
Electron-Deficient Ru(II) Complexes as Catalyst Precursors for Ethylene Hydrophenylation

 , and
, and 
Abstract
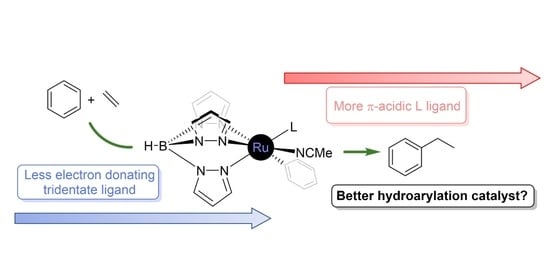
:
1. Introduction
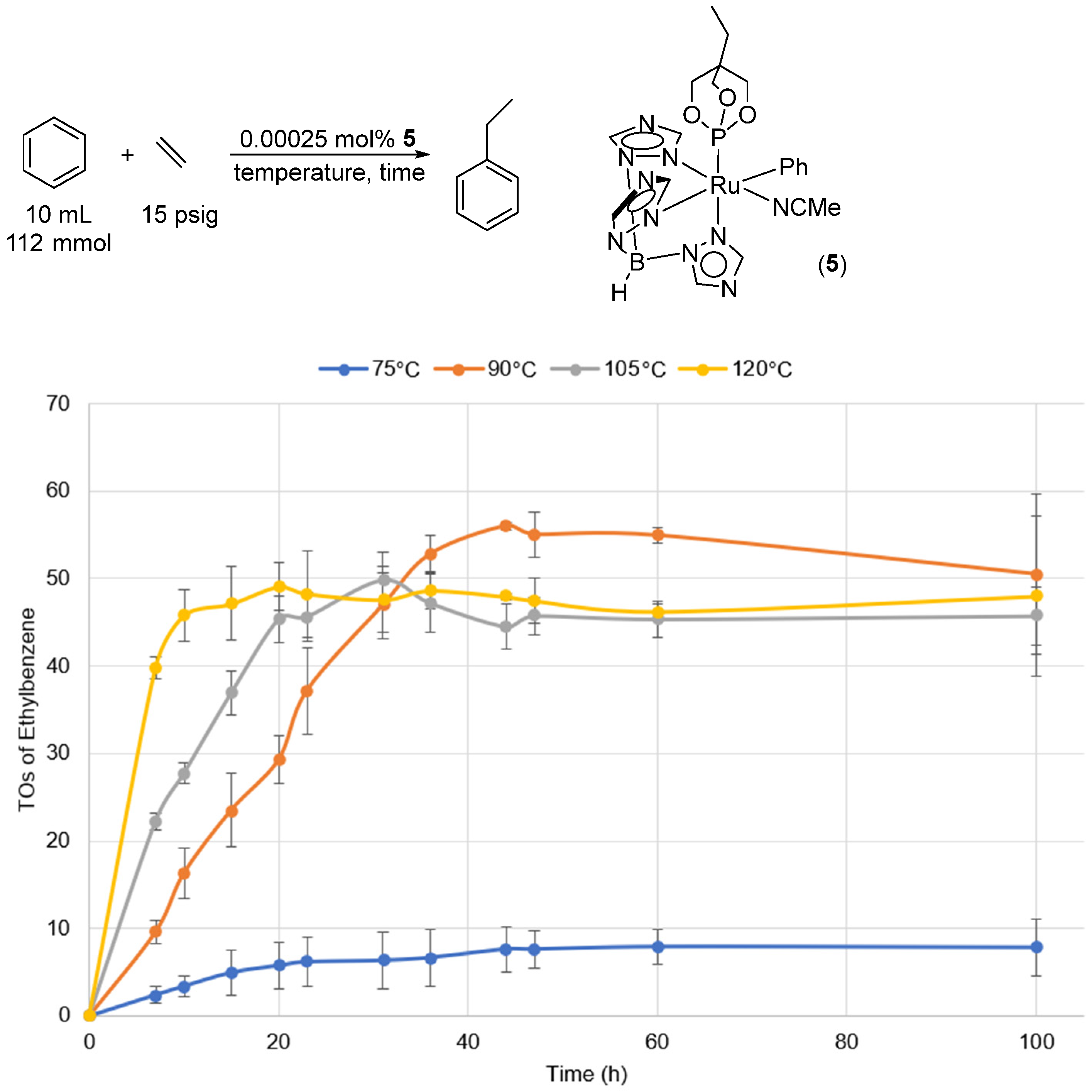
2. Results and Discussion
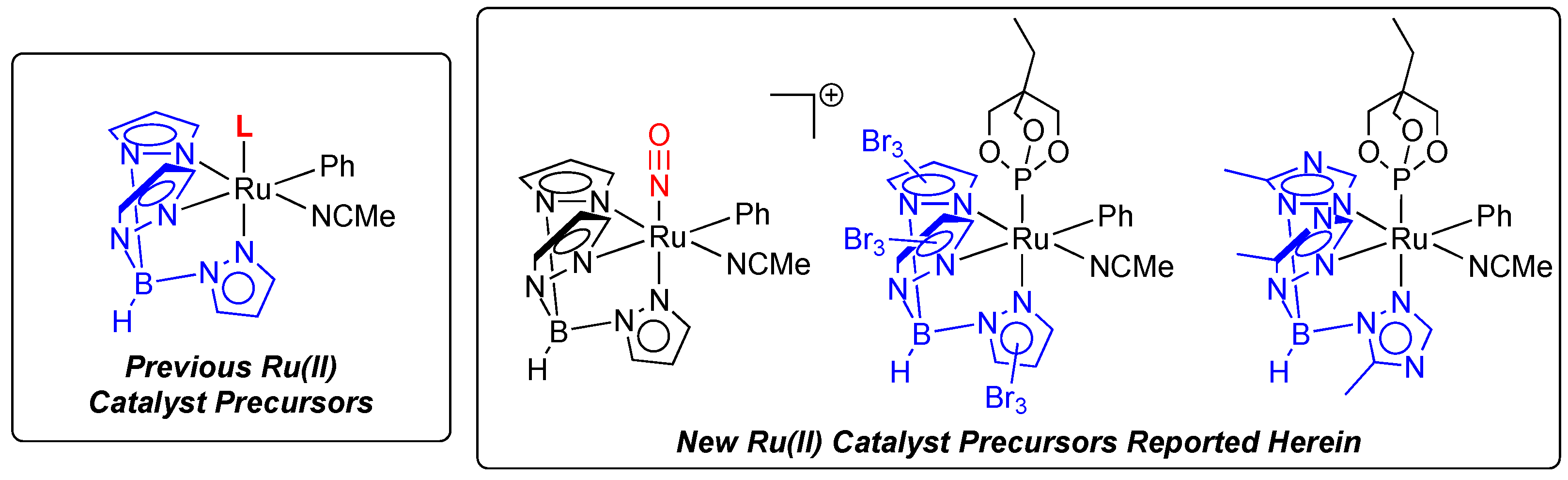
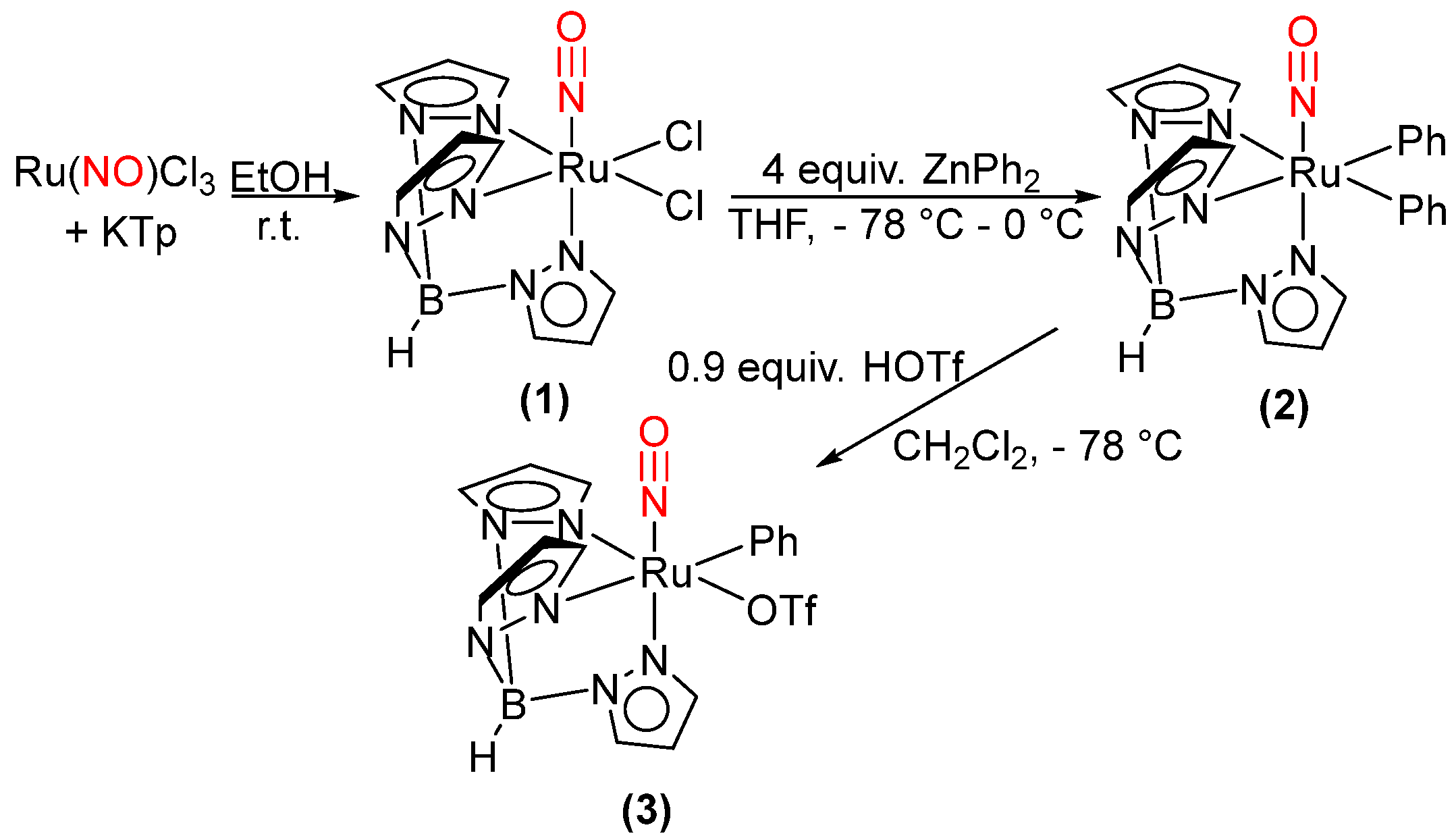
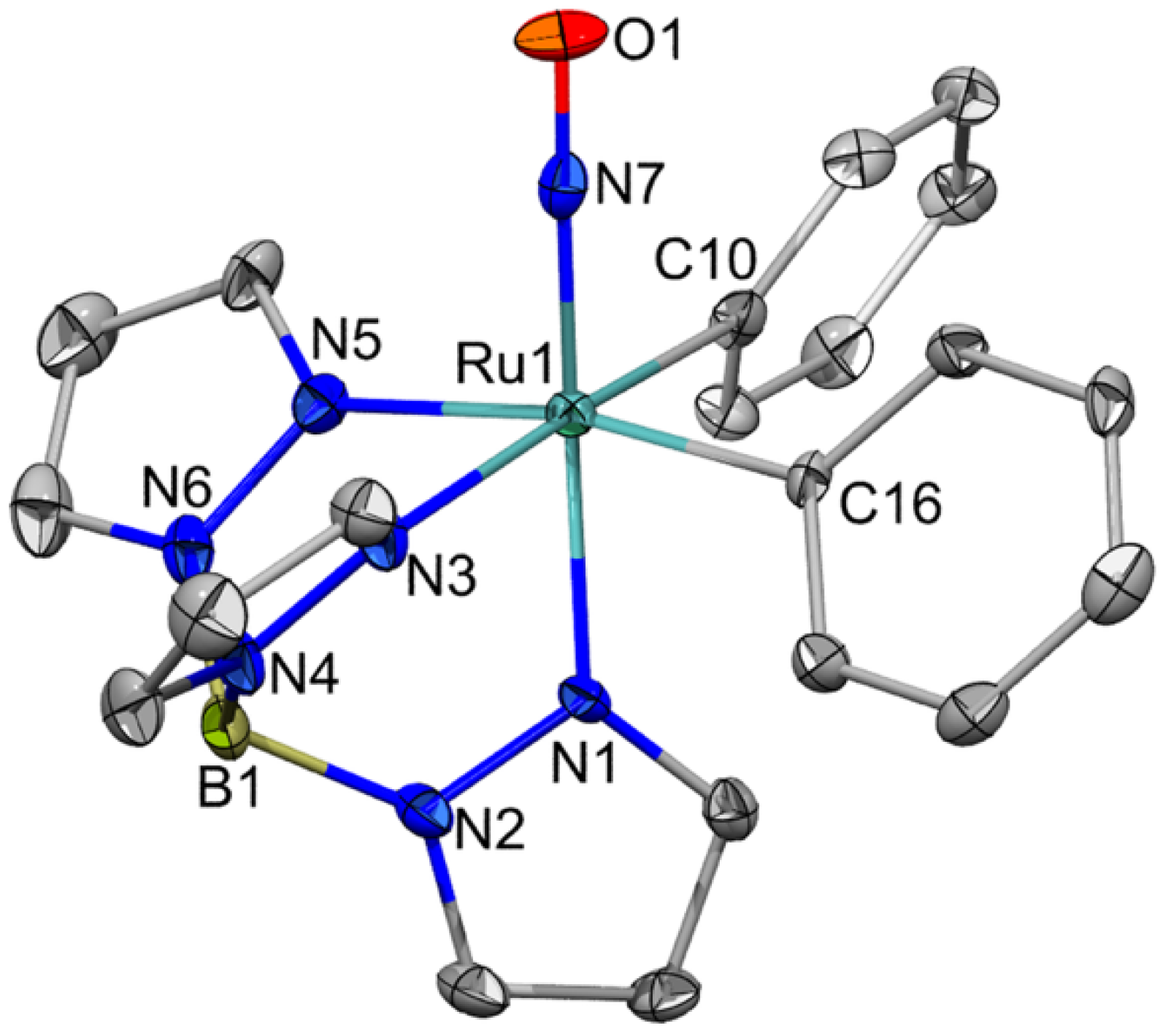
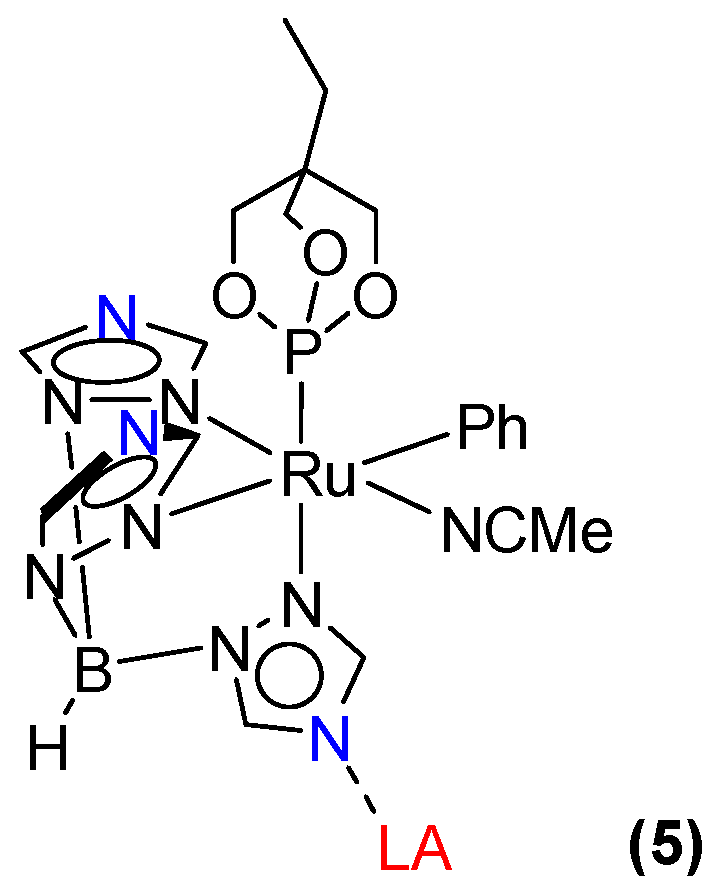
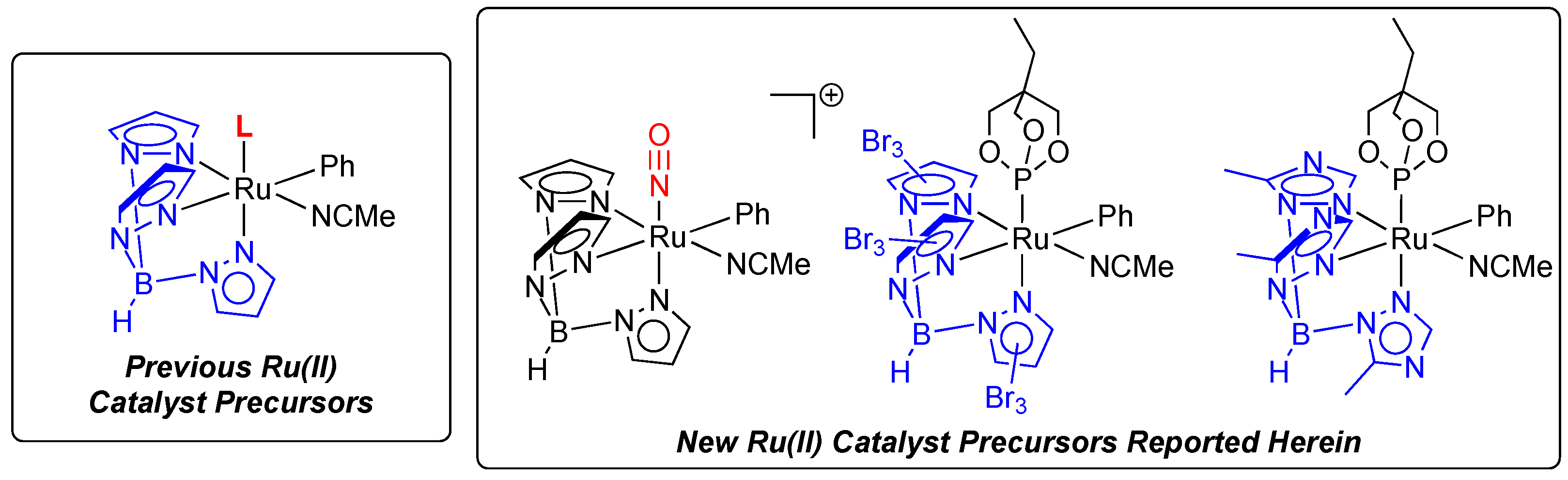
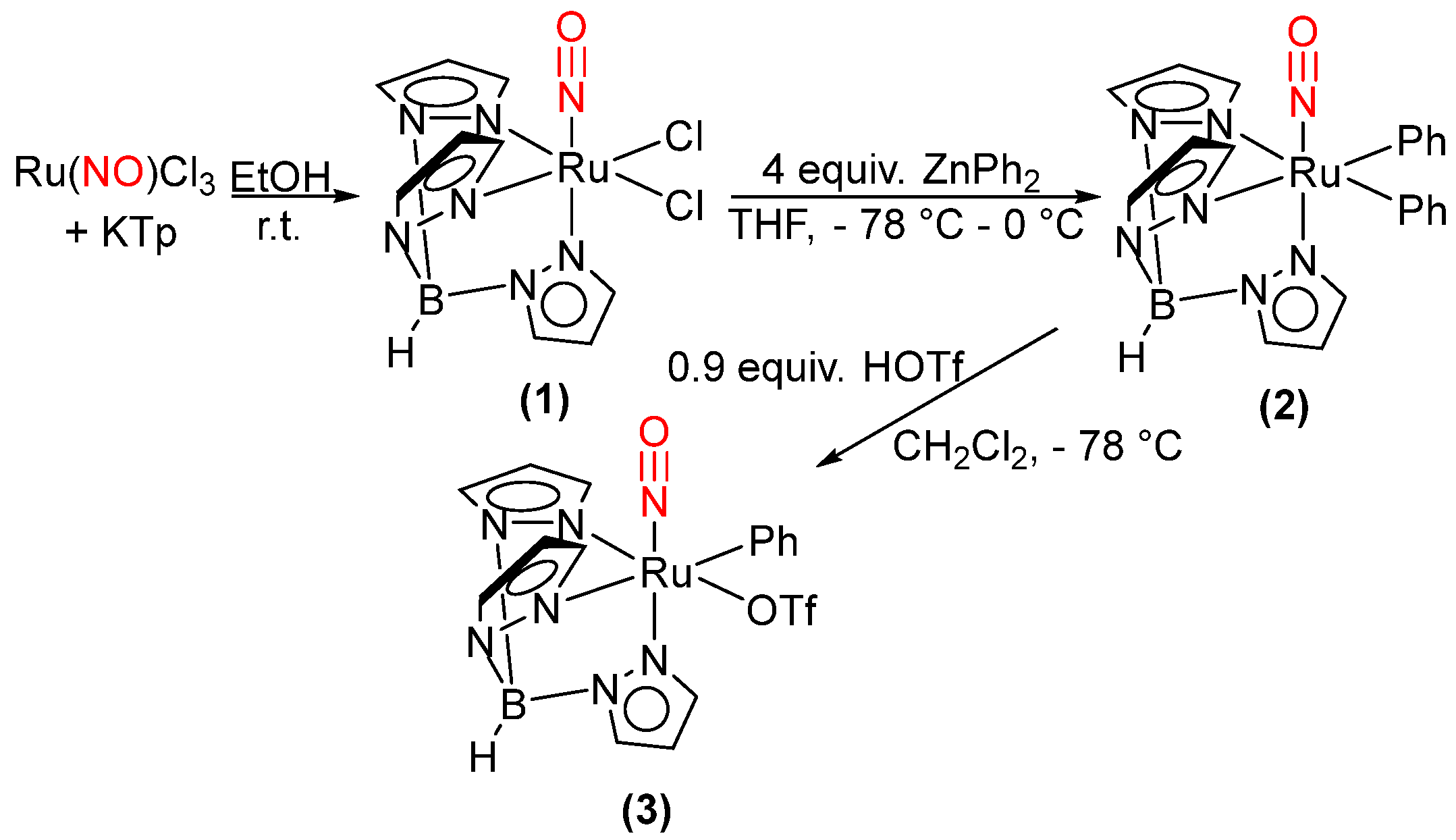
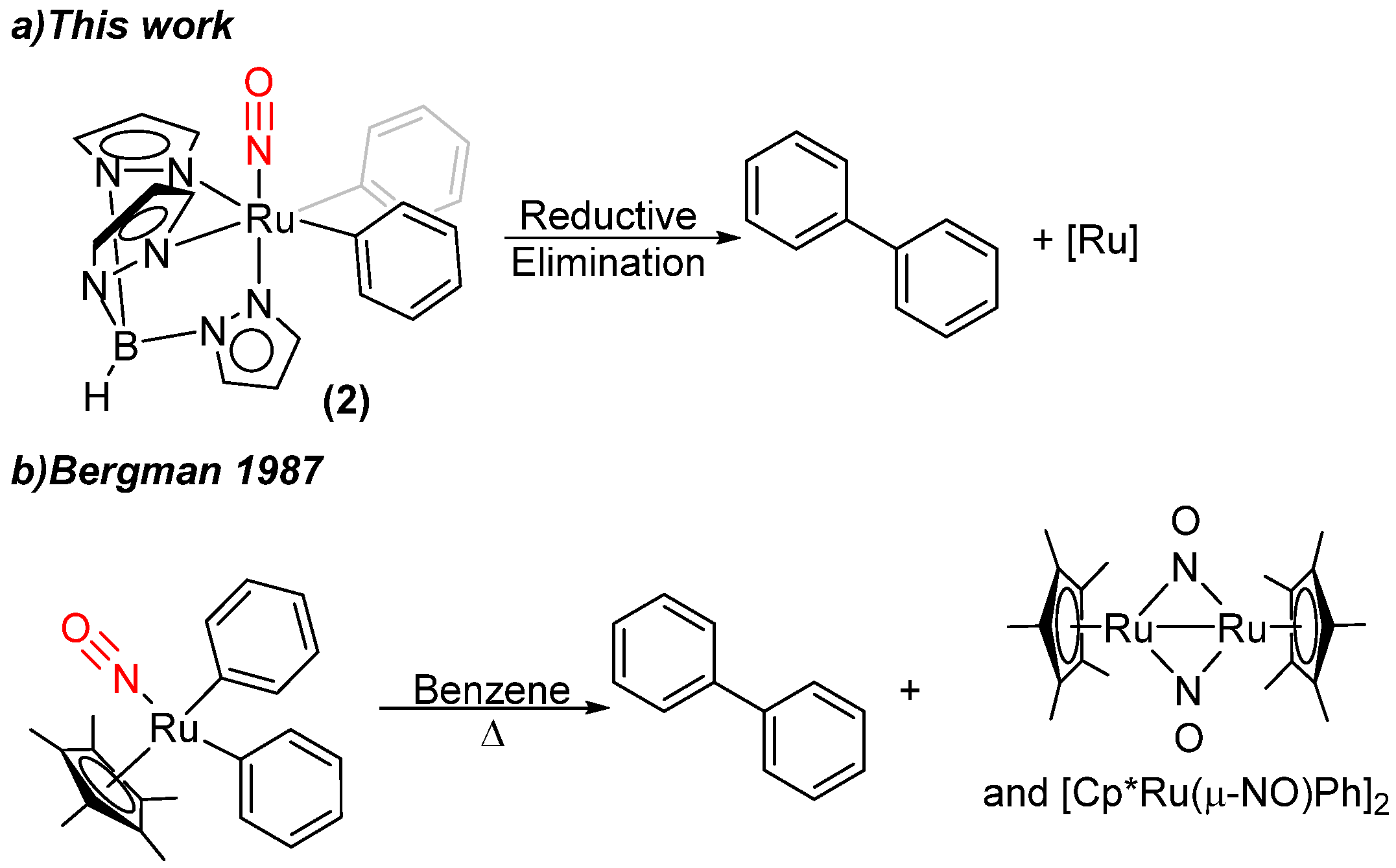
2.1. Attempted Synthesis of Complexes That Provide Access to [TpRu(NO)Ph]+
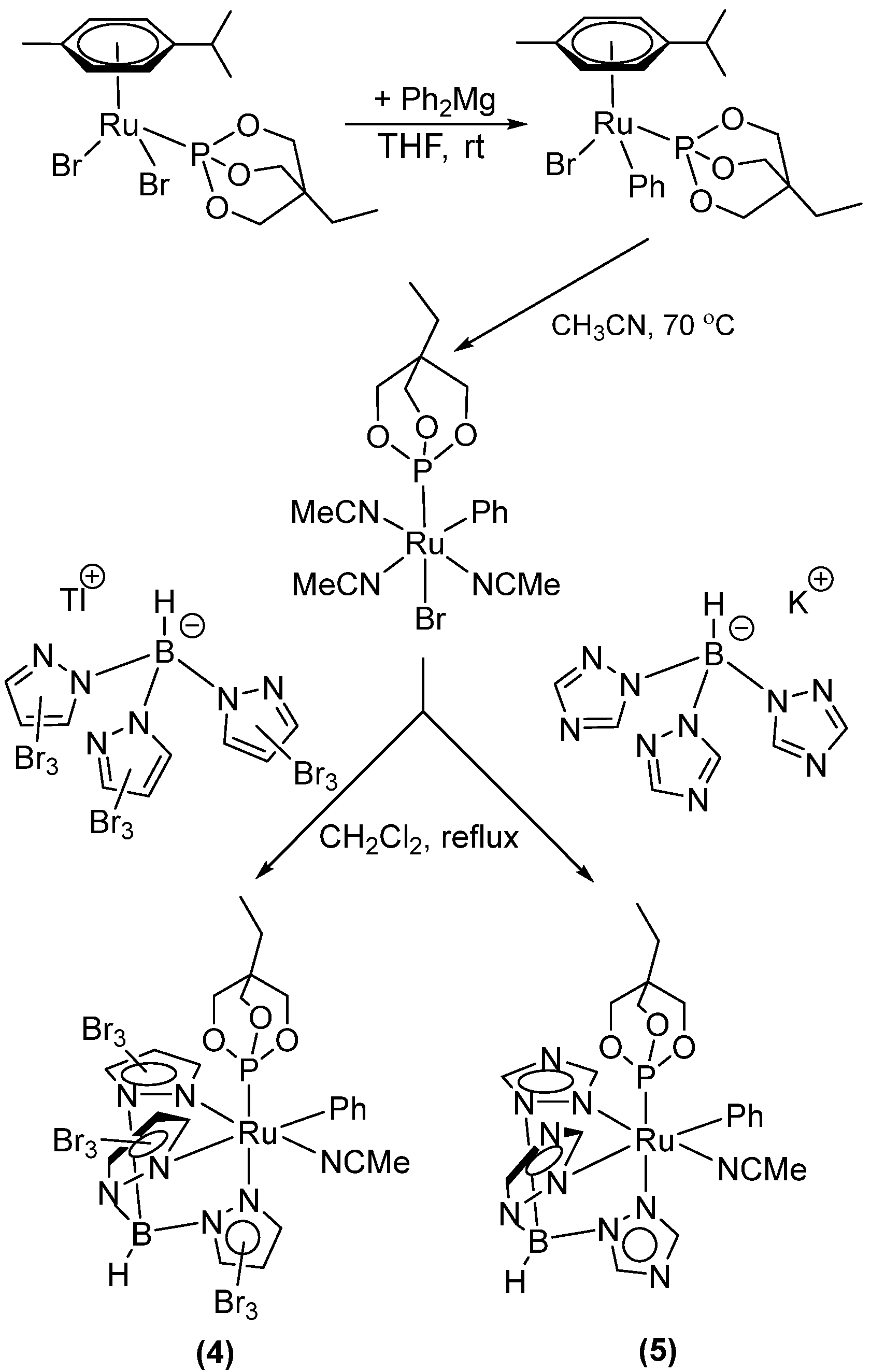
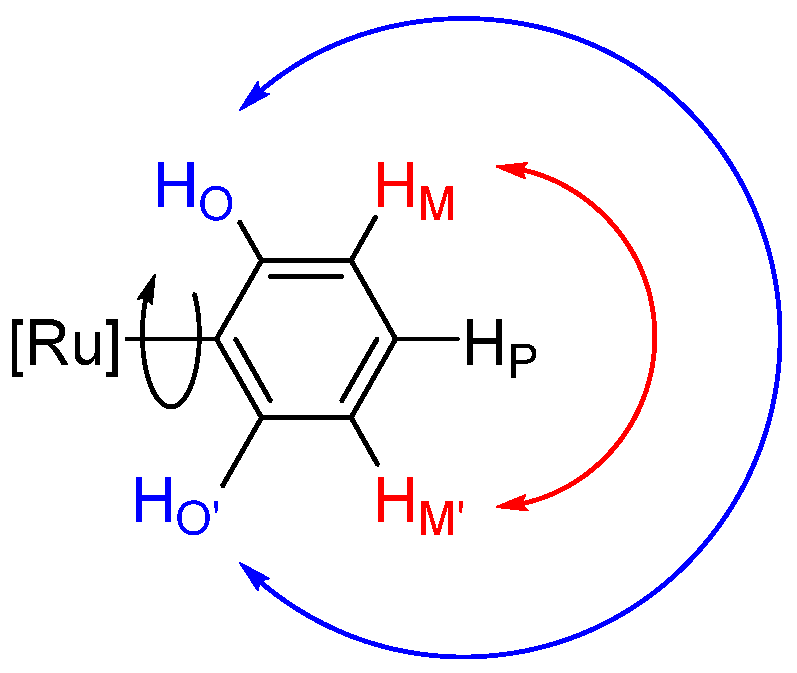
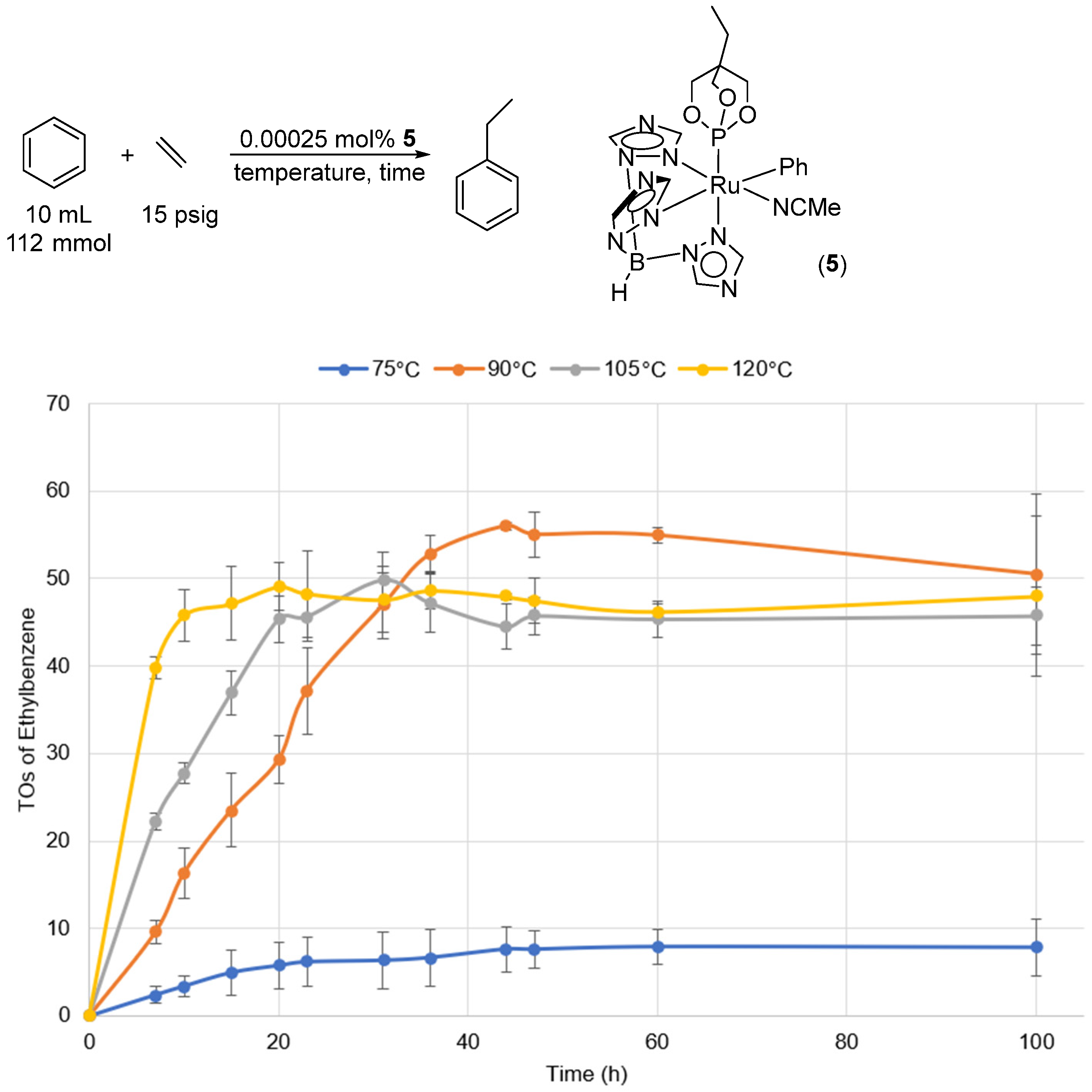
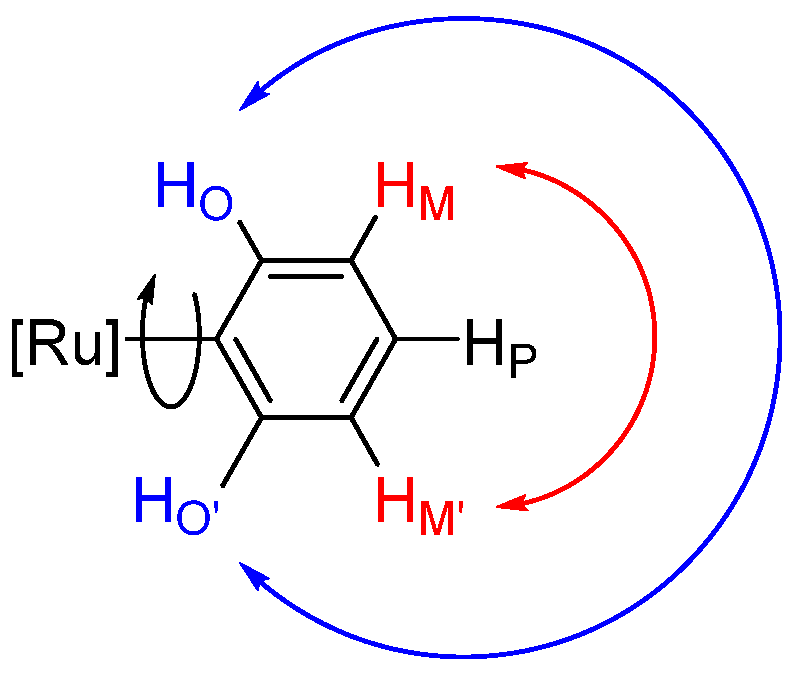
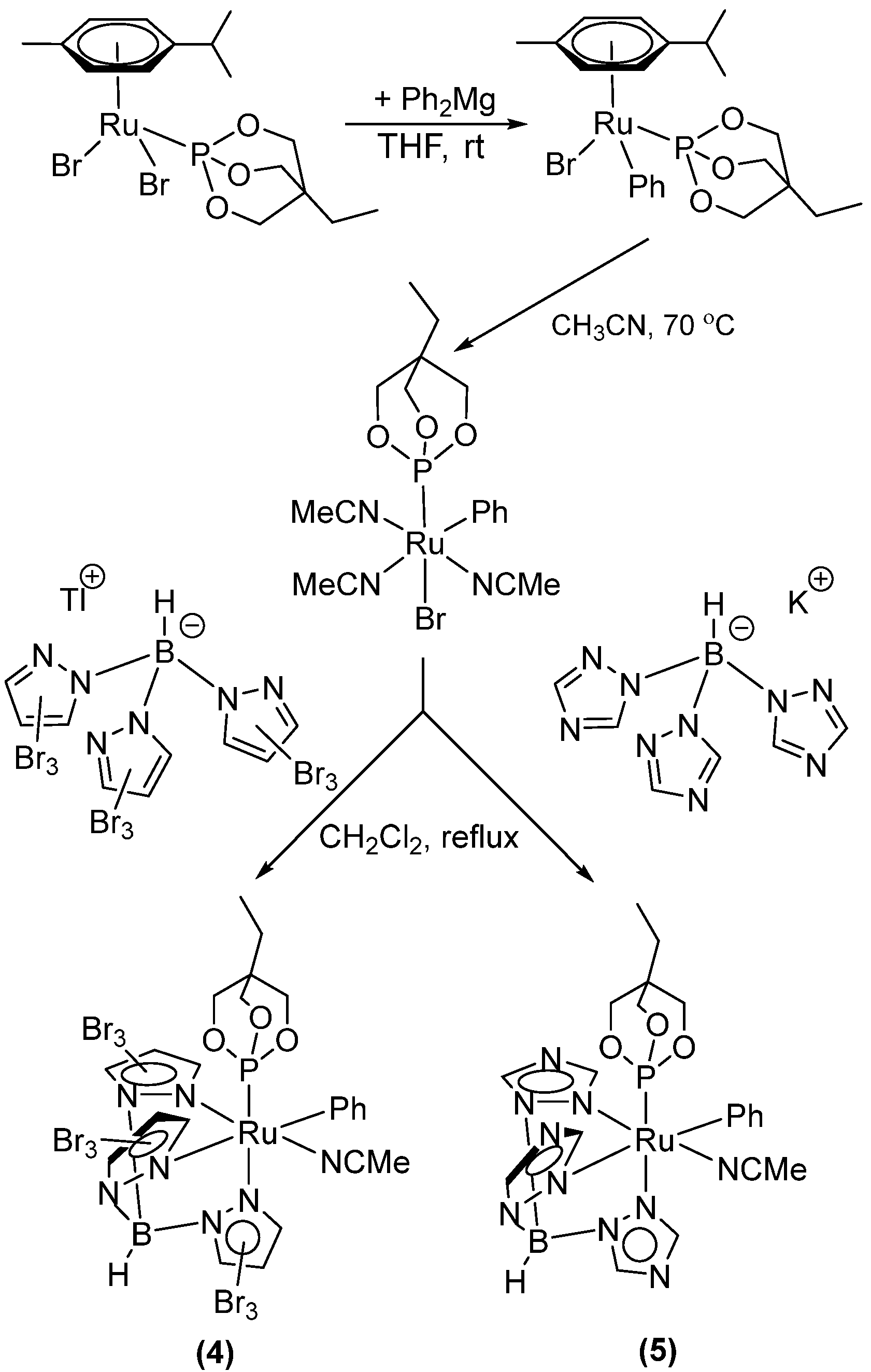
2.2. Ruthenium(II) Complexes Supported by TpBr3 and Ttz Ligands
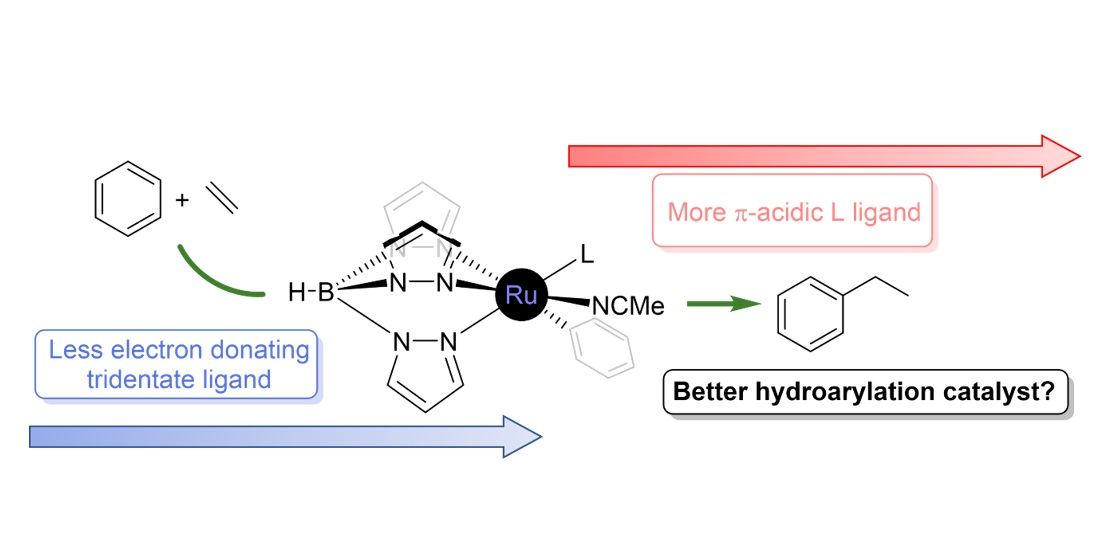
3. Summary and Conclusions
4. Experimental Section
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Garside, M. Guide to the Business of Chemistry; American Chemistry Council: Washington, DC, USA, 2019. [Google Scholar]
- Gerzeliev, I.M.; Khadzhiev, S.N.; Sakharova, I.E. Ethylbenzene synthesis and benzene transalkylation with diethylbenzenes on zeolite catalysts. Pet. Chem. 2011, 51, 39–48. [Google Scholar] [CrossRef]
- Čejka, J.; Wichterlová, B. Acid-Catalyzed Synthesis of Mono- and Dialkyl Benzenes over Zeolites: Active Sites, Zeolite Topology, and Reaction Mechanisms. Catal. Rev. 2002, 44, 375–421. [Google Scholar] [CrossRef]
- Perego, C.; Pollesel, P. Advances in Aromatics Processing Using Zeolite Catalysts. Adv. Nanoporous Mater. 2010, 1, 97–149. [Google Scholar]
- Perego, C.; Ingallina, P. Combining alkylation and transalkylation for alkylaromatic production. Green Chem. 2004, 6, 274–279. [Google Scholar] [CrossRef]
- Perego, C.; Ingallina, P. Recent advances in the industrial alkylation of aromatics: New catalysts and new processes. Catal. Today 2002, 73, 3–22. [Google Scholar] [CrossRef]
- Saper, N.I.; Ohgi, A.; Small, D.W.; Semba, K.; Nakao, Y.; Hartwig, J.F. Nickel-catalysed anti-Markovnikov hydroarylation of unactivated alkenes with unactivated arenes facilitated by non-covalent interactions. Nat. Chem. 2020, 12, 276–283. [Google Scholar] [CrossRef]
- Zhu, W.; Gunnoe, T.B. Advances in Rhodium-Catalyzed Oxidative Arene Alkenylation. Acc. Chem. Res. 2020, 53, 920–936. [Google Scholar] [CrossRef]
- Zhu, W.; Gunnoe, T.B. Advances in Group 10 Transition-Metal-Catalyzed Arene Alkylation and Alkenylation. J. Am. Chem. Soc. 2021, 143, 6746–6766. [Google Scholar] [CrossRef]
- Gunnoe, T.B.; Schinski, W.L.; Jia, X.; Zhu, W. Transition-Metal-Catalyzed Arene Alkylation and Alkenylation: Catalytic Processes for the Generation of Chemical Intermediates. ACS Catal. 2020, 10, 14080–14092. [Google Scholar] [CrossRef]
- Bhalla, G.; Oxgaard, J.; Goddard, W.A.; Periana, R.A. Anti-Markovnikov Hydroarylation of Unactivated Olefins Catalyzed by a Bis-tropolonato Iridium(III) Organometallic Complex. Organometallics 2005, 24, 3229–3232. [Google Scholar] [CrossRef]
- Periana, R.A.; Liu, X.Y.; Bhalla, G. Novel bis-acac-O,O-Ir(III) catalyst for anti-Markovnikov, hydroarylation of olefins operates by arene CH activation. Chem. Commun. 2002, 24, 3000–3001. [Google Scholar] [CrossRef] [PubMed]
- McKeown, B.A.; Foley, N.A.; Lee, J.P.; Gunnoe, T.B. Hydroarylation of Unactivated Olefins Catalyzed by Platinum(II) Complexes. Organometallics 2008, 27, 4031–4033. [Google Scholar] [CrossRef]
- McKeown, B.A.; Gonzalez, H.E.; Friedfeld, M.R.; Brosnahan, A.M.; Gunnoe, T.B.; Cundari, T.R.; Sabat, M. Platinum(II)-Catalyzed Ethylene Hydrophenylation: Switching Selectivity between Alkyl- and Vinylbenzene Production. Organometallics 2013, 32, 2857–2865. [Google Scholar] [CrossRef]
- McKeown, B.A.; Gonzalez, H.E.; Friedfeld, M.R.; Gunnoe, T.B.; Cundari, T.R.; Sabat, M. Mechanistic studies of ethylene hydrophenylation catalyzed by bipyridyl Pt(II) complexes. J. Am. Chem. Soc. 2011, 133, 19131–19152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKeown, B.A.; Gonzalez, H.E.; Gunnoe, T.B.; Cundari, T.R.; Sabat, M. PtII-Catalyzed Ethylene Hydrophenylation: Influence of Dipyridyl Chelate Ring Size on Catalyst Activity and Longevity. ACS Catal. 2013, 3, 1165–1171. [Google Scholar] [CrossRef]
- McKeown, B.A.; Gonzalez, H.E.; Michaelos, T.; Gunnoe, T.B.; Cundari, T.R.; Crabtree, R.H.; Sabat, M. Control of Olefin Hydroarylation Catalysis via a Sterically and Electronically Flexible Platinum(II) Catalyst Scaffold. Organometallics 2013, 32, 3903–3913. [Google Scholar] [CrossRef]
- Clement, M.L.; Grice, K.A.; Luedtke, A.T.; Kaminsky, W.; Goldberg, K.I. Platinum(II) olefin hydroarylation catalysts: Tuning selectivity for the anti-Markovnikov product. Chem. Eur. J 2014, 20, 17287–17291. [Google Scholar] [CrossRef]
- Luedtke, A.T.; Goldberg, K.I. Intermolecular hydroarylation of unactivated olefins catalyzed by homogeneous platinum complexes. Angew. Chem. Int. Ed. Engl. 2008, 47, 7694–7696. [Google Scholar] [CrossRef]
- Andreatta, J.R.; McKeown, B.A.; Gunnoe, T.B. Transition metal catalyzed hydroarylation of olefins using unactivated substrates: Recent developments and challenges. J. Organomet. Chem. 2011, 696, 305–315. [Google Scholar] [CrossRef]
- Burgess, S.A.; Joslin, E.E.; Gunnoe, T.B.; Cundari, T.R.; Sabat, M.; Myers, W.H. Hydrophenylation of ethylene using a cationic Ru(II) catalyst: Comparison to a neutral Ru(ii) catalyst. Chem. Sci. 2014, 5, 4355–4366. [Google Scholar] [CrossRef]
- Foley, N.A.; Ke, Z.; Gunnoe, T.B.; Cundari, T.R.; Petersen, J.L. Aromatic C−H Activation and Catalytic Hydrophenylation of Ethylene by TpRu{P(OCH2)3CEt}(NCMe)Ph. Organometallics 2008, 27, 3007–3017. [Google Scholar] [CrossRef] [Green Version]
- Foley, N.A.; Lail, M.; Gunnoe, T.B.; Cundari, T.R.; Boyle, P.D.; Petersen, J.L. Combined Experimental and Computational Study of TpRu{P(pyr)3}(NCMe)Me (pyr =N-pyrrolyl): Inter- and Intramolecular Activation of C−H Bonds and the Impact of Sterics on Catalytic Hydroarylation of Olefins. Organometallics 2007, 26, 5507–5516. [Google Scholar] [CrossRef] [Green Version]
- Foley, N.A.; Lail, M.; Lee, J.P.; Gunnoe, T.B.; Cundari, T.R.; Petersen, J.L. Comparative reactivity of TpRu(L)(NCMe)Ph (L = CO or PMe3): Impact of ancillary ligand l on activation of carbon-hydrogen bonds including catalytic hydroarylation and hydrovinylation/oligomerization of ethylene. J. Am. Chem. Soc. 2007, 129, 6765–6781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foley, N.A.; Lee, J.P.; Ke, Z.; Gunnoe, T.B.; Cundari, T.R. Ru(II) catalysts supported by hydridotris(pyrazolyl)borate for the hydroarylation of olefins: Reaction scope, mechanistic studies, and guides for the development of improved catalysts. Acc. Chem. Res. 2009, 42, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Joslin, E.E.; McMullin, C.L.; Gunnoe, T.B.; Cundari, T.R.; Sabat, M.; Myers, W.H. Catalytic Hydroarylation of Ethylene Using TpRu(L)(NCMe)Ph (L = 2,6,7-Trioxa-1-phosphabicyclo [2,2,1]heptane): Comparison to TpRu(L′)(NCMe)Ph Systems (L′ = CO, PMe3, P(pyr)3, or P(OCH2)3CEt). Organometallics 2012, 31, 6851–6860. [Google Scholar] [CrossRef]
- Lail, M.; Arrowood, B.N.; Gunnoe, T.B. Addition of arenes to ethylene and propene catalyzed by ruthenium. J. Am. Chem. Soc. 2003, 125, 7506–7507. [Google Scholar] [CrossRef] [PubMed]
- Lail, M.; Bell, C.M.; Conner, D.; Cundari, T.R.; Gunnoe, T.B.; Petersen, J.L. Experimental and Computational Studies of Ruthenium(II)-Catalyzed Addition of Arene C−H Bonds to Olefins. Organometallics 2004, 23, 5007–5020. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, T.; Periana, R.A.; Taube, D.J.; Yoshida, H. Direct Synthesis of Styrene by Rhodium-Catalyzed Oxidative Arylation of Ethylene with Benzene. J. Catal. 2002, 206, 272–280. [Google Scholar] [CrossRef]
- Taube, D.; Periana, R.; Matsumoto, T. Oxidative Coupling of Olefins and Aromatics Using a Rhodium Catalyst and a Copper (Ii) Redox Agent. Google Patents US6127590A, 3 October 2000. [Google Scholar]
- Vaughan, B.A.; Webster-Gardiner, M.S.; Cundari, T.R.; Gunnoe, T.B. Organic chemistry. A rhodium catalyst for single-step styrene production from benzene and ethylene. Science 2015, 348, 421–424. [Google Scholar] [CrossRef] [Green Version]
- Vaughan, B.A.; Khani, S.K.; Gary, J.B.; Kammert, J.D.; Webster-Gardiner, M.S.; McKeown, B.A.; Davis, R.J.; Cundari, T.R.; Gunnoe, T.B. Mechanistic Studies of Single-Step Styrene Production Using a Rhodium(I) Catalyst. J. Am. Chem. Soc. 2017, 139, 1485–1498. [Google Scholar] [CrossRef]
- Zhu, W.; Luo, Z.; Chen, J.; Liu, C.; Yang, L.; Dickie, D.A.; Liu, N.; Zhang, S.; Davis, R.J.; Gunnoe, T.B. Mechanistic Studies of Single-Step Styrene Production Catalyzed by Rh Complexes with Diimine Ligands: An Evaluation of the Role of Ligands and Induction Period. ACS Catal. 2019, 9, 7457–7475. [Google Scholar] [CrossRef]
- Liebov, N.S.; Zhu, W.; Chen, J.; Webster-Gardiner, M.S.; Schinski, W.L.; Gunnoe, T.B. Rhodium-Catalyzed Alkenylation of Toluene Using 1-Pentene: Regioselectivity to Generate Precursors for Bicyclic Compounds. Organometallics 2019, 38, 3860–3870. [Google Scholar] [CrossRef]
- Jia, X.; Frye, L.I.; Zhu, W.; Gu, S.; Gunnoe, T.B. Synthesis of Stilbenes by Rhodium-Catalyzed Aerobic Alkenylation of Arenes via C–H Activation. J. Am. Chem. Soc. 2020, 142, 10534–10543. [Google Scholar] [CrossRef]
- Zhu, W.; Gunnoe, T.B. Rhodium-Catalyzed Arene Alkenylation Using Only Dioxygen as the Oxidant. ACS Catal. 2020, 10, 11519–11531. [Google Scholar] [CrossRef]
- Webster-Gardiner, M.S.; Chen, J.; Vaughan, B.A.; McKeown, B.A.; Schinski, W.; Gunnoe, T.B. Catalytic Synthesis of “Super” Linear Alkenyl Arenes Using an Easily Prepared Rh(I) Catalyst. J. Am. Chem. Soc. 2017, 139, 5474–5480. [Google Scholar] [CrossRef]
- Chen, J.; Nielsen, R.J.; Goddard, W.A., 3rd; McKeown, B.A.; Dickie, D.A.; Gunnoe, T.B. Catalytic Synthesis of Superlinear Alkenyl Arenes Using a Rh(I) Catalyst Supported by a “Capping Arene” Ligand: Access to Aerobic Catalysis. J. Am. Chem. Soc. 2018, 140, 17007–17018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Z.; Whitcomb, C.A.; Kaylor, N.; Zhang, Y.; Zhang, S.; Davis, R.J.; Gunnoe, T.B. Oxidative Alkenylation of Arenes Using Supported Rh Materials: Evidence that Active Catalysts are Formed by Rh Leaching. ChemCatChem 2021, 13, 260–270. [Google Scholar] [CrossRef]
- Jia, X.; Gary, J.B.; Gu, S.; Cundari, T.R.; Gunnoe, T.B. Oxidative Hydrophenylation of Ethylene Using a Cationic Ru(II) Catalyst: Styrene Production with Ethylene as the Oxidant. Isr. J. Chem. 2017, 57, 1037–1046. [Google Scholar] [CrossRef]
- Weissman, H.; Song, X.; Milstein, D. Ru-catalyzed oxidative coupling of arenes with olefins using O2. J. Am. Chem. Soc. 2001, 123, 337–338. [Google Scholar] [CrossRef]
- Matsumoto, T.; Yoshida, H. Oxidative Arylation of Ethylene with Benzene to Produce Styrene. Chem. Lett. 2000, 29, 1064–1065. [Google Scholar] [CrossRef]
- Jia, X.; Foley, A.M.; Liu, C.; Vaughan, B.A.; McKeown, B.A.; Zhang, S.; Gunnoe, T.B. Styrene Production from Benzene and Ethylene Catalyzed by Palladium(II): Enhancement of Selectivity toward Styrene via Temperature-dependent Vinyl Ester Consumption. Organometallics 2019, 38, 3532–3541. [Google Scholar] [CrossRef]
- Bair, J.S.; Schramm, Y.; Sergeev, A.G.; Clot, E.; Eisenstein, O.; Hartwig, J.F. Linear-selective hydroarylation of unactivated terminal and internal olefins with trifluoromethyl-substituted arenes. J. Am. Chem. Soc. 2014, 136, 13098–13101. [Google Scholar] [CrossRef] [Green Version]
- Joslin, E.E.; Quillian, B.; Gunnoe, T.B.; Cundari, T.R.; Sabat, M.; Myers, W.H. C-H activation of pyrazolyl ligands by Ru(II). Inorg. Chem. 2014, 53, 6270–6279. [Google Scholar] [CrossRef] [PubMed]
- Ozkal, E.; Llanes, P.; Bravo, F.; Ferrali, A.; Pericàs, M.A. Fine-Tunable Tris(triazolyl)methane Ligands for Copper(I)- Catalyzed Azide-Alkyne Cycloaddition Reactions. Adv. Synth. Catal. 2014, 356, 857–869. [Google Scholar] [CrossRef]
- Wang, D.; Etienne, L.; Echeverria, M.; Moya, S.; Astruc, D. A highly active and magnetically recoverable tris(triazolyl)-Cu(I) catalyst for alkyne-azide cycloaddition reactions. Chem. Eur. J. 2014, 20, 4047–4054. [Google Scholar] [CrossRef]
- Chen, H.; Hou, S.; Tan, Y. An ‘in-water’ halogen-ion compatible “click” catalyst for cucurbituril guest ligation. Supramol. Chem. 2016, 28, 801–809. [Google Scholar] [CrossRef]
- Arikawa, Y.; Yamaguchi, S.; Haige, R.; Oshiro, E.; Umakoshi, K.; Onishi, M. Methylation of a nitrosylruthenium complex bearing a hydridotris(pyrazolyl)borate ligand. J. Organomet. Chem. 2014, 755, 12–15. [Google Scholar] [CrossRef] [Green Version]
- Onishi, M. Syntheses and Spectral Properties of New Nitrosyl-[poly(1-pyrazolyl)borato]ruthenium Complexes. Bull. Chem. Soc. Jpn. 1991, 64, 3039–3045. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.; Bergman, R.G. Reductive elimination leading to transient (. eta. 5-C5Me5) Ru (NO). Trapping, dimerization and oxidative addition reactions of this intermediate and the effect of structural variation on its generation. J. Am. Chem. Soc. 1987, 109, 4298–4304. [Google Scholar] [CrossRef]
- Tellers, D.M.; Skoog, S.J.; Bergman, R.G.; Gunnoe, T.B.; Harman, W.D. Comparison of the Relative Electron-Donating Abilities of Hydridotris(pyrazolyl)borate and Cyclopentadienyl Ligands: Different Interactions with Different Transition Metals. Organometallics 2000, 19, 2428–2432. [Google Scholar] [CrossRef]
- Bergman, R.G.; Cundari, T.R.; Gillespie, A.M.; Gunnoe, T.B.; Harman, W.D.; Klinckman, T.R.; Temple, M.D.; White, D.P. Computational Study of Methane Activation by TpRe(CO)2 and CpRe(CO)2 with a Stereoelectronic Comparison of Cyclopentadienyl and Scorpionate Ligands. Organometallics 2003, 22, 2331–2337. [Google Scholar] [CrossRef]
- Heyer, A.J.; Shivokevich, P.J.; Hooe, S.L.; Welch, K.D.; Harman, W.D.; Machan, C.W. Reversible modulation of the redox characteristics of acid-sensitive molybdenum and tungsten scorpionate complexes. Dalton Trans. 2018, 47, 6323–6332. [Google Scholar] [CrossRef] [PubMed]
- Bongiovanni, J.L.; Rowe, B.W.; Fadden, P.T.; Taylor, M.T.; Wells, K.R.; Kumar, M.; Papish, E.T.; Yap, G.P.A.; Zeller, M. Synthesis, structural studies and solubility properties of zinc(II), nickel(II) and copper(II) complexes of bulky tris(triazolyl)borate ligands. Inorg. Chim. Acta 2010, 363, 2163–2170. [Google Scholar] [CrossRef]
- Gardner, S.R.; Papish, E.T.; Monillas, W.H.; Yap, G.P.A. Tris(triazolyl)borate ligands of intermediate steric bulk for the synthesis of biomimetic structures with hydrogen bonding and solubility in hydrophilic solvents. J. Inorg. Biochem. 2008, 102, 2179–2183. [Google Scholar] [CrossRef] [PubMed]
- Jernigan, F.E.; Sieracki, N.A.; Taylor, M.T.; Jenkins, A.S.; Engel, S.E.; Rowe, B.W.; Jové, F.A.; Yap, G.P.A.; Papish, E.T.; Ferrence, G.M. Sterically Bulky Tris(triazolyl)borate Ligands as Water-Soluble Analogues of Tris(pyrazolyl)borate. Inorg. Chem. 2007, 46, 360–362. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Papish, E.T.; Zeller, M.; Hunter, A.D. Zinc complexes of TtzR, Me with O and S donors reveal differences between Tp and Ttz ligands: Acid stability and binding to H or an additional metal (TtzR,Me = tris(3-R-5-methyl-1,2,4-triazolyl)borate; R = Ph, tBu). Dalton Trans. 2011, 40, 7517–7533. [Google Scholar] [CrossRef]
- Papish, E.T.; Donahue, T.M.; Wells, K.R.; Yap, G.P.A. How are tris(triazolyl)borate ligands electronically different from tris(pyrazolyl)borate ligands? A study of (TtztBu,Me)CuCO [TtztBu,Me = tris(3-t-butyl-5-methyl-1,2,4-triazolyl)borate]. Dalton Trans. 2008, 22, 2923–2925. [Google Scholar] [CrossRef]
- Dixon, N.A.; McQuarters, A.B.; Kraus, J.S.; Soffer, J.B.; Lehnert, N.; Schweitzer-Stenner, R.; Papish, E.T. Dramatic tuning of ligand donor properties in (Ttz)CuCO through remote binding of H+ (Ttz = hydrotris(triazolyl)borate). Chem. Commun. 2013, 49, 5571–5573. [Google Scholar] [CrossRef]
- Yakelis, N.A.; Bergman, R.G. Safe Preparation and Purification of Sodium Tetrakis [(3, 5-trifluoromethyl) phenyl] borate (NaBArF24): Reliable and Sensitive Analysis of Water in Solutions of Fluorinated Tetraarylborates. Organometallics 2005, 24, 3579–3581. [Google Scholar] [CrossRef] [Green Version]
- Lühder, K.; Nehls, D.; Madeja, K. A Simple Method for the Preparation of Dialkyl-Magnesiumorgano or Diarylmagnesiumorgano Compounds. J. Fur Prakt. Chem. 1983, 325, 1027–1029. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
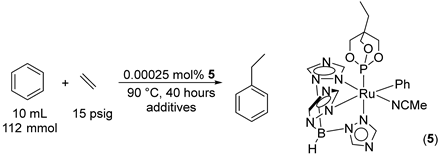
| Entry | Additives a | TON of Ethylbenzene |
|---|---|---|
| 1 | HNTf2 b | − |
| 2 | HBArF’4 | − |
| 3 | LiNTf2 | 120(6) |
| 4 | NaNTf2 | 91(3) |
| 5 | KNTf2 | 54(9) |
| 6 | LiBArF’4 | 147(12) |
| 7 | AlMe3 | − |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, X.; Tian, S.; Shivokevich, P.J.; Harman, W.D.; Dickie, D.A.; Gunnoe, T.B. Electron-Deficient Ru(II) Complexes as Catalyst Precursors for Ethylene Hydrophenylation. Inorganics 2022, 10, 76. https://doi.org/10.3390/inorganics10060076
Jia X, Tian S, Shivokevich PJ, Harman WD, Dickie DA, Gunnoe TB. Electron-Deficient Ru(II) Complexes as Catalyst Precursors for Ethylene Hydrophenylation. Inorganics. 2022; 10(6):76. https://doi.org/10.3390/inorganics10060076
Chicago/Turabian StyleJia, Xiaofan, Songyuan Tian, Philip J. Shivokevich, W. Dean Harman, Diane A. Dickie, and T. Brent Gunnoe. 2022. "Electron-Deficient Ru(II) Complexes as Catalyst Precursors for Ethylene Hydrophenylation" Inorganics 10, no. 6: 76. https://doi.org/10.3390/inorganics10060076
APA StyleJia, X., Tian, S., Shivokevich, P. J., Harman, W. D., Dickie, D. A., & Gunnoe, T. B. (2022). Electron-Deficient Ru(II) Complexes as Catalyst Precursors for Ethylene Hydrophenylation. Inorganics, 10(6), 76. https://doi.org/10.3390/inorganics10060076