Scorrano, G.; Prato, M. Intramolecular electron transfer in fullerene/ferrocene based donor−bridge−acceptor dyads. J. Am. Chem. Soc. 1997, 119, 974. [Google Scholar] [CrossRef]
- Sixt, T.; Sieger, M.; Krafft, M.J.; Bubrin, D.; Fiedler, J.; Kaim, W. Ambi-valence taken literally: Ruthenium vs iron oxidation in (1,1′-diphosphinoferrocene) ruthenium (II) hydride and chloride complexes as deduced from spectroelectrochemistry of the heterodimetallic “mixed-valent” intermediates. Organometallics 2010, 29, 5511. [Google Scholar] [CrossRef]
- Nguyen, P.; Elipe, P.G.; Manners, I. Organometallic polymers with transition metals in the main chain. Chem. Rev. 1999, 99, 1515. [Google Scholar] [CrossRef] [PubMed]
- MacDowell, D.W.H.; Jourdenais, R.A.; Naylor, R.W.; Wisowaty, J.C. The Reaction of Thiophene-3,4-dicarbonyl Chloride with Aluminum Chloride and Benzene. J. Org. Chem. 1972, 37, 4406. [Google Scholar] [CrossRef]
- El-Gaby, M.S.A.; Zahran, M.A.; Ismail, M.M.F.; Ammar, Y.A. A novel synthesis of dibenzo [c,f] chromenes, dibenzo [c,h] chromenes and benzo [7,8] chromeno [3,4-f] isoindoles as antimicrobial agents. II Farmaco. 2000, 55, 227. [Google Scholar]
- Meyer, C.; Neue, B.; Schepmann, D.; Yanagisawa, S.; Yamaguchi, J.; Würthwein, E.-U.; Itami, K.; Wünsch, B. Exploitation of an additional hydrophobic pocket of σ 1 receptors: Late-stage diverse modifications of spirocyclic thiophenes by C–H bond functionalization. Org. Biomol. Chem. 2011, 9, 8016. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Schepmann, D.; Yanagisawa, S.; Yamaguchi, J.; Itami, K.; Wünsch, B. Late-Stage C–H Bond Arylation of Spirocyclic σ1 Ligands for Analysis of Complementary σ1 Receptor Surface. Eur. J. Org. Chem. 2012, 2012, 5972. [Google Scholar] [CrossRef]
- Meyer, C.; Schepmann, D.; Yanagisawa, S.; Yamaguchi, J.; Col, V.D.; Laurini, E.; Itami, K.; Pricl, S.; Wünsch, B.J. Pd-catalyzed direct C–H bond functionalization of spirocyclic σ1 ligands: Generation of a pharmacophore model and analysis of the reverse binding mode by docking into a 3D homology model of the σ1 receptor. Med. Chem. 2012, 55, 8047. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Neue, B.; Schepmann, D.; Yanagisawa, S.; Yamaguchi, J.; Würthwein, E.-U.; Itami, K.; Wünsch, B. Improvement of σ1 receptor affinity by late-stage C–H-bond arylation of spirocyclic lactones. Bioorg. Med. Chem. 2013, 21, 1844. [Google Scholar] [CrossRef]
- Al-Mousawi, S.M.; El-Apasery, M.A. Synthesis of some monoazo disperse dyes derived from aminothienochromene. Molecules 2013, 18, 8837. [Google Scholar] [CrossRef]
- Fuse, S.; Takahashi, R.; Takahashi, T. Facile, One-Step Synthesis of 5-Substituted Thieno [3,4-c] pyrrole-4,6-dione by Palladium-Catalyzed Carbonylative Amidation. Eur. J. Org. Chem. 2015, 16, 3430. [Google Scholar] [CrossRef]
- Kim, J.E.; Lee, J.; Yun, H.; Baek, Y.; Lee, P.H. Rhodium-catalyzed intramolecular transannulation reaction of alkynyl thiadiazole enabled 5, n-fused thiophenes. J. Org. Chem. 2017, 82, 1437–1447. [Google Scholar] [CrossRef] [PubMed]
- Benachenhou, F.; Mesli, M.A.; El Borai, M.; Hanquet, B.; Guilard, R.J. Synthesis and structure elucidation of the condensation products between thiophene dicarbaldehydes and aromatic amines. Potential analgesic and anti-inflammatory agents. Heterocycl. Chem. 1988, 25, 1531. [Google Scholar] [CrossRef]
- Nielsen, C.B.; Bjørnholm, T. New regiosymmetrical dioxopyrrolo-and dihydropyrrolo-functionalized polythiophenes. Org. Lett. 2004, 6, 3381. [Google Scholar] [CrossRef] [PubMed]
- Crayston, J.A.; Iraqi, A.; Mallon, P.; Walton, J.C. Preparation and characterisation of thienonaphthoquinones and their radical ions. J. Chem. Soc. Perkin Trans. 1993, 2, 1589. [Google Scholar] [CrossRef]
- Vishnumurthy, K.; Makriyannis, A.J. Novel and Efficient One-Step Parallel Synthesis of Dibenzopyranones via Suzuki− Miyaura Cross Coupling. Comb. Chem. 2010, 12, 664. [Google Scholar] [CrossRef][Green Version]
- Taher, D.; Awwadi, F.F.; Pfaff, U.; Speck, J.M.; Rüffer, T.; Lang, H. A series of Se-ferrocenyl thiophene carboselenoates–Synthesis, solid-state structure and electrochemistry. J. Organomet. Chem. 2013, 736, 9. [Google Scholar] [CrossRef]
- Taher, D.; Awwadi, F.F.; Speck, J.M.; Korb, M.; Schaarschmidt, D.; Weheabby, S.; Habashneh, A.Y.; Al-Noaimi, M.; El-Khateeb, M.; Abu-Orabi, S.T.; et al. Heterocyclic-based ferrocenyl carboselenolates: Synthesis, solid-state structure and electrochemical investigations. J. Organomet. Chem. 2017, 845, 55. [Google Scholar] [CrossRef]
- Taher, D.; Awwadi, F.F.; Speck, J.M.; Korb, M.; Wagner, C.; Hamed, E.M.; Al-Noaimi, M.; Habashneh, A.Y.; El-khateeb, M.; Abu-Orabi, S.T.; et al. Ferrocenyl thiocarboxylates: Synthesis, solid-state structure and electrochemical investigations. J. Organomet. Chem. 2017, 847, 59. [Google Scholar] [CrossRef]
- Taher, D.; Awwadi, F.F.; Speck, J.M.; Korb, M.; Schaarschmidt, D.; Wagner, C.; Amarne, H.; Merzweiler, K.; van Koten, G.; Lang, H.J. From ferrocenyl selenoesters to diferrocenyl methanols. Organomet. Chem. 2018, 863, 1. [Google Scholar] [CrossRef]
- Taher, D.; Corrigan, J.F. Aryl (trimethylsilyl) selenides as Reagents for the Synthesis of Mono-and Diselenoesters. Organometallics 2011, 30, 5943. [Google Scholar] [CrossRef]
- Ghazzy, A.; Taher, D.; Helal, W.; Korbb, M.; Al-Shewikib, R.K.; Weheabby, S.; Al-Saidd, N.; Abu-Orabi, S.T.; Lang, H. Aryl ferrocenylmethylesters: Synthesis, solid-state structure and electrochemical investigations. Arab. J. Chem. 2020, 13, 3546. [Google Scholar] [CrossRef]
- Taher, D. Synthesis and reactivity of cyclopentadienyl ruthenium complexes containing ferrocenylselenolates. Transit. Met. Chem. 2009, 34, 641. [Google Scholar] [CrossRef]
- Taher, D.; Wallbank, A.I.; Turner, E.A.; Cuthbert, H.L.; Corrigan, J.F. Alk-2-ynyl Trimethylsilyl Chalcogenoethers by Nucleophilic Substitution of Propargyl Bromides. Eur. J. Inor. Chem. 2006, 22, 4616. [Google Scholar] [CrossRef]
- Taher, D.; Awwadi, F.; El-khateeb, M.; Lang, H. Synthesis and reactivity of cyclopentadienyl iron complexes containing ferrocenyl selenolates. Transit. Met. Chem. 2012, 37, 601. [Google Scholar] [CrossRef]
- Taher, D.; Ghazzy, A.; Awwadi, F.F.; Helal, W.; al Khalyfeh, K.; Korb, M.; Hildebrandt, A.; Kovalski, E.; Lang, H. Ferrocenylmethyl-functionalized 5-membered heterocycles: Synthesis, solid-state structure and electrochemical investigations. Polyhedron 2018, 152, 188. [Google Scholar] [CrossRef]
- Hildebrandt, A.; Miesel, D.; Lang, H. Electrostatic interactions within mixed-valent compounds. Coord. Chem. Rev. 2018, 371, 56. [Google Scholar] [CrossRef]
- Lang, H. Organometallic π-tweezers and 1,1′-bis (diphenylphosphanyl) ferrocene as bidentate chelating ligands for the synthesis of heteromultimetallic compounds. Polyhedron 2018, 139, 50. [Google Scholar] [CrossRef]
- Heinze, K.; Lang, H. Ferrocene-Beauty and Function. Organometallics 2013, 20, 5623. [Google Scholar] [CrossRef]
- Naik, V.S.; Pragasam, A.; Jayanna, H.S.; Vinitha, G. Structural, linear optical, second and third-order nonlinear optical properties of two halogenated chalcone derivatives containing thiophene moiety. Chem. Phys. Lett. 2020, 761, 138051. [Google Scholar] [CrossRef]
- Terpstra, J.W.; van Leusen, A.M. A new synthesis of benzo [b] thiophenes and benzo [c] thiophenes by annulation of disubstituted thiophenes. J. Org. Chem. 1986, 51, 230–238. [Google Scholar] [CrossRef]
- Swarts, J.C.; Nafady, A.; Roudebush, J.H.; Trupia, S.; Geiger, W.E. One-electron oxidation of ruthenocene: Reactions of the ruthenocenium ion in gentle electrolyte media. Inorg. Chem. 2009, 48, 2156. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Jin, B.; Liu, P.; Cheng, L.; Cheng, W.; Zhang, S. Synthesis, electrochemistry and IR spectroelectrochemistry of bisferrocenyl bridged benzene derivates. J. Organomet. Chem. 2012, 697, 57. [Google Scholar] [CrossRef]
- Hildebrandt, A.; al Khalyfeh, K.; Nawroth, J.F.; Jordan, R. Electron transfer studies on conjugated ferrocenyl-containing oligomers. Organometallics 2016, 35, 3713. [Google Scholar] [CrossRef]
- Kalita, D.; Morisue, M.; Kobuke, Y. Synthesis and electrochemical properties of slipped-cofacial porphyrin dimers of ferrocene-functionalized Zn-imidazolyl-porphyrins as potential terminal electron donors in photosynthetic models. New J. Chem. 2006, 30, 77. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Spackman, M.A.; Mitchell, A.S. Novel tools for visualizing and exploring intermolecular interactions in molecular crystals. Acta Crystallogr. B 2004, 60, 627–668. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystals. Cryst. Eng. Comm. 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Liu, X.; Du, L.; Zhang, W.; Li, R.; Feng, F.; Feng, X.J. Syntheses, structures and hirshfeld surface analyses of two 3D supramolecules based on nitrogen-heterocyclic tricarboxylate ligand. Mol. Struct. 2019, 1194, 138–143. [Google Scholar] [CrossRef]
- Gritzner, G.; Kuta, J. Recommendations on reporting electrode potentials in nonaqueous solvents. Pure Appl. Chem. 1984, 56, 461. [Google Scholar] [CrossRef]
- Nafady, A.; Geiger, W.E. Characterization of the successive one-electron oxidation products of the dicobalt fulvalenediyl (Fv) compound Co2Fv (CO) 4 and its phosphine-substituted product. Organometallics 2008, 27, 5624. [Google Scholar] [CrossRef]
- Guillot, B.; Enrique, E.; Huder, L.; Jelsch, C. MS19. O01. Acta Cryst. A 2014, 70, C279. [Google Scholar] [CrossRef]
- Jelsch, C.; Ejsmont, K.; Huder, L. The enrichment ratio of atomic contacts in crystals, an indicator derived from the Hirshfeld surface analysis. IUCrJ 2014, 1, 119. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.L.; Jotani, M.M.; Tiekink, E.R.T. Utilizing Hirshfeld surface calculations, non-covalent interaction (NCI) plots and the calculation of interaction energies in the analysis of molecular packing. Acta Crystallogr. E 2019, 75, 308. [Google Scholar] [CrossRef] [PubMed]
- Claus, R.; Lewtak, J.P.; Müller, T.J.; Swarts, J.C. Structural influences on the electrochemistry of 1, 1′-di (hydroxyalkyl) ferrocenes. Structure of [Fe {η5-C5H4–CH (OH)–(CH2) 3OH} 2]. J. Organomet. Chem. 2013, 740, 61. [Google Scholar] [CrossRef]
- Braga, D.; Maini, L.; Paganelli, F.; Tagliavini, E.; Casolari, S.; Grepioni, F.J. Organometallic building blocks for crystal engineering. Synthesis, structure and hydrogen bonding interactions in [Fe (η5-C5H4-CH2 (CH3) OH) 2],[Fe (η5-C5H3 (CH3) COOH) 2], [Fe (η5-C5H4CH (CH3) NH (η5-C5H4CH (CH3))] and in the diaminecyclohexane salt [Fe (η5-C5H4COO) 2] 2−[(1S, 2S)-(NH3) 2C6H10] 2+·2 [H2O]. Organomet. Chem. 2001, 609, 637–639. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
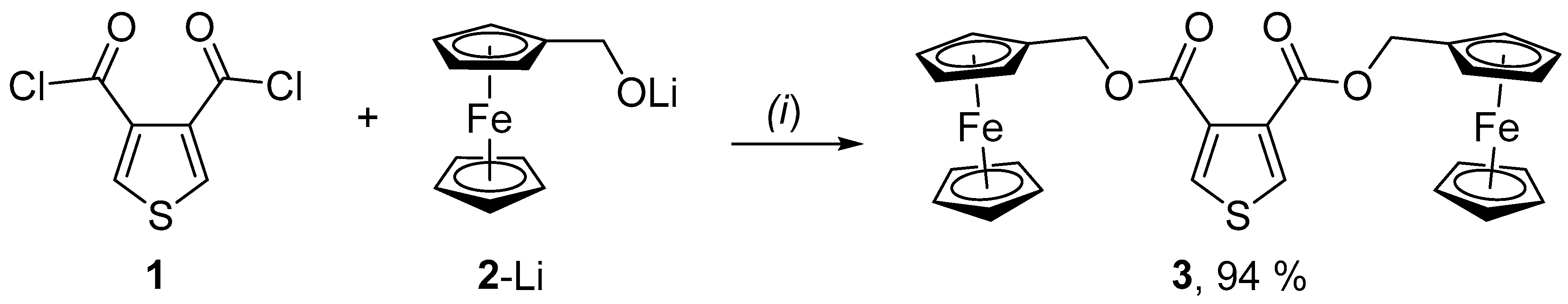{kind=link}
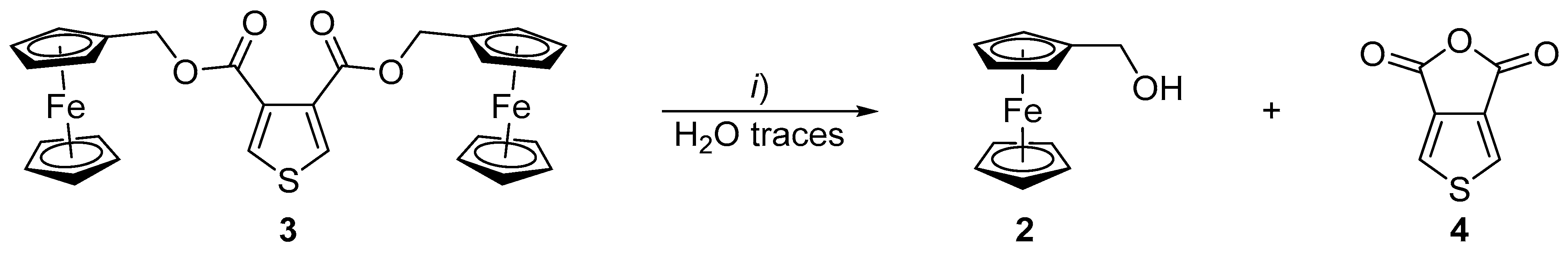{kind=link}
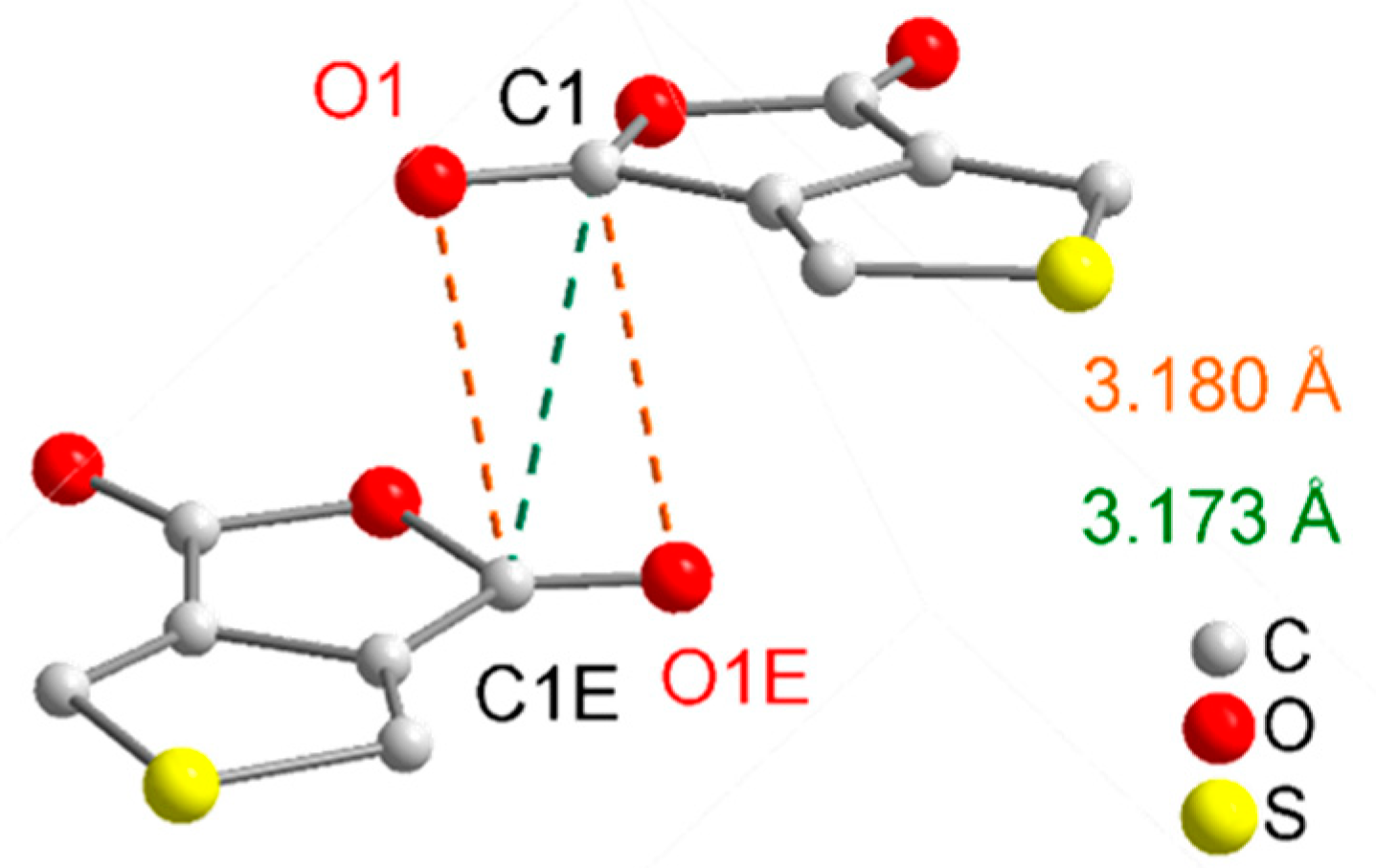
| D–H···A | D–H | H···A | D···A | D–H···A |
|---|---|---|---|---|
| C6–H7···O2i | 0.93 | 2.54 | 3.2708(4) | 136 |
| C6–H7···O1ii | 0.93 | 2.57 | 3.1211(4) | 118 |
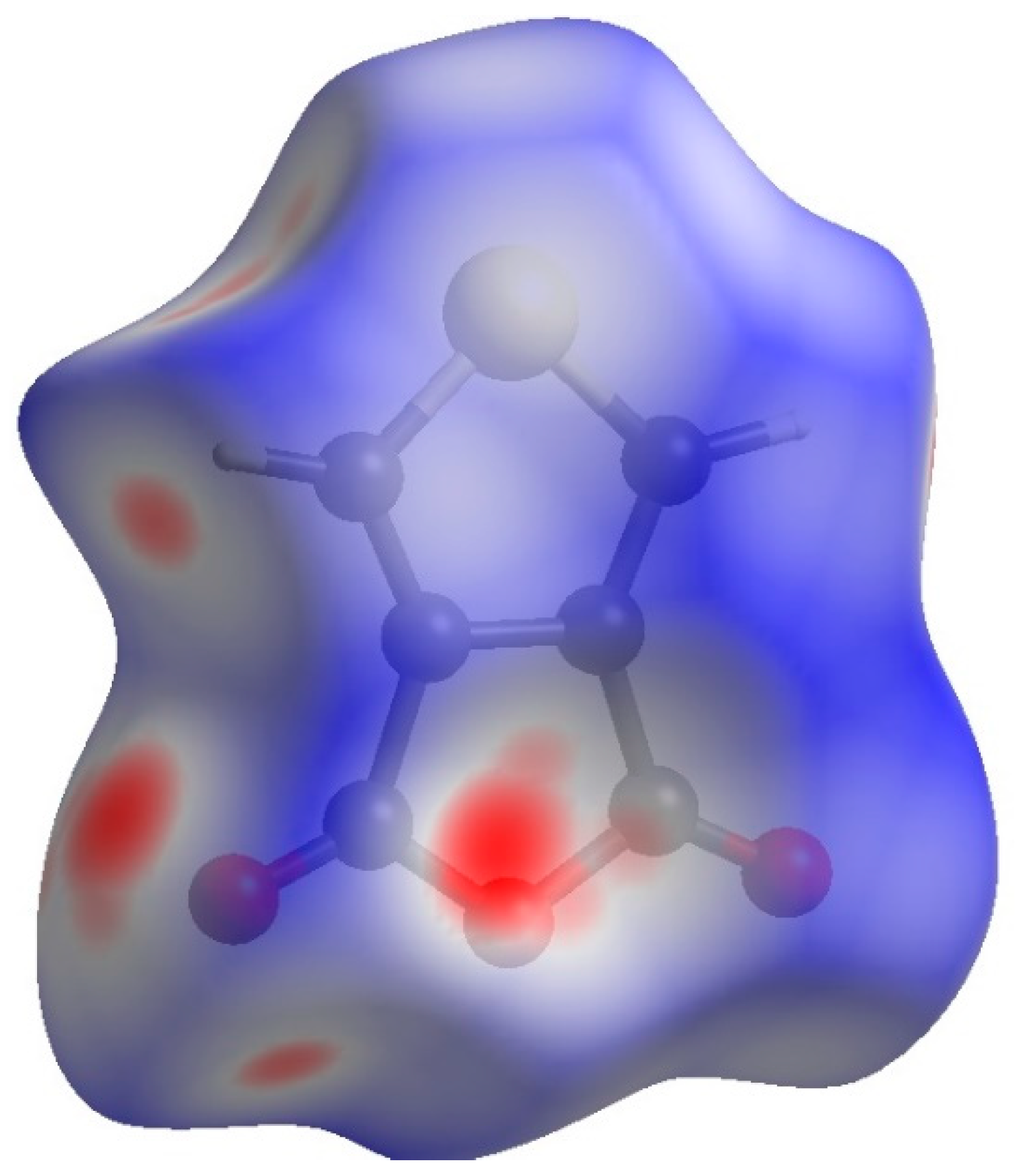
| Atoms | H | C | O | S |
|---|---|---|---|---|
| Surface interior (%) | 17.53 | 30.67 | 31.41 | 20.39 |
| Surface exterior (%) | 17.86 | 28.16 | 33.22 | 20.76 |
| Actual contacts merged (%) | ||||
| H | 0.68 | - | - | - |
| C | 6.37 | 10.00 | - | - |
| O | 24.98 | 22.97 | 2.50 | - |
| S | 2.68 | 9.48 | 11.69 | 8.65 |
| Equiprobable contacts merged (%) | ||||
| H | 3.13 | - | - | - |
| C | 10.41 | 8.64 | - | - |
| O | 11.43 | 19.03 | 10.43 | - |
| S | 7.28 | 12.11 | 13.30 | 4.23 |
| Enrichment reciprocal contacts merged (%) | ||||
| H | 0.22 | - | - | - |
| C | 0.61 | 1.16 | - | - |
| O | 2.18 | 1.21 | 0.24 | - |
| S | 0.37 | 0.78 | 0.88 | 2.04 |
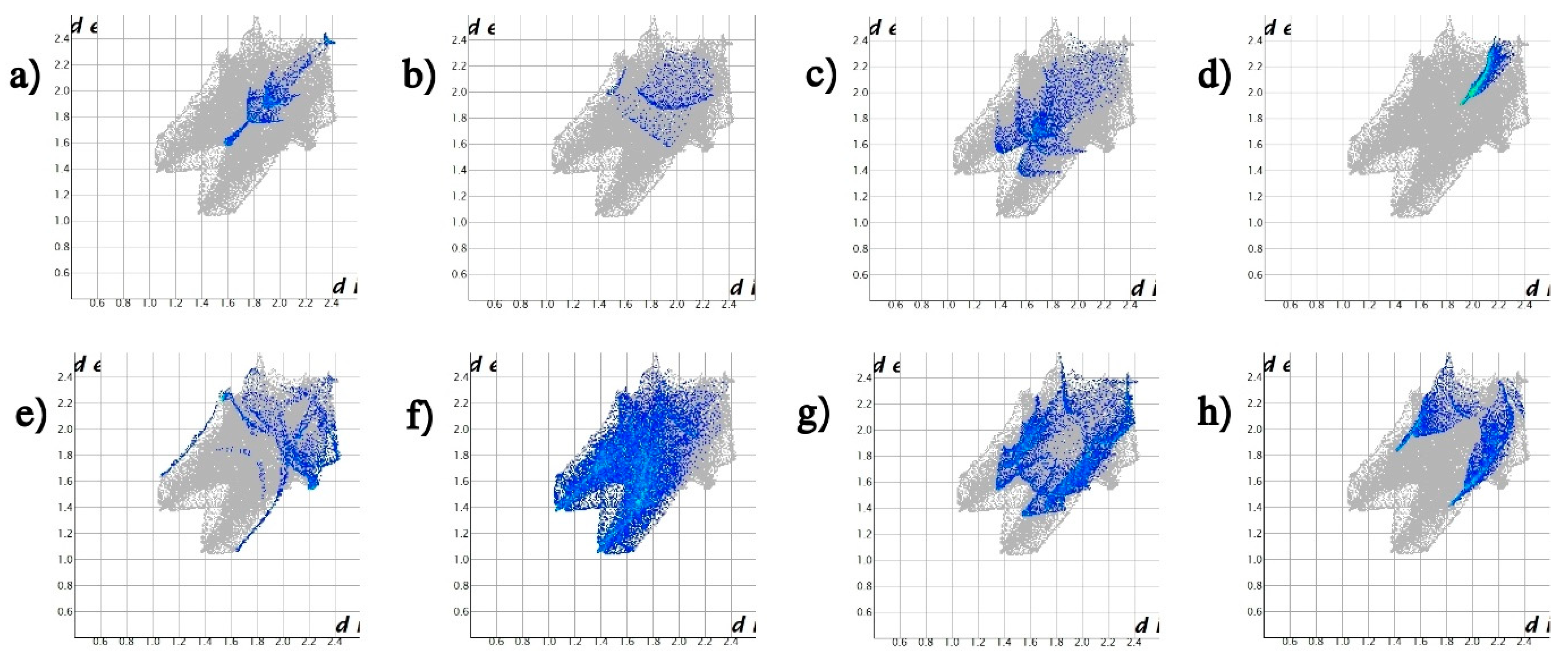
| Contacts | Symmetry Operation | E_ele | E_pol | E_dis | E_rep | E_tot |
|---|---|---|---|---|---|---|
| O···O, O···H, and O···C | y, −x + 1/2, −z + 1/2 | −25.7 | −5.0 | −17.6 | 22.2 | −32.5 |
| S···O, O···H, and O···C | x + 1/2, y + 1/2, −z | −13.4 | −2.8 | −6.6 | 9.5 | −16.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghazzy, A.; Taher, D.; Korb, M.; Al Khalyfeh, K.; Helal, W.; Amarne, H.; Rüffer, T.; Ishtaiwi, Z.; Lang, H. Rearrangement of Diferrocenyl 3,4-Thiophene Dicarboxylate. Inorganics 2022, 10, 96. https://doi.org/10.3390/inorganics10070096
Ghazzy A, Taher D, Korb M, Al Khalyfeh K, Helal W, Amarne H, Rüffer T, Ishtaiwi Z, Lang H. Rearrangement of Diferrocenyl 3,4-Thiophene Dicarboxylate. Inorganics. 2022; 10(7):96. https://doi.org/10.3390/inorganics10070096
Chicago/Turabian StyleGhazzy, Asma, Deeb Taher, Marcus Korb, Khaled Al Khalyfeh, Wissam Helal, Hazem Amarne, Tobias Rüffer, Zakariyya Ishtaiwi, and Heinrich Lang. 2022. "Rearrangement of Diferrocenyl 3,4-Thiophene Dicarboxylate" Inorganics 10, no. 7: 96. https://doi.org/10.3390/inorganics10070096
APA StyleGhazzy, A., Taher, D., Korb, M., Al Khalyfeh, K., Helal, W., Amarne, H., Rüffer, T., Ishtaiwi, Z., & Lang, H. (2022). Rearrangement of Diferrocenyl 3,4-Thiophene Dicarboxylate. Inorganics, 10(7), 96. https://doi.org/10.3390/inorganics10070096