
Mitochondrial De Novo Assembly of Iron–Sulfur Clusters in Mammals: Complex Matters in a Complex That Matters


Abstract
:1. Introduction
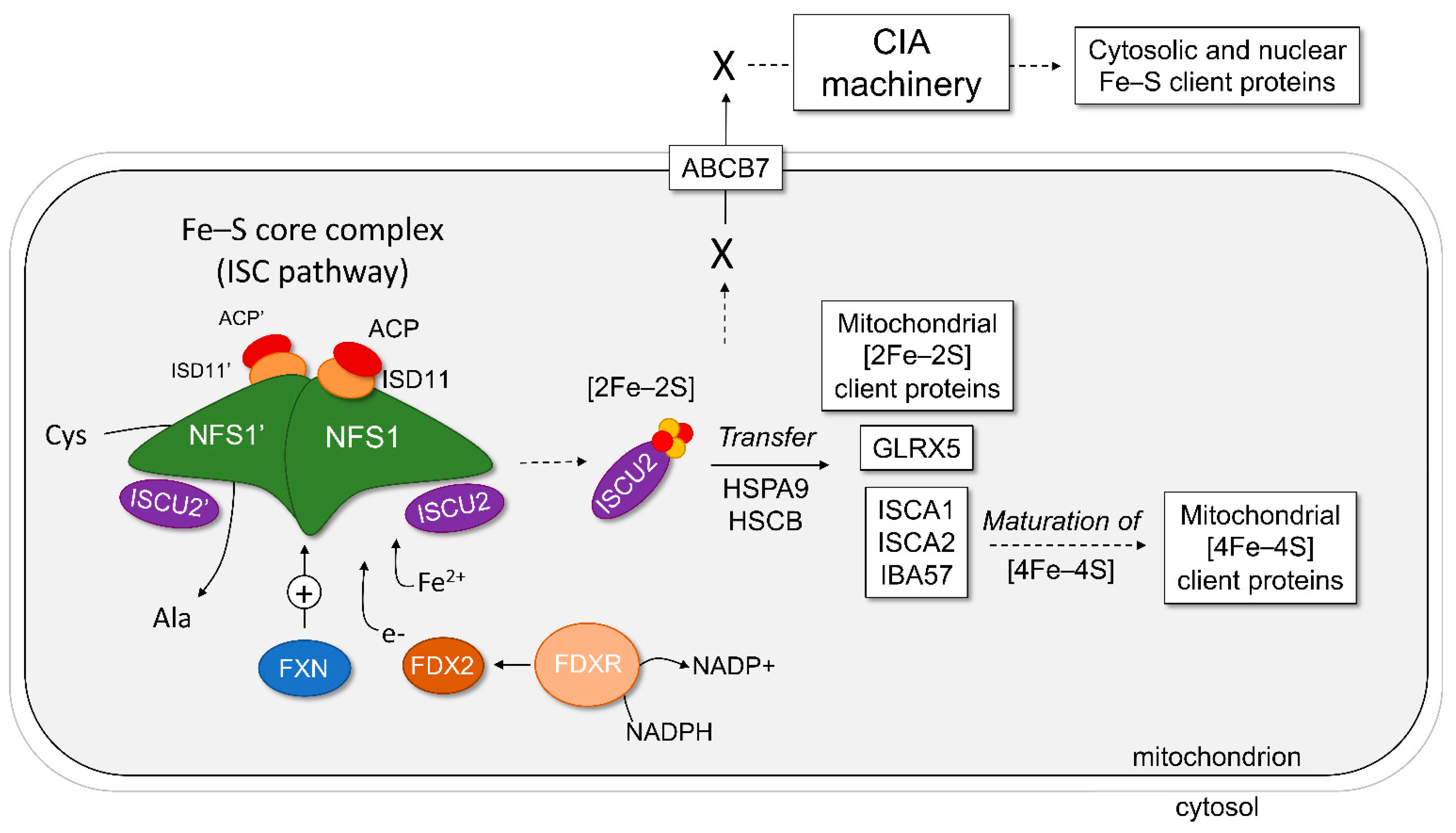
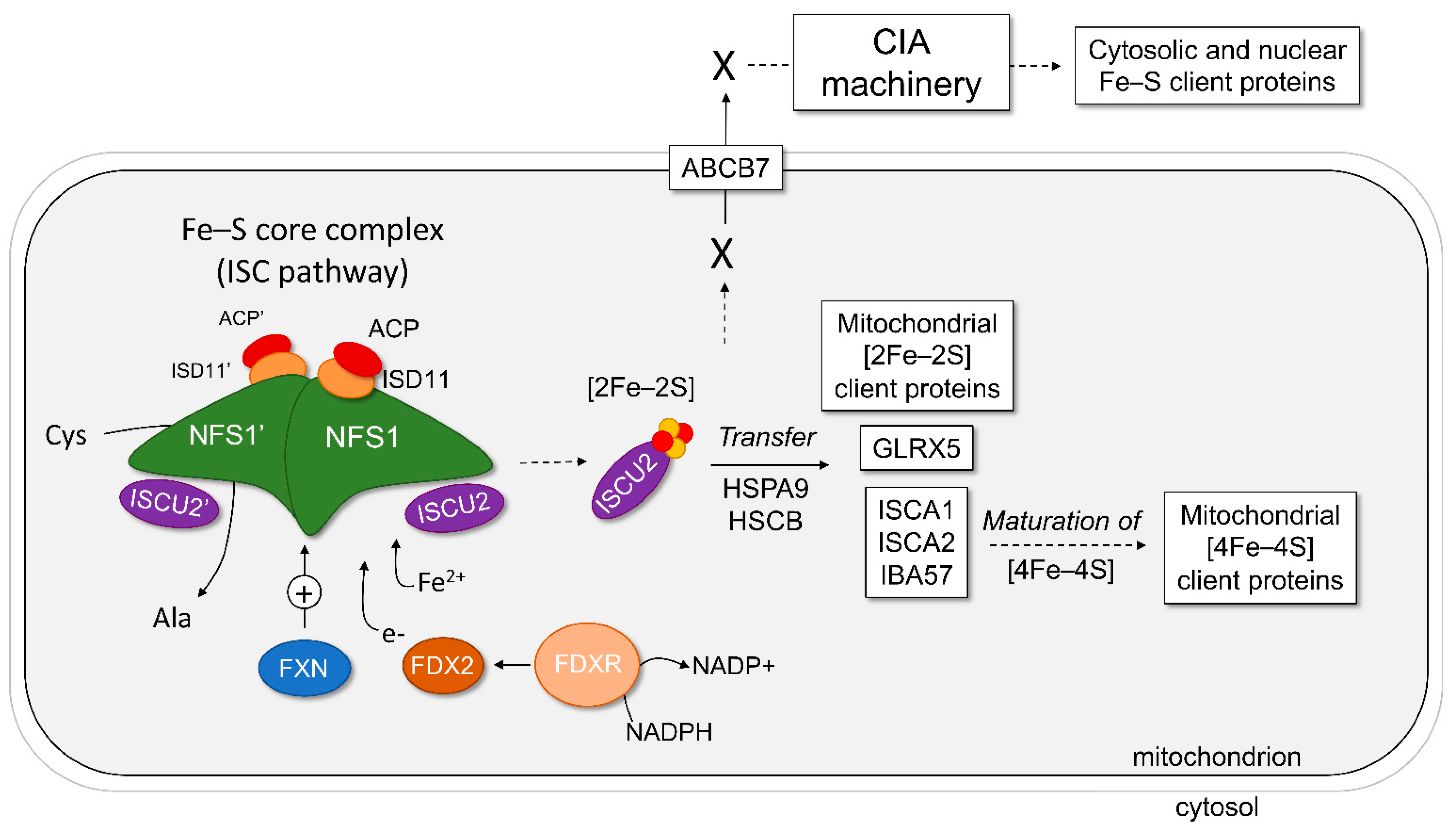
2. The Core Complex of the Mitochondrial ISC Pathway
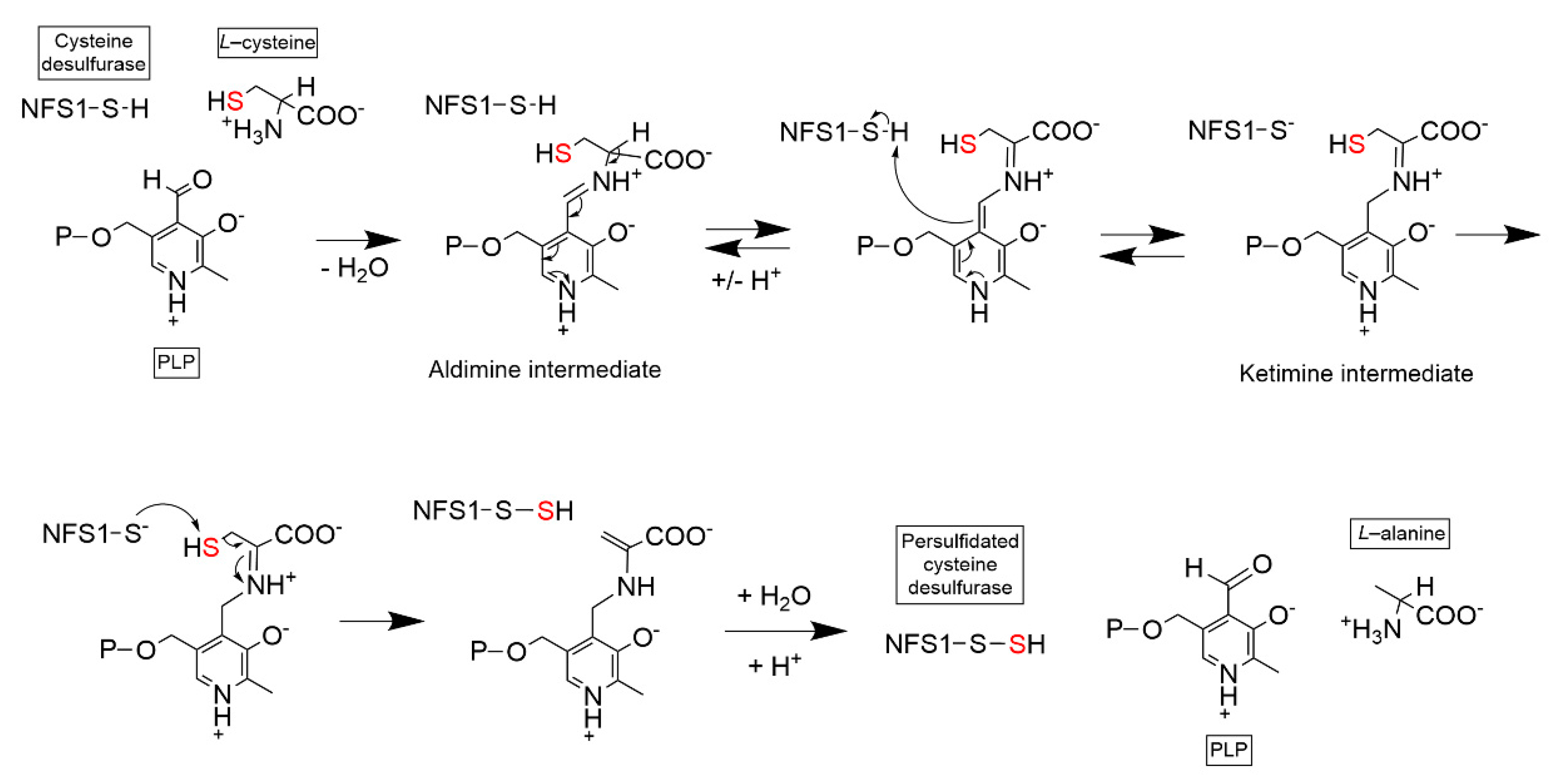
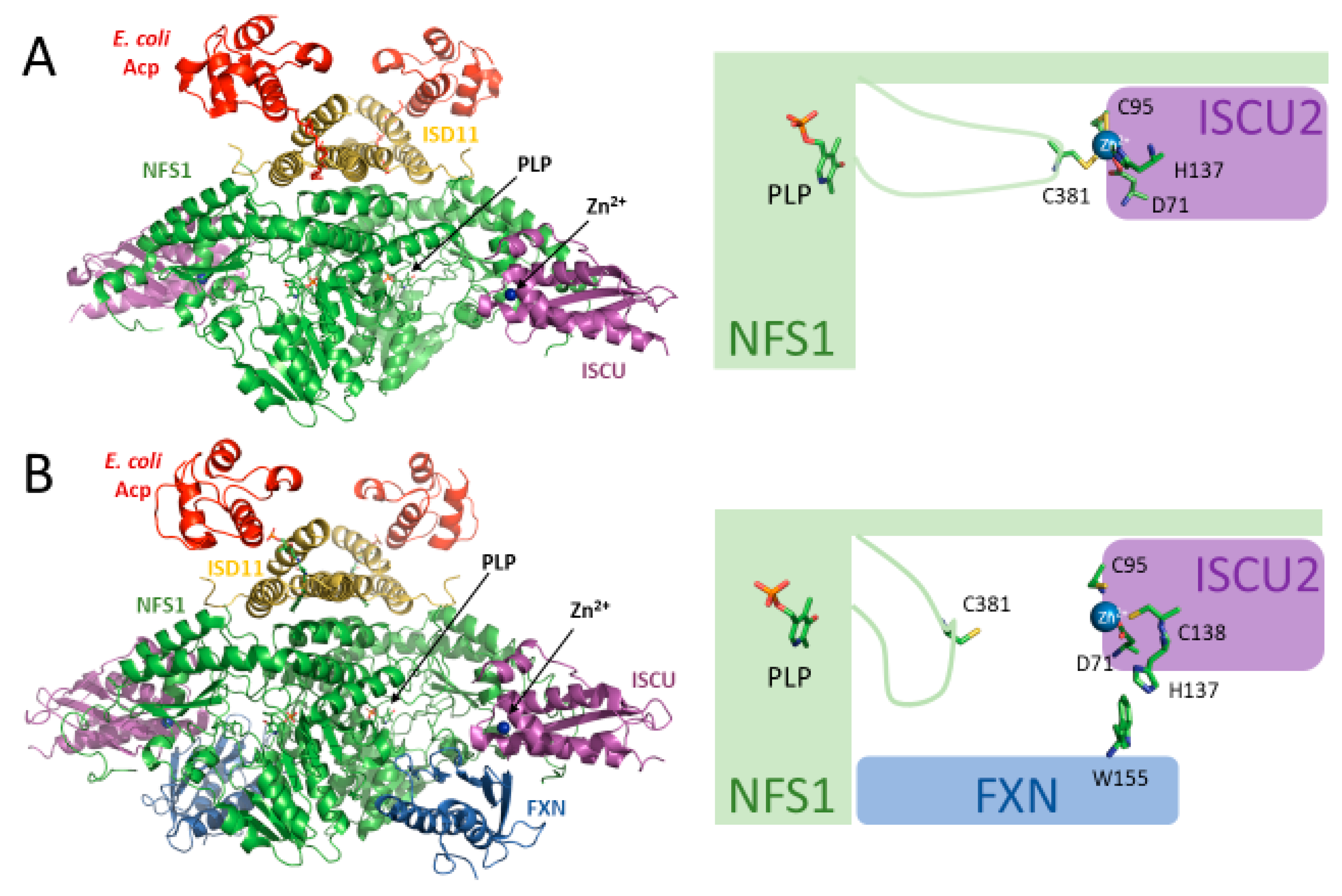
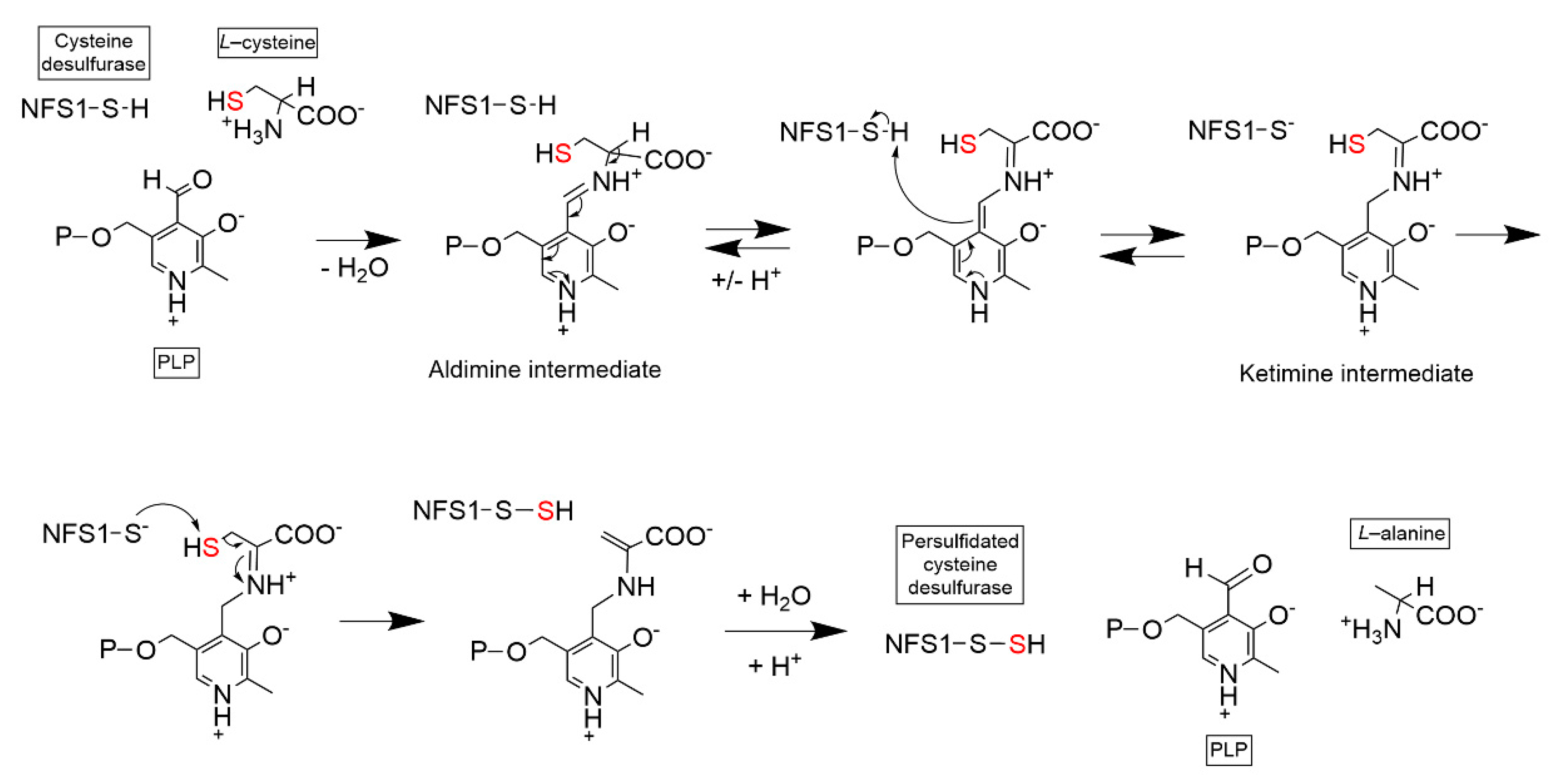
2.1. Cysteine Desulfurase (NFS1)
2.2. LYRM Accessory Protein (ISD11)
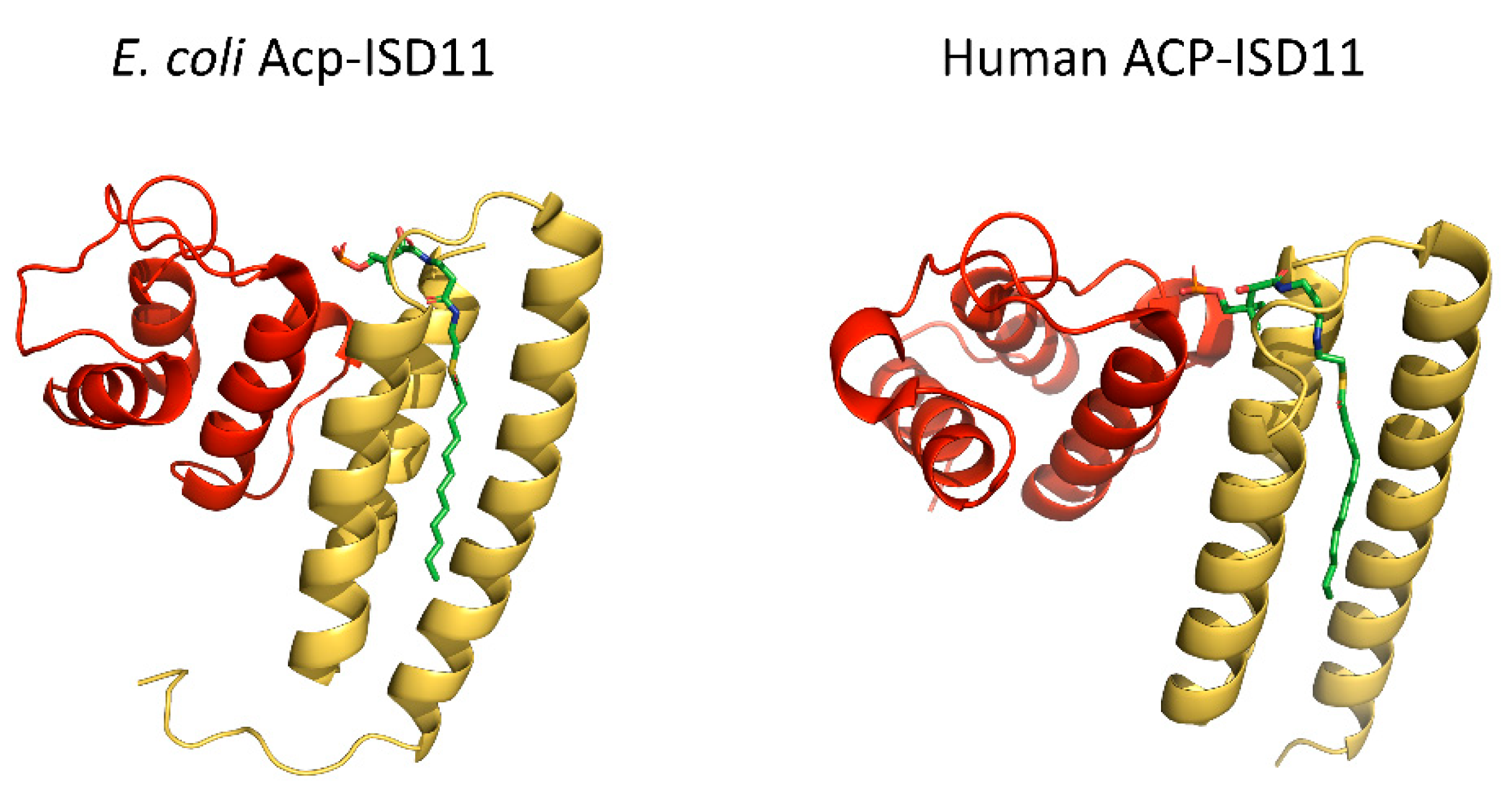
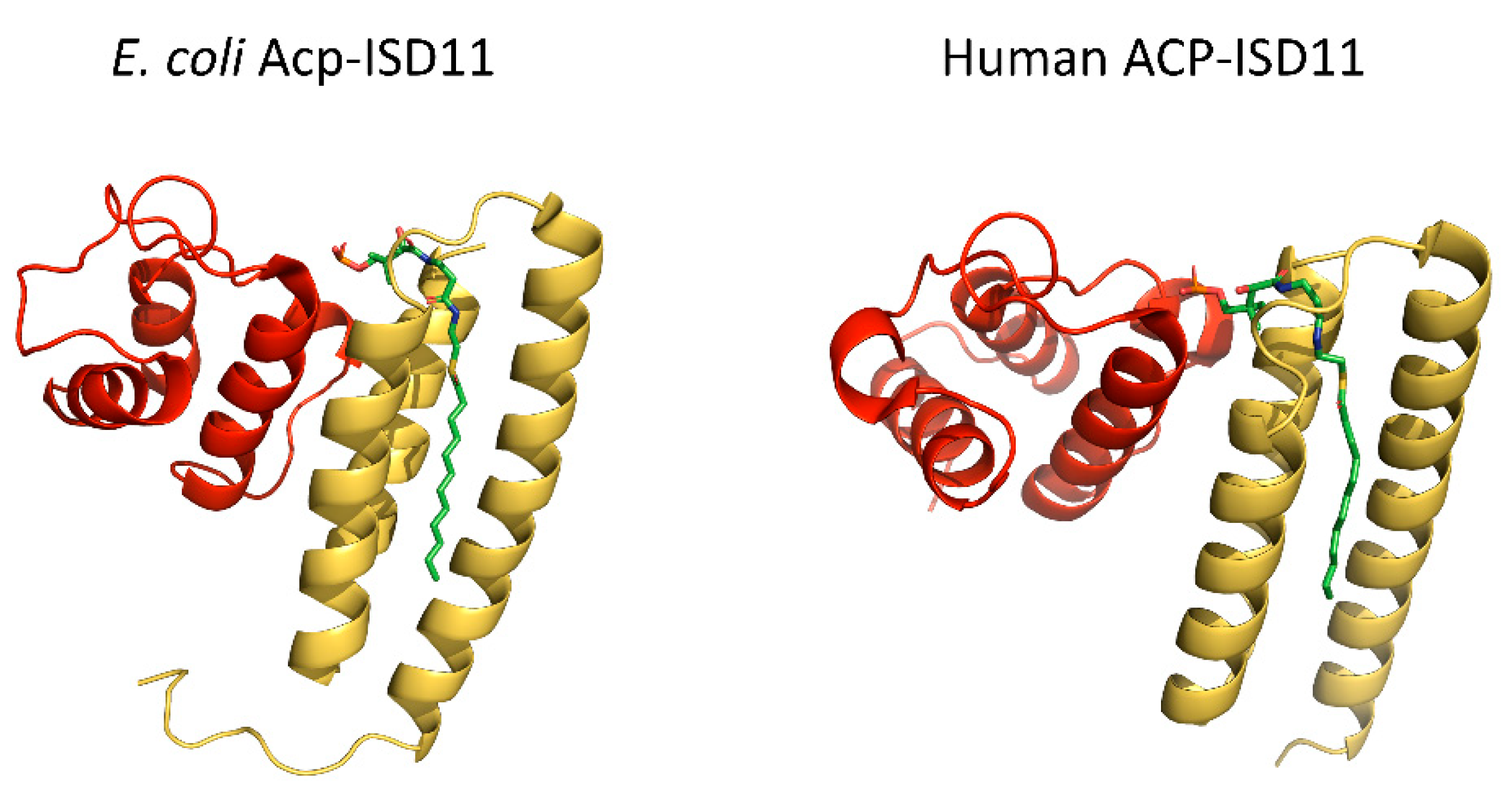
2.3. Acyl Carrier Protein (ACP)
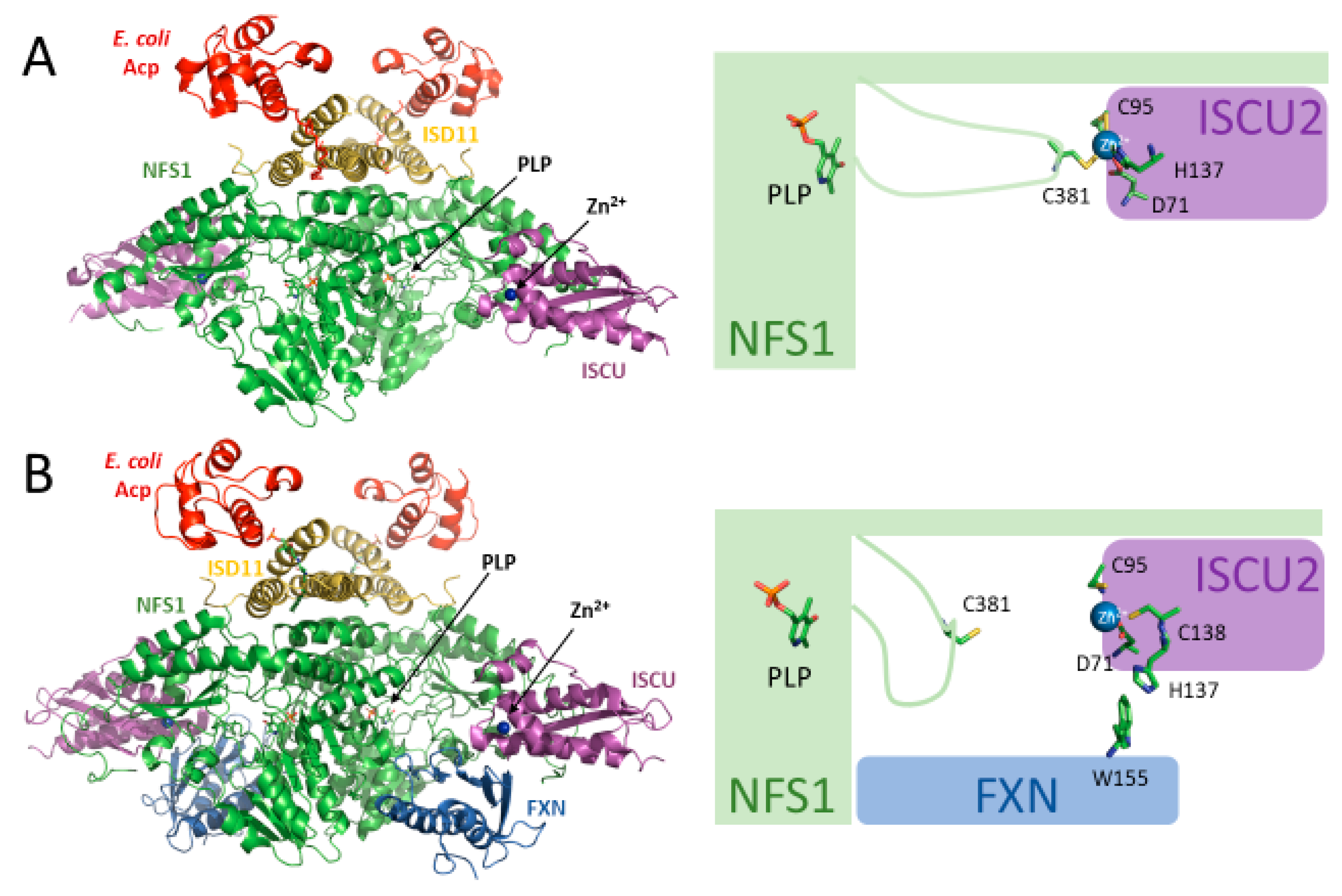
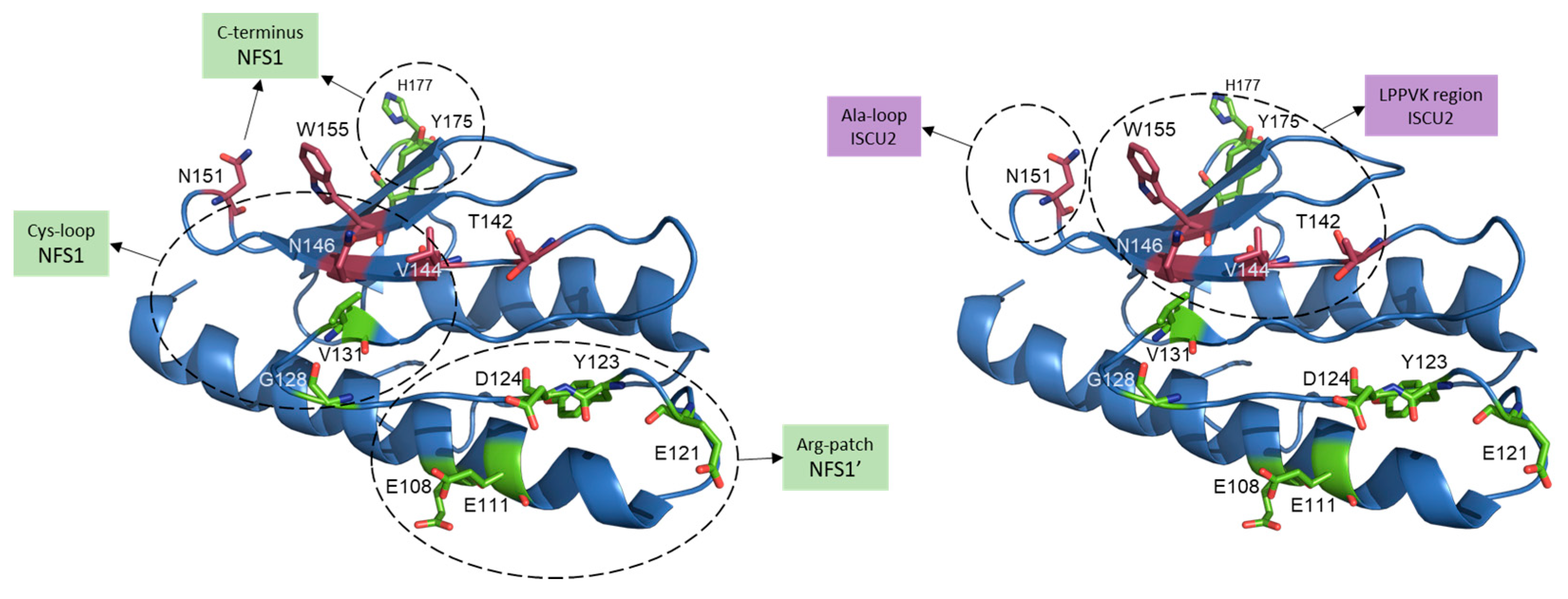
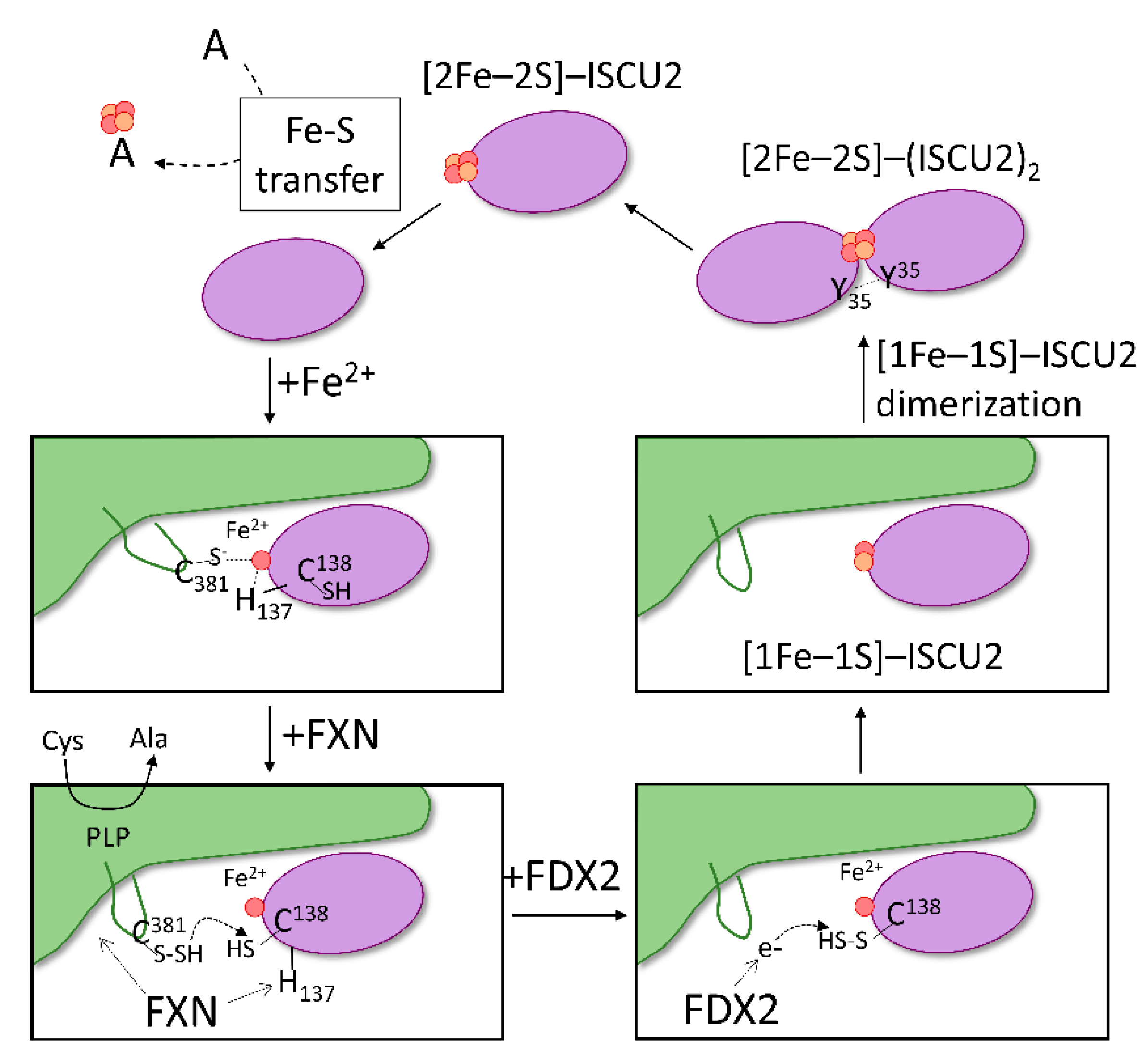
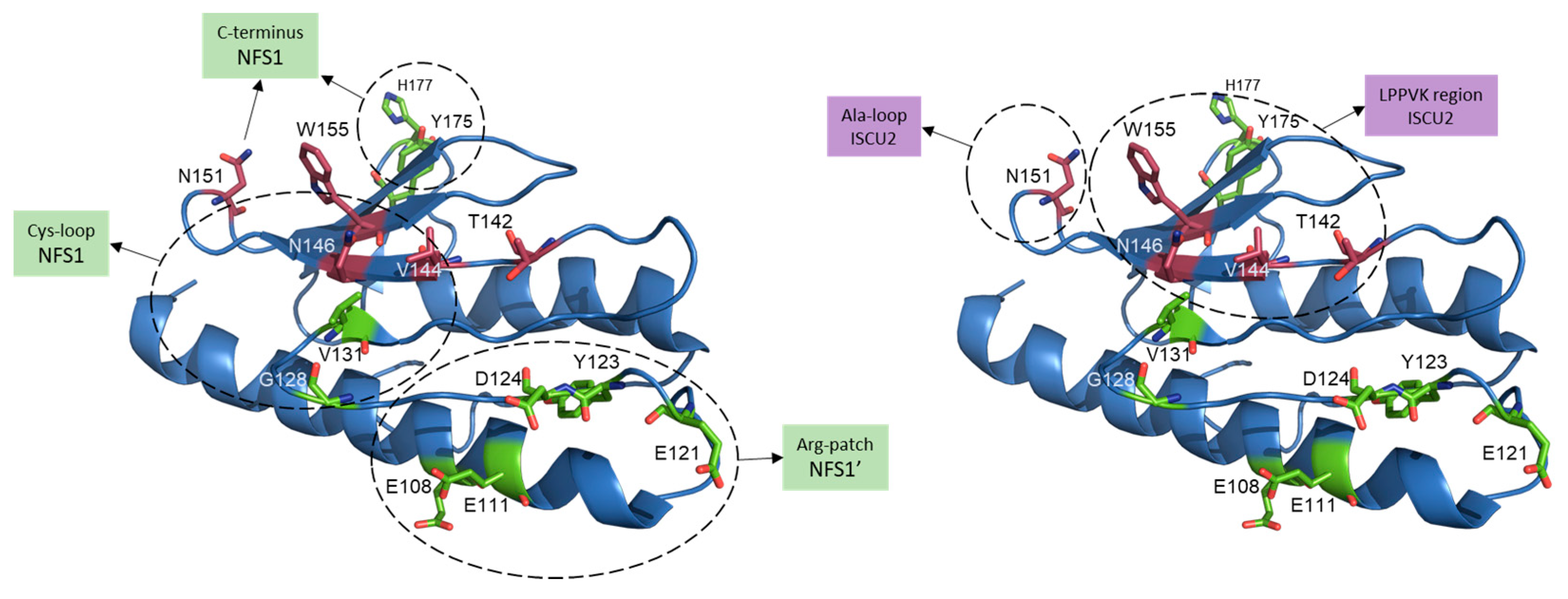
2.4. Scaffold Protein (ISCU2)
2.5. Frataxin (FXN)
2.6. Ferredoxin-Ferredoxin Reductase (FDX2-FDXR)
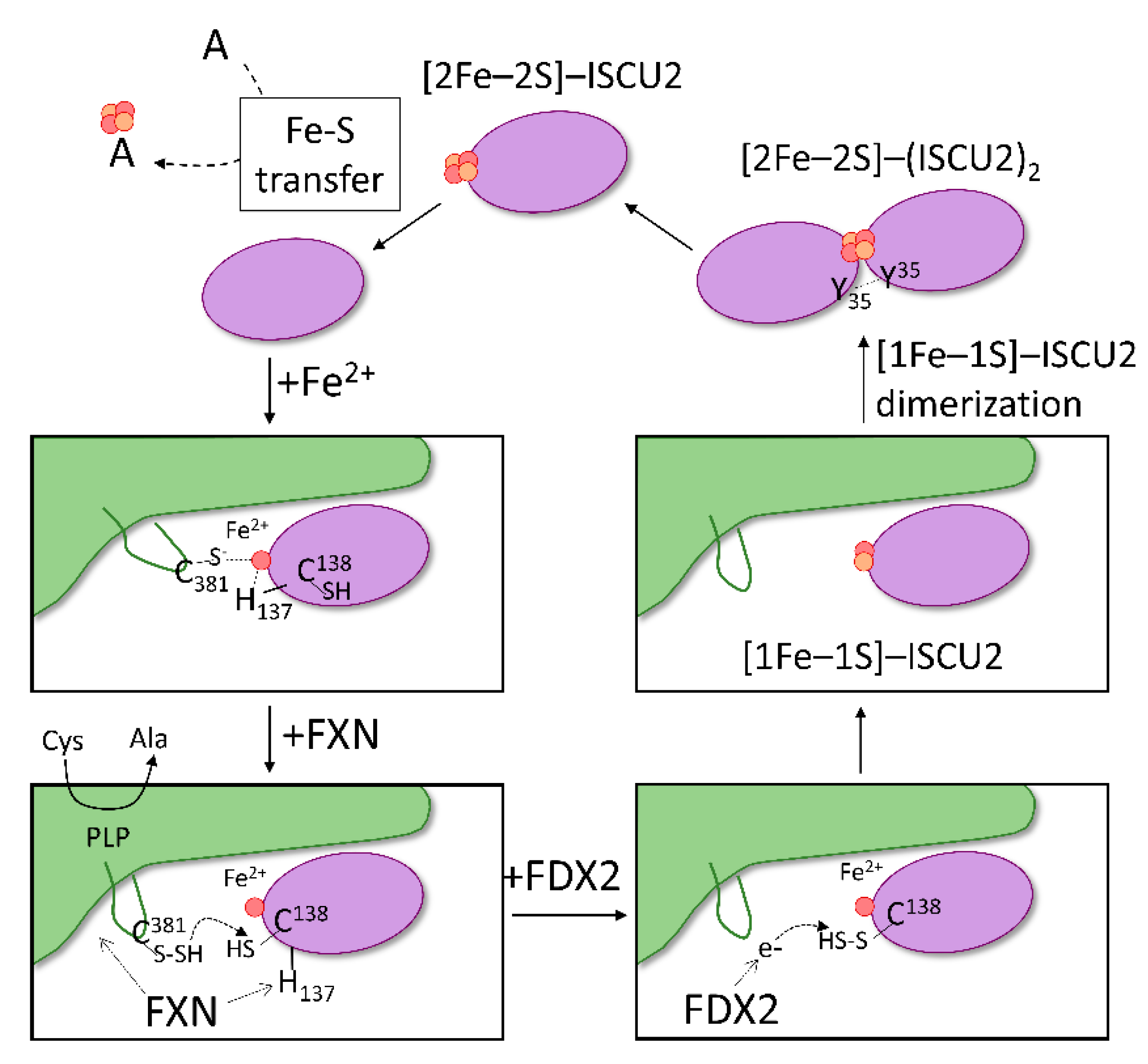
2.7. Model of Fe–S Assembly within the Core Complex
3. A Complex That Matters: Human Genetic Diseases Affecting the Mitochondrial Fe–S Core Complex
4. Further Reading on Fe–S
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Peters, J.W.; Broderick, J.B. Emerging paradigms for complex iron–sulfur cofactor assembly and insertion. Annu. Rev. Biochem. 2012, 81, 429–450. [Google Scholar] [CrossRef] [PubMed]
- Schonauer, M.S.; Kastaniotis, A.J.; Kursu, V.A.; Hiltunen, J.K.; Dieckmann, C.L. Lipoic acid synthesis and attachment in yeast mitochondria. J. Biol. Chem. 2009, 284, 23234–23242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowland, E.A.; Snowden, C.K.; Cristea, I.M. Protein lipoylation: An evolutionarily conserved metabolic regulator of health and disease. Curr. Opin. Chem. Biol. 2018, 42, 76–85. [Google Scholar] [CrossRef] [PubMed]
- McLain, A.L.; Szweda, P.A.; Szweda, L.I. α-Ketoglutarate dehydrogenase: A mitochondrial redox sensor. Free Radic. Res. 2011, 45, 29–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, L.; Tórtora, V.; Mansilla, S.; Radi, R. Aconitases: Non-redox Iron–Sulfur Proteins Sensitive to Reactive Species. Acc. Chem. Res. 2019, 52, 2609–2619. [Google Scholar] [CrossRef] [PubMed]
- Rouault, T.A.; Maio, N. Biogenesis and functions of mammalian Iron–sulfur proteins in the regulation of iron homeostasis and pivotal metabolic pathways. J. Biol. Chem. 2017, 292, 12744–12753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Vizarra, E.; Zeviani, M. Mitochondrial complex III Rieske Fe–S protein processing and assembly. Cell Cycle 2018, 17, 681–687. [Google Scholar] [CrossRef] [Green Version]
- Rudolf, J.; Makrantoni, V.; Ingledew, W.J.; Stark, M.J.; White, M.F. The DNA repair helicases XPD and FancJ have essential iron–sulfur domains. Mol. Cell 2006, 23, 801–808. [Google Scholar] [CrossRef]
- Netz, D.J.; Stith, C.M.; Stumpfig, M.; Kopf, G.; Vogel, D.; Genau, H.M.; Stodola, J.L.; Lill, R.; Burgers, P.M.; Pierik, A.J. Eukaryotic DNA polymerases require an iron-sulfur cluster for the formation of active complexes. Nat. Chem. Biol. 2011, 8, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Čavužić, M.; Liu, Y. Biosynthesis of Sulfur-Containing tRNA Modifications: A Comparison of Bacterial, Archaeal, and Eukaryotic Pathways. Biomolecules 2017, 7, 27. [Google Scholar] [CrossRef] [Green Version]
- Lill, R.; Dutkiewicz, R.; Freibert, S.A.; Heidenreich, T.; Mascarenhas, J.; Netz, D.J.; Paul, V.D.; Pierik, A.J.; Richter, N.; Stumpfig, M.; et al. The role of mitochondria and the CIA machinery in the maturation of cytosolic and nuclear iron-sulfur proteins. Eur. J. Cell Biol. 2015, 94, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Ciofi-Baffoni, S.; Nasta, V.; Banci, L. Protein networks in the maturation of human iron-sulfur proteins. Metallomics 2018, 10, 49–72. [Google Scholar] [CrossRef] [PubMed]
- Braymer, J.J.; Freibert, S.A.; Rakwalska-Bange, M.; Lill, R. Mechanistic concepts of iron-sulfur protein biogenesis in Biology. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118863. [Google Scholar] [CrossRef]
- Andreini, C.; Rosato, A.; Banci, L. The Relationship between Environmental Dioxygen and Iron-Sulfur Proteins Explored at the Genome Level. PLoS ONE 2017, 12, e0171279. [Google Scholar] [CrossRef] [PubMed]
- Lill, R.; Freibert, S.A. Mechanisms of Mitochondrial Iron-Sulfur Protein Biogenesis. Annu. Rev. Biochem. 2020, 89, 471–499. [Google Scholar] [CrossRef]
- Wiedemann, N.; Pfanner, N. Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem. 2017, 86, 685–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmucker, S.; Argentini, M.; Carelle-Calmels, N.; Martelli, A.; Puccio, H. The in vivo mitochondrial two-step maturation of human frataxin. Hum. Mol. Genet. 2008, 17, 3521–3531. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.; Nakamura, M. Functional Assignment of the ORF2-iscS-iscU-iscA-hscB-hscA-fdx-0RF3 Gene Cluster Involved in the Assembly of Fe–S Clusters in Escherichia coli. J. Biochem. 1999, 126, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Muhlenhoff, U.; Gerber, J.; Richhardt, N.; Lill, R. Components involved in assembly and dislocation of iron-sulfur clusters on the scaffold protein Isu1p. EMBO J. 2003, 22, 4815–4825. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; White, R.H.; Cash, V.L.; Jack, R.F.; Dean, D.R. Cysteine desulfurase activity indicates a role for NIFS in metallocluster biosynthesis. Proc. Natl. Acad. Sci. USA 1993, 90, 2754–2758. [Google Scholar] [CrossRef] [Green Version]
- Fox, N.G.; Yu, X.; Feng, X.; Bailey, H.J.; Martelli, A.; Nabhan, J.F.; Strain-Damerell, C.; Bulawa, C.; Yue, W.W.; Han, S. Structure of the human frataxin-bound iron-sulfur cluster assembly complex provides insight into its activation mechanism. Nat. Commun. 2019, 10, 2210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boniecki, M.T.; Freibert, S.A.; Muhlenhoff, U.; Lill, R.; Cygler, M. Structure and functional dynamics of the mitochondrial Fe/S cluster synthesis complex. Nat. Commun. 2017, 8, 1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freibert, S.A.; Boniecki, M.T.; Stumpfig, C.; Schulz, V.; Krapoth, N.; Winge, D.R.; Muhlenhoff, U.; Stehling, O.; Cygler, M.; Lill, R. N-terminal tyrosine of ISCU2 triggers [2Fe–2S] cluster synthesis by ISCU2 dimerization. Nat. Commun. 2021, 12, 6902. [Google Scholar] [CrossRef] [PubMed]
- Biederbick, A.; Stehling, O.; Rosser, R.; Niggemeyer, B.; Nakai, Y.; Elsasser, H.P.; Lill, R. Role of human mitochondrial Nfs1 in cytosolic iron-sulfur protein biogenesis and iron regulation. Mol. Cell Biol. 2006, 26, 5675–5687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cory, S.A.; Van Vranken, J.G.; Brignole, E.J.; Patra, S.; Winge, D.R.; Drennan, C.L.; Rutter, J.; Barondeau, D.P. Structure of human Fe–S assembly subcomplex reveals unexpected cysteine desulfurase architecture and acyl-ACP–ISD11 interactions. Proc. Natl. Acad. Sci. USA 2017, 114, E5325–E5334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.X.; Thompson, K.; Taylor, R.W.; Olahova, M. Mitochondrial OXPHOS Biogenesis: Co-Regulation of Protein Synthesis, Import, and Assembly Pathways. Int. J. Mol. Sci. 2020, 21, 3820. [Google Scholar] [CrossRef] [PubMed]
- Dibley, M.G.; Formosa, L.E.; Lyu, B.; Reljic, B.; McGann, D.; Muellner-Wong, L.; Kraus, F.; Sharpe, A.J.; Stroud, D.A.; Ryan, M.T. The Mitochondrial Acyl-carrier Protein Interaction Network Highlights Important Roles for LYRM Family Members in Complex I and Mitoribosome Assembly. Mol. Cell Proteom. 2020, 19, 65–77. [Google Scholar] [CrossRef]
- Adam, A.C.; Bornhovd, C.; Prokisch, H.; Neupert, W.; Hell, K. The Nfs1 interacting protein Isd11 has an essential role in Fe/S cluster biogenesis in mitochondria. EMBO J. 2006, 25, 174–183. [Google Scholar] [CrossRef] [Green Version]
- Wiedemann, N.; Urzica, E.; Guiard, B.; Müller, H.; Lohaus, C.; Meyer, H.E.; Ryan, M.T.; Meisinger, C.; Mühlenhoff, U.; Lill, R.; et al. Essential role of Isd11 in mitochondrial iron–sulfur cluster synthesis on Isu scaffold proteins. EMBO J. 2006, 25, 184–195. [Google Scholar] [CrossRef]
- Shi, Y.; Ghosh, M.C.; Tong, W.H.; Rouault, T.A. Human ISD11 is essential for both iron-sulfur cluster assembly and maintenance of normal cellular iron homeostasis. Hum. Mol. Genet. 2009, 18, 3014–3025. [Google Scholar] [CrossRef]
- Van Vranken, J.G.; Jeong, M.Y.; Wei, P.; Chen, Y.C.; Gygi, S.P.; Winge, D.R.; Rutter, J. The mitochondrial acyl carrier protein (ACP) coordinates mitochondrial fatty acid synthesis with iron sulfur cluster biogenesis. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Majmudar, J.D.; Feng, X.; Fox, N.G.; Nabhan, J.F.; Towle, T.; Ma, T.; Gooch, R.; Bulawa, C.; Yue, W.W.; Martelli, A. 4’-Phosphopantetheine and long acyl chain-dependent interactions are integral to human mitochondrial acyl carrier protein function. Medchemcomm 2019, 10, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Masud, A.J.; Kastaniotis, A.J.; Rahman, M.T.; Autio, K.J.; Hiltunen, J.K. Mitochondrial acyl carrier protein (ACP) at the interface of metabolic state sensing and mitochondrial function. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118540. [Google Scholar] [CrossRef]
- Herrera, M.G.; Noguera, M.E.; Sewell, K.E.; Agudelo Suarez, W.A.; Capece, L.; Klinke, S.; Santos, J. Structure of the Human ACP-ISD11 Heterodimer. Biochemistry 2019, 58, 4596–4609. [Google Scholar] [CrossRef] [PubMed]
- Webert, H.; Freibert, S.A.; Gallo, A.; Heidenreich, T.; Linne, U.; Amlacher, S.; Hurt, E.; Muhlenhoff, U.; Banci, L.; Lill, R. Functional reconstitution of mitochondrial Fe/S cluster synthesis on Isu1 reveals the involvement of ferredoxin. Nat. Commun. 2014, 5, 5013. [Google Scholar] [CrossRef] [Green Version]
- Agar, J.N.; Zheng, L.; Cash, V.L.; Dean, D.R.; Johnson, M.K. Role of the IscU Protein in Iron−Sulfur Cluster Biosynthesis: IscS-mediated Assembly of a [Fe2S2] Cluster in IscU. J. Am. Chem. Soc. 2000, 122, 2136–2137. [Google Scholar] [CrossRef]
- Bridwell-Rabb, J.; Fox, N.G.; Tsai, C.L.; Winn, A.M.; Barondeau, D.P. Human frataxin activates Fe–S cluster biosynthesis by facilitating sulfur transfer chemistry. Biochemistry 2014, 53, 4904–4913. [Google Scholar] [CrossRef] [Green Version]
- Parent, A.; Elduque, X.; Cornu, D.; Belot, L.; Le Caer, J.P.; Grandas, A.; Toledano, M.B.; D’Autreaux, B. Mammalian frataxin directly enhances sulfur transfer of NFS1 persulfide to both ISCU and free thiols. Nat. Commun. 2015, 6, 5686. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Fuzery, A.K.; Tonelli, M.; Ta, D.T.; Westler, W.M.; Vickery, L.E.; Markley, J.L. Structure and dynamics of the iron-sulfur cluster assembly scaffold protein IscU and its interaction with the cochaperone HscB. Biochemistry 2009, 48, 6062–6071. [Google Scholar] [CrossRef] [Green Version]
- Cai, K.; Frederick, R.O.; Kim, J.H.; Reinen, N.M.; Tonelli, M.; Markley, J.L. Human mitochondrial chaperone (mtHSP70) and cysteine desulfurase (NFS1) bind preferentially to the disordered conformation, whereas co-chaperone (HSC20) binds to the structured conformation of the iron-sulfur cluster scaffold protein (ISCU). J. Biol. Chem. 2013, 288, 28755–28770. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Kelly, G.; Pastore, A. The Scaffold Protein IscU Retains a Structured Conformation in the FeS Cluster Assembly Complex. ChemBioChem 2014, 15, 1682–1686. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Tonelli, M.; Kim, T.; Markley, J.L. Three-dimensional structure and determinants of stability of the iron-sulfur cluster scaffold protein IscU from Escherichia coli. Biochemistry 2012, 51, 5557–5563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, N.G.; Martelli, A.; Nabhan, J.F.; Janz, J.; Borkowska, O.; Bulawa, C.; Yue, W.W. Zinc(II) binding on human wild-type ISCU and Met140 variants modulates NFS1 desulfurase activity. Biochimie 2018, 152, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Iannuzzi, C.; Adrover, M.; Puglisi, R.; Yan, R.; Temussi, P.A.; Pastore, A. The role of zinc in the stability of the marginally stable IscU scaffold protein. Protein Sci. 2014, 23, 1208–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gervason, S.; Larkem, D.; Mansour, A.B.; Botzanowski, T.; Muller, C.S.; Pecqueur, L.; Le Pavec, G.; Delaunay-Moisan, A.; Brun, O.; Agramunt, J.; et al. Physiologically relevant reconstitution of iron-sulfur cluster biosynthesis uncovers persulfide-processing functions of ferredoxin-2 and frataxin. Nat. Commun. 2019, 10, 3566. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.I.; Dizin, E.; Yoon, T.; Cowan, J.A. Kinetic and structural characterization of human mortalin. Protein Expr. Purif. 2010, 72, 75–81. [Google Scholar] [CrossRef] [Green Version]
- Hoff, K.G.; Silberg, J.J.; Vickery, L.E. Interaction of the iron-sulfur cluster assembly protein IscU with the Hsc66/Hsc20 molecular chaperone system of Escherichia coli. Proc. Natl. Acad. Sci. USA 2000, 97, 7790–7795. [Google Scholar] [CrossRef] [Green Version]
- Hoff, K.G.; Cupp-Vickery, J.R.; Vickery, L.E. Contributions of the LPPVK motif of the iron-sulfur template protein IscU to interactions with the Hsc66-Hsc20 chaperone system. J. Biol. Chem. 2003, 278, 37582–37589. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, N.; Yuda, E.; Fujishiro, T.; Hirabayashi, K.; Wada, K.; Takahashi, Y. Identification of IscU residues critical for de novo iron–sulfur cluster assembly. Mol. Microbiol. 2019, 112, 1769–1783. [Google Scholar] [CrossRef]
- Cook, J.D.; Bencze, K.Z.; Jankovic, A.D.; Crater, A.K.; Busch, C.N.; Bradley, P.B.; Stemmler, A.J.; Spaller, M.R.; Stemmler, T.L. Monomeric Yeast Frataxin Is an Iron-Binding Protein†. Biochemistry 2006, 45, 7767–7777. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Gakh, O.; Mooney, S.M.; Isaya, G. The Ferroxidase Activity of Yeast Frataxin. J. Biol. Chem. 2002, 277, 38589–38595. [Google Scholar] [CrossRef] [Green Version]
- Yoon, T.; Cowan, J.A. Iron−Sulfur Cluster Biosynthesis. Characterization of Frataxin as an Iron Donor for Assembly of [2Fe–2S] Clusters in ISU-Type Proteins. J. Am. Chem. Soc. 2003, 125, 6078–6084. [Google Scholar] [CrossRef] [PubMed]
- Bou-Abdallah, F.; Adinolfi, S.; Pastore, A.; Laue, T.M.; Dennis Chasteen, N. Iron binding and oxidation kinetics in frataxin CyaY of Escherichia coli. J. Mol. Biol. 2004, 341, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Yoon, T.; Dizin, E.; Cowan, J.A. N-terminal iron-mediated self-cleavage of human frataxin: Regulation of iron binding and complex formation with target proteins. J. Biol. Inorg. Chem. 2007, 12, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Dizin, E.; Cowan, J.A. Mapping iron binding sites on human frataxin: Implications for cluster assembly on the ISU Fe–S cluster scaffold protein. J. Biol. Inorg. Chem. 2008, 13, 825–836. [Google Scholar] [CrossRef]
- Gerber, J.; Muhlenhoff, U.; Lill, R. An interaction between frataxin and Isu1/Nfs1 that is crucial for Fe/S cluster synthesis on Isu1. EMBO Rep. 2003, 4, 906–911. [Google Scholar] [CrossRef] [Green Version]
- Cavadini, P.; O’Neill, H.A.; Benada, O.; Isaya, G. Assembly and iron-binding properties of human frataxin, the protein deficient in Friedreich ataxia. Hum. Mol. Genet. 2002, 11, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Gakh, O.; O’Neill, H.A.; Mangravita, A.; Nichol, H.; Ferreira, G.C.; Isaya, G. Yeast frataxin sequentially chaperones and stores iron by coupling protein assembly with iron oxidation. J. Biol. Chem. 2003, 278, 31340–31351. [Google Scholar] [CrossRef] [Green Version]
- Aloria, K.; Schilke, B.; Andrew, A.; Craig, E.A. Iron-induced oligomerization of yeast frataxin homologue Yfh1 is dispensable in vivo. EMBO Rep. 2004, 5, 1096–1101. [Google Scholar] [CrossRef] [Green Version]
- Seguin, A.; Sutak, R.; Bulteau, A.L.; Garcia-Serres, R.; Oddou, J.L.; Lefevre, S.; Santos, R.; Dancis, A.; Camadro, J.M.; Latour, J.M.; et al. Evidence that yeast frataxin is not an iron storage protein in vivo. Biochim. Biophys. Acta 2010, 1802, 531–538. [Google Scholar] [CrossRef]
- Tsai, C.L.; Barondeau, D.P. Human frataxin is an allosteric switch that activates the Fe–S cluster biosynthetic complex. Biochemistry 2010, 49, 9132–9139. [Google Scholar] [CrossRef] [PubMed]
- Schmucker, S.; Martelli, A.; Colin, F.; Page, A.; Wattenhofer-Donze, M.; Reutenauer, L.; Puccio, H. Mammalian frataxin: An essential function for cellular viability through an interaction with a preformed ISCU/NFS1/ISD11 iron-sulfur assembly complex. PLoS ONE 2011, 6, e16199. [Google Scholar] [CrossRef] [PubMed]
- Patra, S.; Barondeau, D.P. Mechanism of activation of the human cysteine desulfurase complex by frataxin. Proc. Natl. Acad. Sci. USA 2019, 116, 19421–19430. [Google Scholar] [CrossRef] [Green Version]
- Colin, F.; Martelli, A.; Clemancey, M.; Latour, J.M.; Gambarelli, S.; Zeppieri, L.; Birck, C.; Page, A.; Puccio, H.; Ollagnier de Choudens, S. Mammalian frataxin controls sulfur production and iron entry during de novo Fe4S4 cluster assembly. J. Am. Chem. Soc. 2013, 135, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Huichalaf, C.; Perfitt, T.L.; Kuperman, A.; Gooch, R.; Kovi, R.C.; Brenneman, K.A.; Chen, X.; Hirenallur-Shanthappa, D.; Ma, T.; Assaf, B.T.; et al. In vivo overexpression of frataxin causes toxicity mediated by iron-sulfur cluster deficiency. Mol. Ther.-Methods Clin. Dev. 2022. [Google Scholar] [CrossRef]
- Yoon, H.; Knight, S.A.; Pandey, A.; Pain, J.; Zhang, Y.; Pain, D.; Dancis, A. Frataxin-bypassing Isu1: Characterization of the bypass activity in cells and mitochondria. Biochem. J. 2014, 459, 71–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, H.; Knight, S.A.; Pandey, A.; Pain, J.; Turkarslan, S.; Pain, D.; Dancis, A. Turning Saccharomyces cerevisiae into a Frataxin-Independent Organism. PLoS Genet. 2015, 11, e1005135. [Google Scholar] [CrossRef]
- Yoon, H.; Golla, R.; Lesuisse, E.; Pain, J.; Donald, J.E.; Lyver, E.R.; Pain, D.; Dancis, A. Mutation in the Fe–S scaffold protein Isu bypasses frataxin deletion. Biochem. J. 2012, 441, 473–480. [Google Scholar] [CrossRef] [Green Version]
- Das, D.; Patra, S.; Bridwell-Rabb, J.; Barondeau, D.P. Mechanism of frataxin “bypass” in human iron-sulfur cluster biosynthesis with implications for Friedreich’s ataxia. J. Biol. Chem. 2019, 294, 9276–9284. [Google Scholar] [CrossRef]
- Bruschi, M.; Guerlesquin, F. Structure, function and evolution of bacterial ferredoxins. FEMS Microbiol. Rev. 1988, 4, 155–175. [Google Scholar] [CrossRef]
- Li, J.; Saxena, S.; Pain, D.; Dancis, A. Adrenodoxin reductase homolog (Arh1p) of yeast mitochondria required for iron homeostasis. J. Biol. Chem. 2001, 276, 1503–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, H.; Kaut, A.; Kispal, G.; Lill, R. A mitochondrial ferredoxin is essential for biogenesis of cellular iron-sulfur proteins. Proc. Natl. Acad. Sci. USA 2000, 97, 1050–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheftel, A.D.; Stehling, O.; Pierik, A.J.; Elsasser, H.P.; Muhlenhoff, U.; Webert, H.; Hobler, A.; Hannemann, F.; Bernhardt, R.; Lill, R. Humans possess two mitochondrial ferredoxins, Fdx1 and Fdx2, with distinct roles in steroidogenesis, heme, and Fe/S cluster biosynthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 11775–11780. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Adinolfi, S.; Pastore, A. Ferredoxin, in conjunction with NADPH and ferredoxin-NADP reductase, transfers electrons to the IscS/IscU complex to promote iron–sulfur cluster assembly. Biochim. Biophys. Acta-Proteins Proteom. 2015, 1854, 1113–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, R.; Konarev, P.V.; Iannuzzi, C.; Adinolfi, S.; Roche, B.; Kelly, G.; Simon, L.; Martin, S.R.; Py, B.; Barras, F.; et al. Ferredoxin competes with bacterial frataxin in binding to the desulfurase IscS. J. Biol. Chem. 2013, 288, 24777–24787. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Frederick, R.O.; Reinen, N.M.; Troupis, A.T.; Markley, J.L. [2Fe–2S]-Ferredoxin Binds Directly to Cysteine Desulfurase and Supplies an Electron for Iron–Sulfur Cluster Assembly but Is Displaced by the Scaffold Protein or Bacterial Frataxin. J. Am. Chem. Soc. 2013, 135, 8117–8120. [Google Scholar] [CrossRef] [PubMed]
- Uzarska, M.A.; Grochowina, I.; Soldek, J.; Jelen, M.; Schilke, B.; Marszalek, J.; Craig, E.A.; Dutkiewicz, R. During FeS-cluster biogenesis ferredoxin and frataxin use overlapping bindings sites on yeast cysteine desulfurase Nfs1. J. Biol. Chem. 2022, 298, 101570. [Google Scholar] [CrossRef]
- Beilschmidt, L.K.; Ollagnier de Choudens, S.; Fournier, M.; Sanakis, I.; Hograindleur, M.A.; Clemancey, M.; Blondin, G.; Schmucker, S.; Eisenmann, A.; Weiss, A.; et al. ISCA1 is essential for mitochondrial Fe4S4 biogenesis in vivo. Nat. Commun. 2017, 8, 15124. [Google Scholar] [CrossRef]
- Weiler, B.D.; Brück, M.-C.; Kothe, I.; Bill, E.; Lill, R.; Mühlenhoff, U. Mitochondrial [4Fe–4S] protein assembly involves reductive [2Fe–2S] cluster fusion on ISCA1–ISCA2 by electron flow from ferredoxin FDX2. Proc. Natl. Acad. Sci. USA 2020, 117, 20555–20565. [Google Scholar] [CrossRef]
- Hershkovitz, T.; Kurolap, A.; Tal, G.; Paperna, T.; Mory, A.; Staples, J.; Brigatti, K.W.; Regeneron Genetics, C.; Gonzaga-Jauregui, C.; Dumin, E.; et al. A recurring NFS1 pathogenic variant causes a mitochondrial disorder with variable intra-familial patient outcomes. Mol. Genet. Metab. Rep. 2021, 26, 100699. [Google Scholar] [CrossRef]
- Farhan, S.M.K.; Wang, J.; Robinson, J.F.; Lahiry, P.; Siu, V.M.; Prasad, C.; Kronick, J.B.; Ramsay, D.A.; Rupar, C.A.; Hegele, R.A. Exome sequencing identifies NFS 1 deficiency in a novel Fe–S cluster disease, infantile mitochondrial complex II/III deficiency. Mol. Genet. Genom. Med. 2014, 2, 73–80. [Google Scholar] [CrossRef]
- Lim, S.C.; Friemel, M.; Marum, J.E.; Tucker, E.J.; Bruno, D.L.; Riley, L.G.; Christodoulou, J.; Kirk, E.P.; Boneh, A.; DeGennaro, C.M.; et al. Mutations in LYRM4, encoding iron-sulfur cluster biogenesis factor ISD11, cause deficiency of multiple respiratory chain complexes. Hum. Mol. Genet. 2013, 22, 4460–4473. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, R.; Saada, A.; Halvardson, J.; Soiferman, D.; Shaag, A.; Edvardson, S.; Horovitz, Y.; Khayat, M.; Shalev, S.A.; Feuk, L.; et al. Deleterious mutation in FDX1L gene is associated with a novel mitochondrial muscle myopathy. Eur. J. Hum. Genet. 2014, 22, 902–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drugge, U.; Holmberg, M.; Holmgren, G.; Almay, B.G.; Linderholm, H. Hereditary myopathy with lactic acidosis, succinate dehydrogenase and aconitase deficiency in northern Sweden: A genealogical study. J. Med. Genet. 1995, 32, 344–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haller, R.G.; Henriksson, K.G.; Jorfeldt, L.; Hultman, E.; Wibom, R.; Sahlin, K.; Areskog, N.H.; Gunder, M.; Ayyad, K.; Blomqvist, C.G.; et al. Deficiency of skeletal muscle succinate dehydrogenase and aconitase. Pathophysiology of exercise in a novel human muscle oxidative defect. J. Clin. Investig. 1991, 88, 1197–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsson, A.; Lind, L.; Thornell, L.E.; Holmberg, M. Myopathy with lactic acidosis is linked to chromosome 12q23.3-24.11 and caused by an intron mutation in the ISCU gene resulting in a splicing defect. Hum. Mol. Genet. 2008, 17, 1666–1672. [Google Scholar] [CrossRef] [PubMed]
- Mochel, F.; Knight, M.A.; Tong, W.H.; Hernandez, D.; Ayyad, K.; Taivassalo, T.; Andersen, P.M.; Singleton, A.; Rouault, T.A.; Fischbeck, K.H.; et al. Splice mutation in the iron-sulfur cluster scaffold protein ISCU causes myopathy with exercise intolerance. Am. J. Hum. Genet. 2008, 82, 652–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kollberg, G.; Tulinius, M.; Melberg, A.; Darin, N.; Andersen, O.; Holmgren, D.; Oldfors, A.; Holme, E. Clinical manifestation and a new ISCU mutation in iron-sulphur cluster deficiency myopathy. Brain 2009, 132, 2170–2179. [Google Scholar] [CrossRef] [Green Version]
- Nordin, A.; Larsson, E.; Thornell, L.E.; Holmberg, M. Tissue-specific splicing of ISCU results in a skeletal muscle phenotype in myopathy with lactic acidosis, while complete loss of ISCU results in early embryonic death in mice. Hum. Genet. 2011, 129, 371–378. [Google Scholar] [CrossRef]
- Beilschmidt, L.K.; Puccio, H.M. Mammalian Fe–S cluster biogenesis and its implication in disease. Biochimie 2014, 100, 48–60. [Google Scholar] [CrossRef]
- Herman, D.; Jenssen, K.; Burnett, R.; Soragni, E.; Perlman, S.L.; Gottesfeld, J.M. Histone deacetylase inhibitors reverse gene silencing in Friedreich’s ataxia. Nat. Chem. Biol. 2006, 2, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Saveliev, A.; Everett, C.; Sharpe, T.; Webster, Z.; Festenstein, R. DNA triplet repeats mediate heterochromatin-protein-1-sensitive variegated gene silencing. Nature 2003, 422, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, V.; Montermini, L.; Molto, M.D.; Pianese, L.; Cossee, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef]
- Tsai, C.L.; Bridwell-Rabb, J.; Barondeau, D.P. Friedreich’s ataxia variants I154F and W155R diminish frataxin-based activation of the iron-sulfur cluster assembly complex. Biochemistry 2011, 50, 6478–6487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridwell-Rabb, J.; Winn, A.M.; Barondeau, D.P. Structure-function analysis of Friedreich’s ataxia mutants reveals determinants of frataxin binding and activation of the Fe–S assembly complex. Biochemistry 2011, 50, 7265–7274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cossee, M.; Durr, A.; Schmitt, M.; Dahl, N.; Trouillas, P.; Allinson, P.; Kostrzewa, M.; Nivelon-Chevallier, A.; Gustavson, K.H.; Kohlschutter, A.; et al. Friedreich’s ataxia: Point mutations and clinical presentation of compound heterozygotes. Ann. Neurol. 1999, 45, 200–206. [Google Scholar] [CrossRef]
- Harding, I.H.; Lynch, D.R.; Koeppen, A.H.; Pandolfo, M. Central Nervous System Therapeutic Targets in Friedreich Ataxia. Hum. Gene Ther. 2020, 31, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Pandolfo, M. Friedreich ataxia: The clinical picture. J. Neurol. 2009, 256 (Suppl. 1), 3–8. [Google Scholar] [CrossRef]
- Cnop, M.; Mulder, H.; Igoillo-Esteve, M. Diabetes in Friedreich ataxia. J. Neurochem. 2013, 126 (Suppl. 1), 94–102. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, F.; Stork, S.; Liu, D.; Hu, K.; Herrmann, S.; Ertl, G.; Niemann, M. Cardiomyopathy of Friedreich ataxia. J. Neurochem. 2013, 126 (Suppl. 1), 88–93. [Google Scholar] [CrossRef] [PubMed]
- Puccio, H.; Simon, D.; Cossee, M.; Criqui-Filipe, P.; Tiziano, F.; Melki, J.; Hindelang, C.; Matyas, R.; Rustin, P.; Koenig, M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe–S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 2001, 27, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.; Wattenhofer-Donze, M.; Schmucker, S.; Bouvet, S.; Reutenauer, L.; Puccio, H. Frataxin is essential for extramitochondrial Fe–S cluster proteins in mammalian tissues. Hum. Mol. Genet. 2007, 16, 2651–2658. [Google Scholar] [CrossRef] [PubMed]
- Seznec, H.; Simon, D.; Bouton, C.; Reutenauer, L.; Hertzog, A.; Golik, P.; Procaccio, V.; Patel, M.; Drapier, J.C.; Koenig, M.; et al. Friedreich ataxia: The oxidative stress paradox. Hum. Mol. Genet. 2005, 14, 463–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piguet, F.; de Montigny, C.; Vaucamps, N.; Reutenauer, L.; Eisenmann, A.; Puccio, H. Rapid and Complete Reversal of Sensory Ataxia by Gene Therapy in a Novel Model of Friedreich Ataxia. Mol. Ther. 2018, 26, 1940–1952. [Google Scholar] [CrossRef] [Green Version]
- Hinton, T.V.; Batelu, S.; Gleason, N.; Stemmler, T.L. Molecular characteristics of proteins within the mitochondrial Fe–S cluster assembly complex. Micron 2021, 153, 103181. [Google Scholar] [CrossRef] [PubMed]
- Baussier, C.; Fakroun, S.; Aubert, C.; Dubrac, S.; Mandin, P.; Py, B.; Barras, F. Making iron-sulfur cluster: Structure, regulation and evolution of the bacterial ISC system. Adv. Microb. Physiol. 2020, 76, 1–39. [Google Scholar] [CrossRef]
- Srour, B.; Gervason, S.; Monfort, B.; D’Autreaux, B. Mechanism of Iron-Sulfur Cluster Assembly: In the Intimacy of Iron and Sulfur Encounter. Inorganics 2020, 8, 55. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mitochondrial Compartment | ||||
|---|---|---|---|---|
| Fe–S Protein | Full Name | Pathway | Cluster | Function |
| SDHB | Succinate dehydrogenase iron–sulfur subunit B | Cellular Respiration, TCA Cycle | [4Fe–4S] [3Fe–4S] [2Fe–2S] | Subunit of SDH that is involved in Complex II function |
| Rieske protein UQCRFS1/RISP | Cytochrome b-c1 complex subunit Rieske | Cellular Respiration | [2Fe–2S] | Component of Complex III |
| NDUFS1 | NADH-ubiquinone oxidoreductase, 75 kDa subunit | Cellular Respiration | [4Fe–4S] [2Fe–2S] | Subunit of Complex I |
| NDUFS8 | NADH dehydrogenase iron–sulfur protein 8 | Cellular Respiration | [4Fe–4S] | Subunit of Complex I |
| NDUFS7 | NADH dehydrogenase iron–sulfur protein 7 | Cellular Respiration | [4Fe–4S] | Subunit of Complex I |
| NDUFV2 | NADH dehydrogenase flavoprotein 2 | Cellular Respiration | [2Fe–2S] | Subunit of Complex I |
| NDUFV1 | NADH dehydrogenase flavoprotein 1 | Cellular Respiration | [4Fe–4S] | Subunit of Complex I |
| ACO2 | Aconitate hydratase, mitochondrial | TCA Cycle | [4Fe–4S] | Enzyme that generates isocitrate in step 2 of TCA cycle |
| ETFDH | Electron transfer flavoprotein-ubiquinone oxidoreductase, mitochondrial | Fatty acid oxidation | [2Fe–2S] | Accepts electrons from ETF, reduces ubiquinone |
| LIAS | Lipoyl synthase | Lipoic acid synthesis | [4Fe–4S] | Converts octanoylated domains into lipoylated derivatives |
| MOCS1 | Molybdenum cofactor biosynthesis protein 1 | MoCo biosynthesis | [4Fe–4S] | Catalyzes the conversion of 5′-GTP to cyclic pyranopterin monophosphate |
| GLRX2 | Glutaredoxin-2 | Redox homeostasis | [2Fe–2S] | Maintains mitochondrial redox homeostasis upon induction of apoptosis |
| IND1 | Mitochondrial P-loop NTPase | Complex I assembly | [4Fe–4S] | Scaffold for transfer of Fe–S to Complex I |
| Nuclear Compartment | ||||
|---|---|---|---|---|
| Fe–S Protein | Full Name | Pathway | Cluster | Function |
| MUTYH | Adenine DNA glycosylase | DNA repair | [4Fe–4S] | Involved in oxidative DNA damage repair |
| EXO5 | Exonuclease V | DNA repair | [4Fe–4S] | Exonuclease involved in DNA repair |
| NTHL1 | Endonuclease III-like protein 1 | DNA repair | [4Fe–4S] | Catalyzes the first step in base excision repair |
| PRIM2 | DNA primase large subunit | DNA replication | [4Fe–4S] | DNA primase |
| POLA1 | DNA polymerase alpha catalytic subunit | DNA replication | [4Fe–4S] | Catalytic subunit of DNA polymerase alpha |
| POLD1 | DNA polymerase delta catalytic subunit | DNA replication | [4Fe–4S] | Catalytic subunit of DNA polymerase delta |
| POLE | DNA polymerase epsilon catalytic subunit A | DNA replication | [4Fe–4S] | Catalytic subunit of DNA polymerase epsilon |
| POLZ | DNA polymerase zeta catalytic subunit | Translesion DNA synthesis | [4Fe–4S] | Catalytic subunit of DNA polymerase zeta |
| DDX11 | ATP-dependent DNA helicase DDX11 | DNA replication and repair | [4Fe–4S] | DNA-dependent ATPase and ATP-dependent DNA helicase |
| Cytosolic Compartment | ||||
| Fe–S Protein | Full Name | Pathway | Cluster | Function |
| IRP1/ACO1 | Iron regulatory protein 1,cytosolic aconitase 1 | Iron metabolism, TCA cycle | [4Fe–4S] | Regulates iron levels through mRNA binding |
| KIF4A | Chromosome-associated kinesin | Mitosis | [4Fe–4S] | Motor protein involved in metaphase to anaphase to transition |
| TYW1 | S-adenosyl-L-methionine-dependent tRNA 4-demethylwyosine synthase | tRNA modification | [4Fe–4S] | Enzyme involved in tRNA(Phe) biosynthesis |
| CDKAL1 | Threonylcarbamoyladenosine tRNA methylthiotransferase | tRNA modification | [4Fe–4S] | Catalyzes the methylthiolation of t6A |
| ELP3 | Elongator complex protein 3 | Transcription | [4Fe–4S] | Catalytic histone acetyltransferase subunit of the RNA polymerase II elongator complex |
| GPAT | Amidophosphoribosyltransferase, Atase | Nucleotide metabolism | [4Fe–4S] | Involved in the synthesis of N(1)-(5-phospho-D-ribosyl)glycinamide during IMP biosynthesis |
| DPYD | Dihydropyrimidine dehydrogenase [NADP(+)] | Nucleotide metabolism | [4Fe–4S] | Catalyzes the reduction of uracil and thymine |
| XDH | Xanthine dehydrogenase | Nucleotide metabolism | [2Fe–2S] | Involved in purine degradation |
| ABCE1/RLI1 | ATP-binding cassette sub-family E member 1 | Protein translation | [4Fe–4S] | Splitting of ribosome subunits during translation termination |
| DPH1/2 | 2-(3-amino-3-carboxypropyl)histidine synthase subunit 1/2 | Protein translation | [4Fe–4S] | Involved in the post-translational modification of histidine to diphthamide |
| Protein | Human Protein | Yeast Protein | Bacterial Protein | Other Names | Required Cofactor | Function in Fe–S Core Complex |
|---|---|---|---|---|---|---|
| Cysteine Desulfurase | NFS1 | Nfs1 | IscS | - | PLP | Persulfide production and transfer to scaffold |
| LYRM Protein | ISD11 | Isd11 | - | LYRM4 | - | Stabilizes cysteine desulfurase complex |
| Acyl Carrier Protein | NDUFAB1 | Acp1 | Acp | ACP1, ACPm | 4′PP, acyl chain | Binds to LYRM protein for complex stability |
| Scaffold Protein | ISCU | Isu1/2 | IscU | - | - | Receives persulfide from cysteine desulfurase and assembles Fe–S |
| Frataxin | FXN | Yfh1 | CyaY | - | - | Modulates persulfide transfer rate |
| Ferredoxin | FDX2 | Yah1 | Fdx | FDX1L | [2Fe–2S] | Provides electrons to reduce persulfide |
| Ferredoxin Reductase | FDXR | Arh1 | Fpr | - | NADPH+ | Reduces [2Fe–2S]-ferredoxin |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perfitt, T.L.; Martelli, A. Mitochondrial De Novo Assembly of Iron–Sulfur Clusters in Mammals: Complex Matters in a Complex That Matters. Inorganics 2022, 10, 31. https://doi.org/10.3390/inorganics10030031
Perfitt TL, Martelli A. Mitochondrial De Novo Assembly of Iron–Sulfur Clusters in Mammals: Complex Matters in a Complex That Matters. Inorganics. 2022; 10(3):31. https://doi.org/10.3390/inorganics10030031
Chicago/Turabian StylePerfitt, Tyler L., and Alain Martelli. 2022. "Mitochondrial De Novo Assembly of Iron–Sulfur Clusters in Mammals: Complex Matters in a Complex That Matters" Inorganics 10, no. 3: 31. https://doi.org/10.3390/inorganics10030031
APA StylePerfitt, T. L., & Martelli, A. (2022). Mitochondrial De Novo Assembly of Iron–Sulfur Clusters in Mammals: Complex Matters in a Complex That Matters. Inorganics, 10(3), 31. https://doi.org/10.3390/inorganics10030031