
[Hexaamminecobalt(III)] Dichloride Permanganate—Structural Features and Heat-Induced Transformations into (CoII,MnII)(CoIII,MnIII)2O4 Spinels

 , , , ,
, , , ,  and
and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Synthesis and Properties of Compound 1
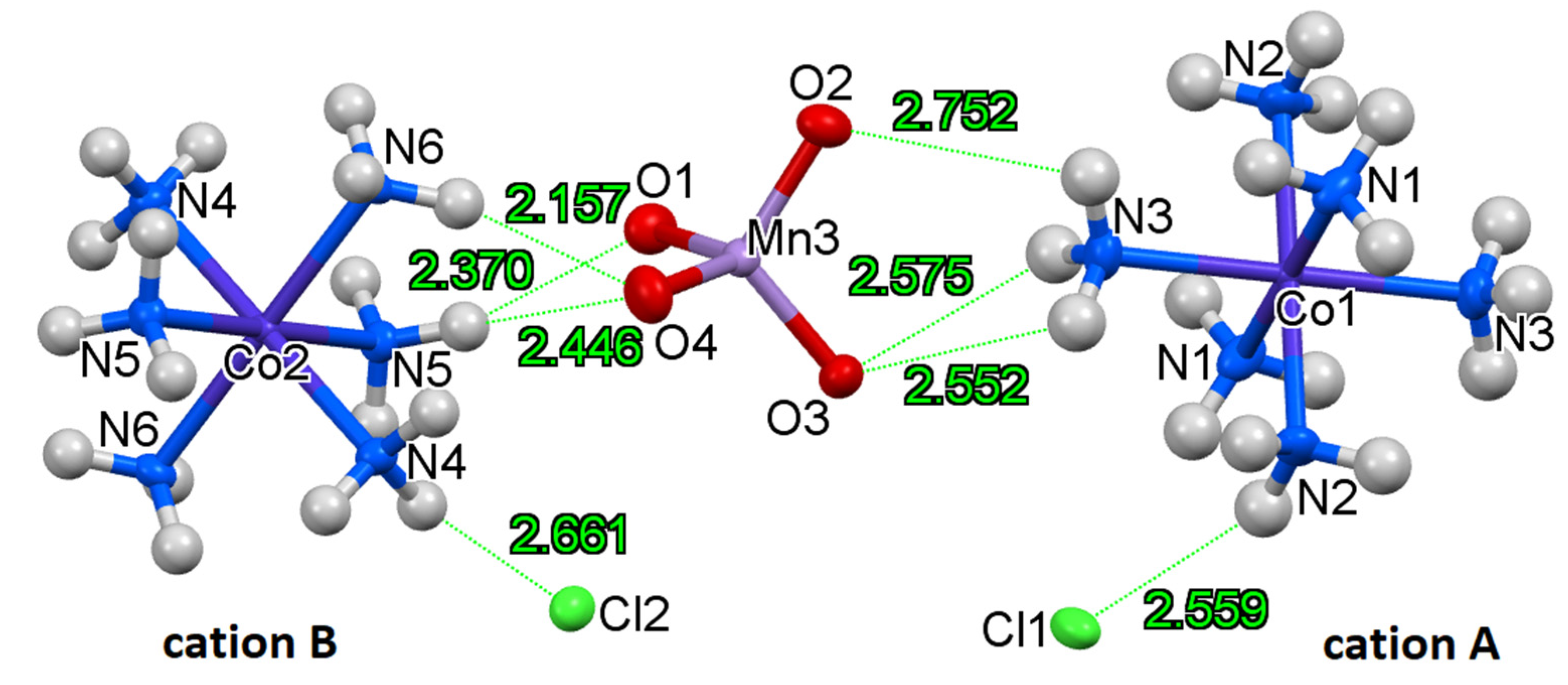
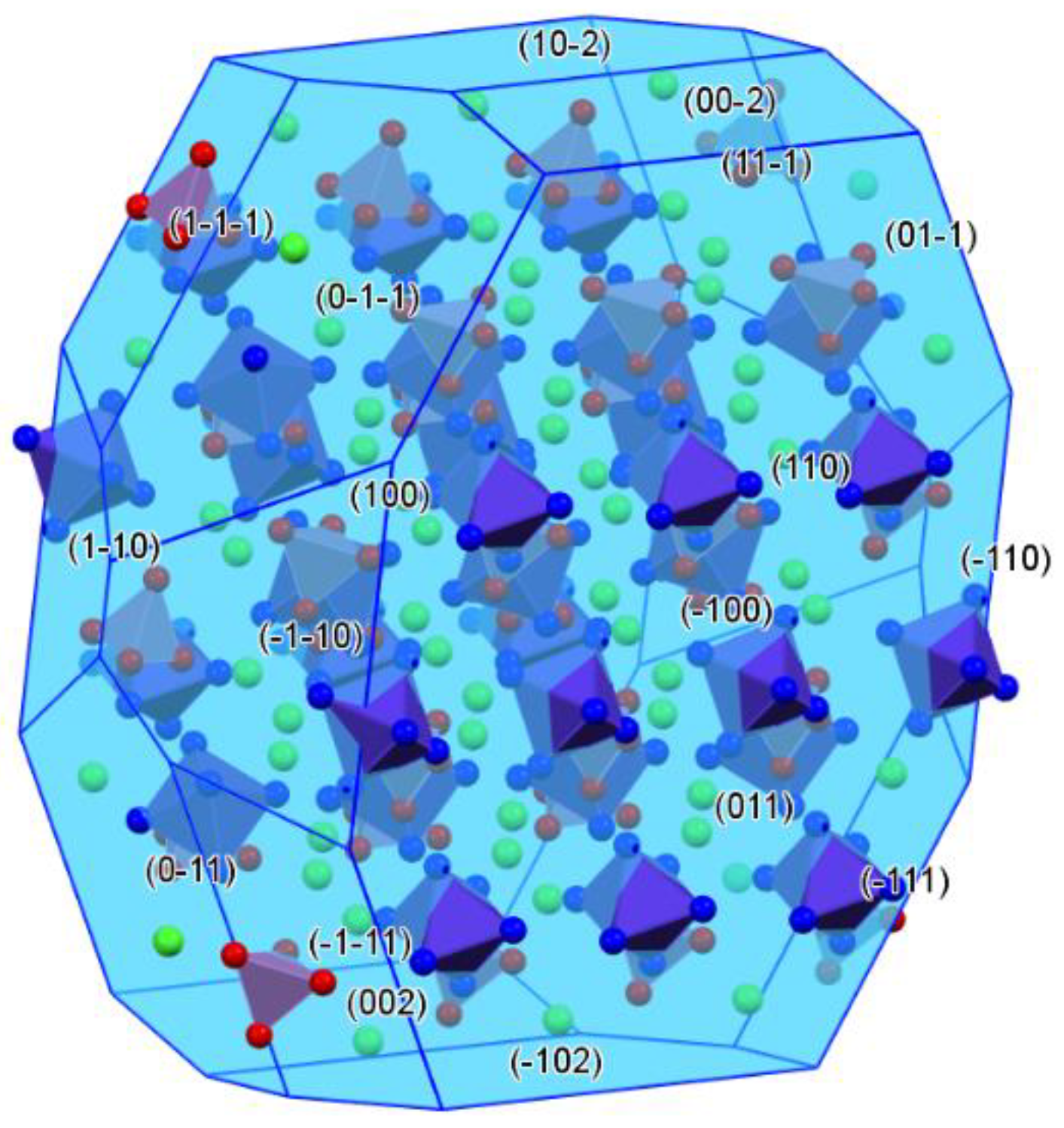
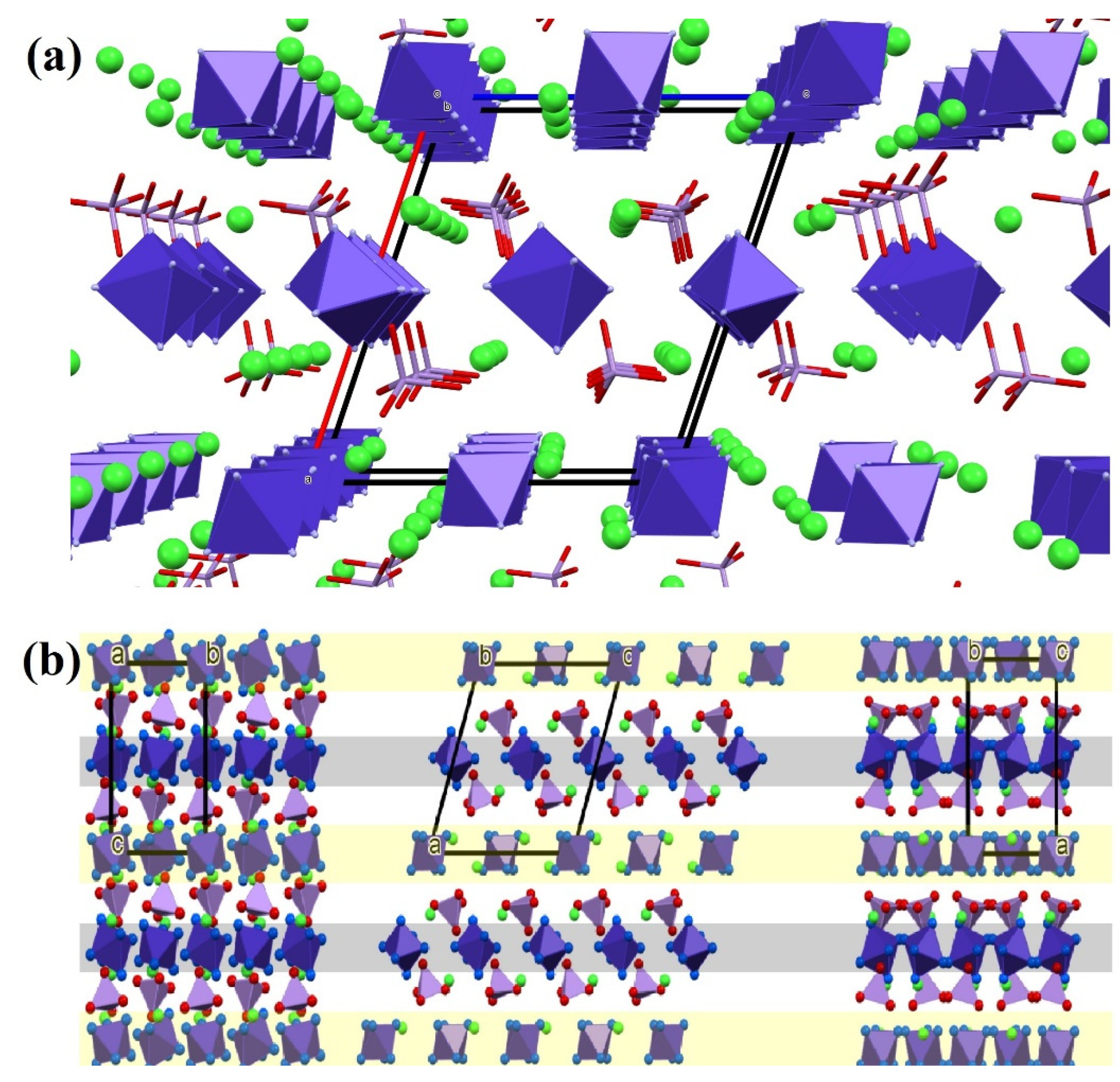
2.2. Structure of Compound 1
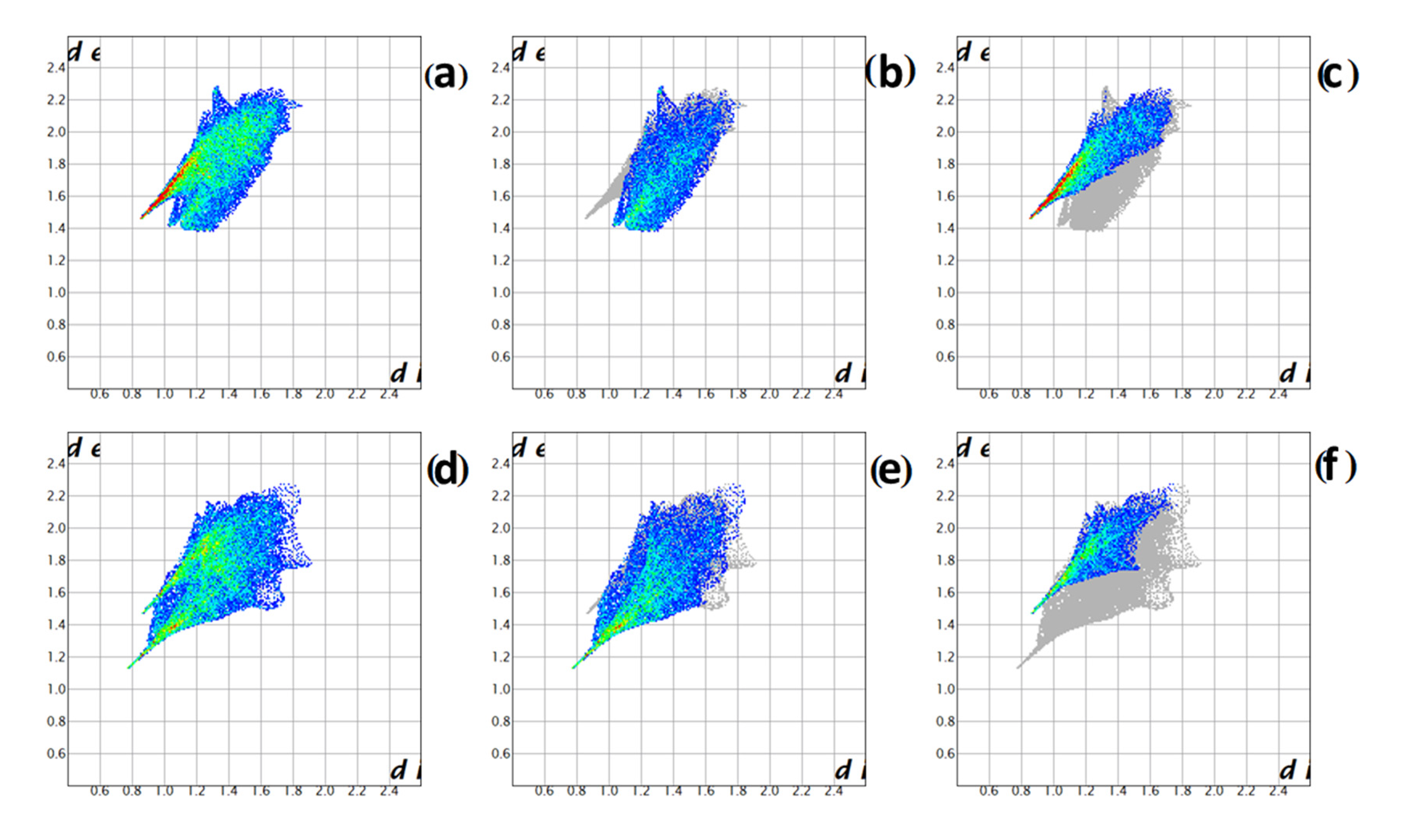
2.3. Spectroscopic Properties of Compound 1
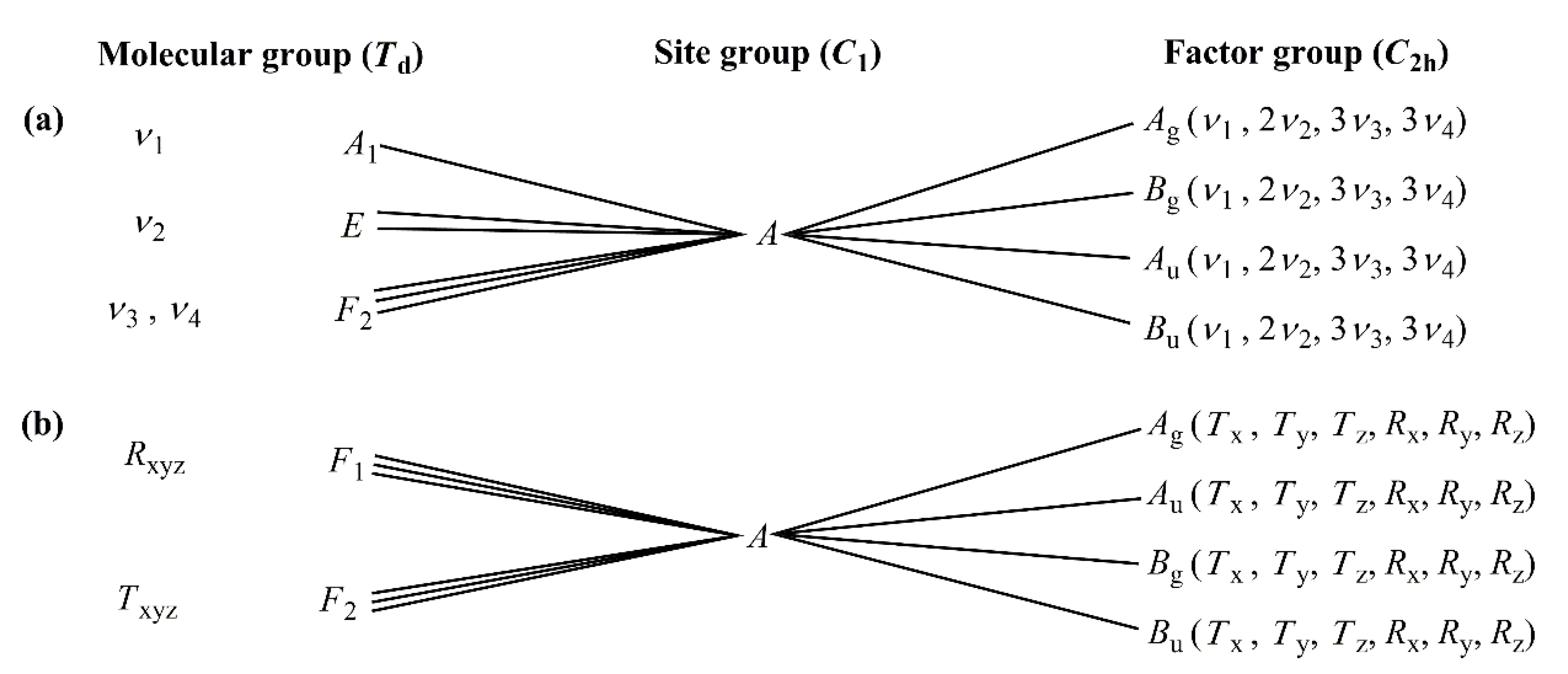
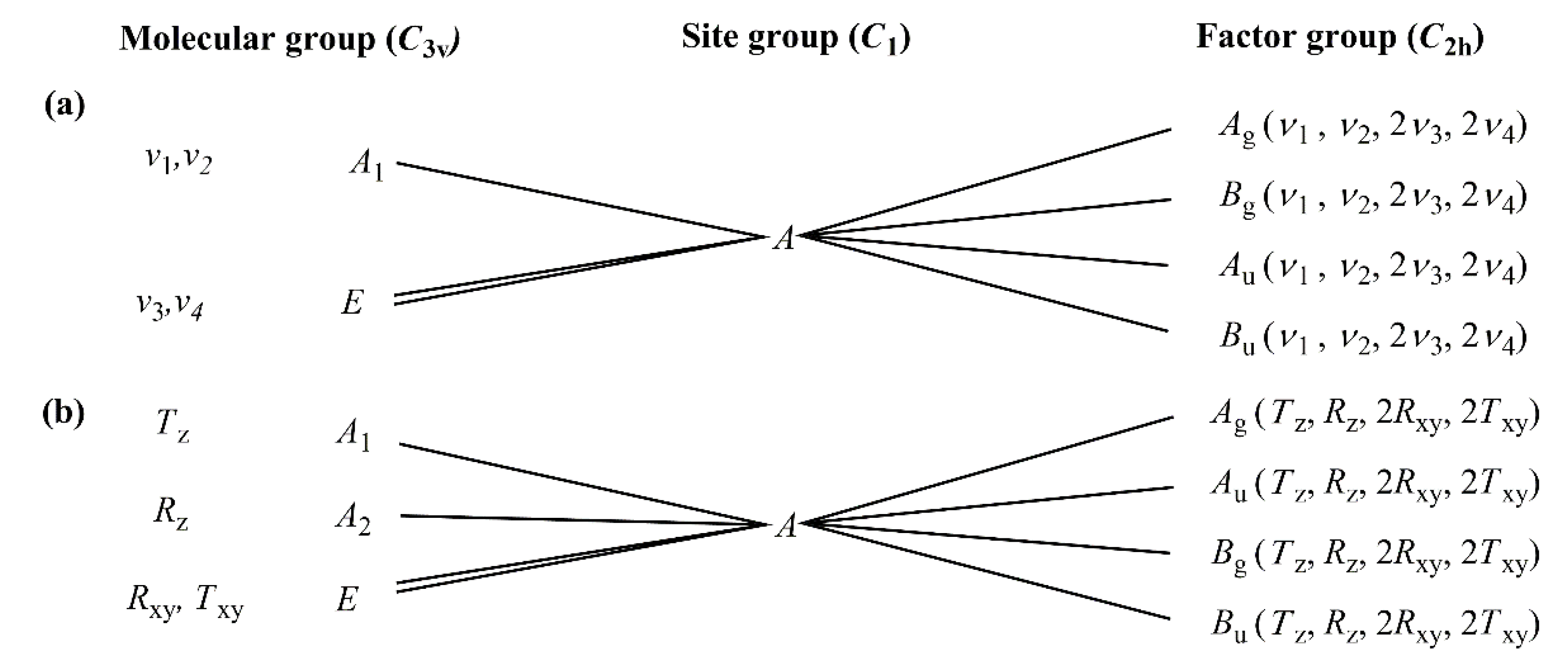
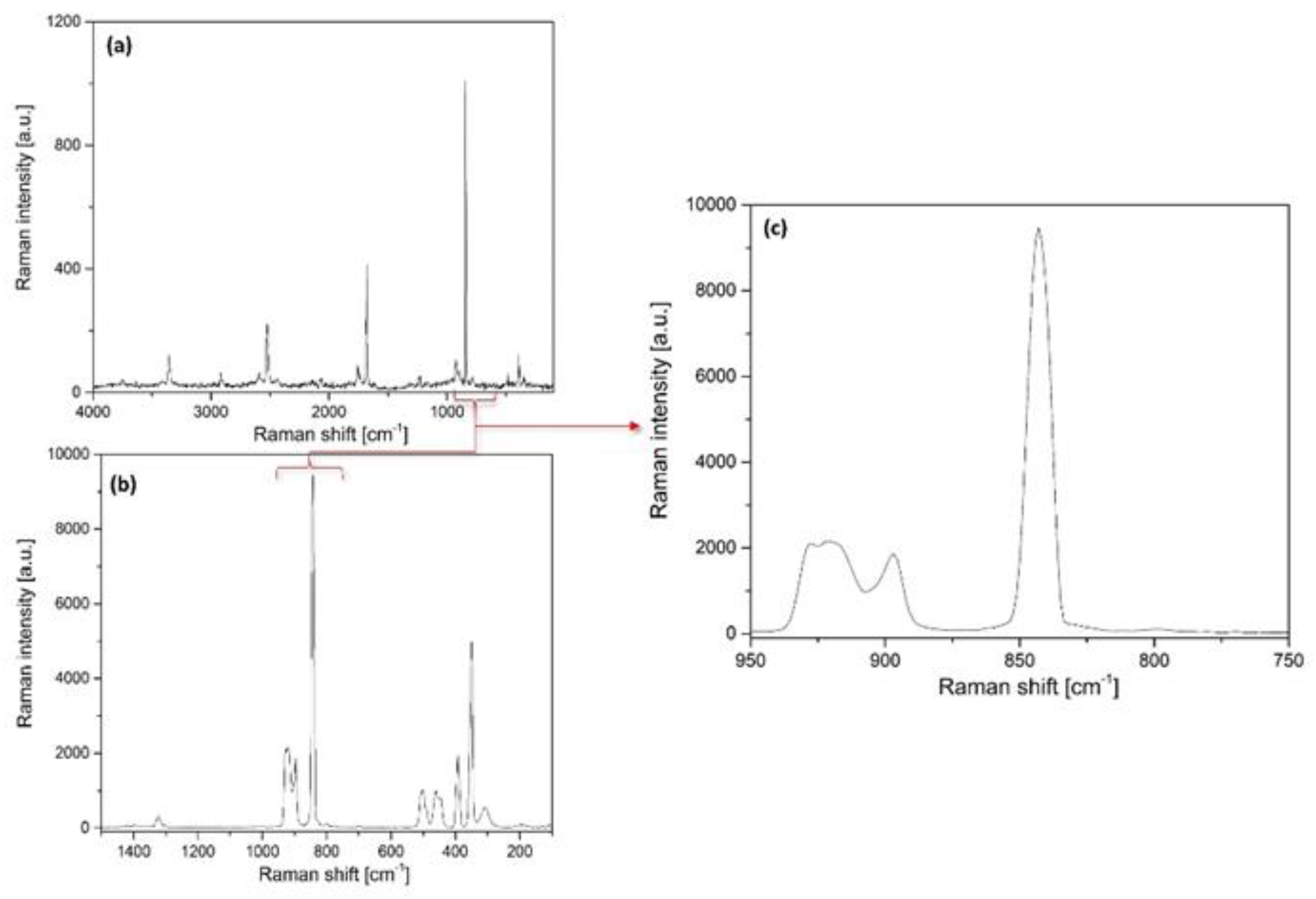
2.3.1. Vibrational Modes of the Permanganate Anion in Compound 1
2.3.2. Vibrational Modes of the [Hexaamminecobalt(III)] Cation in Compound 1
2.4. UV−VIS Spectroscopy
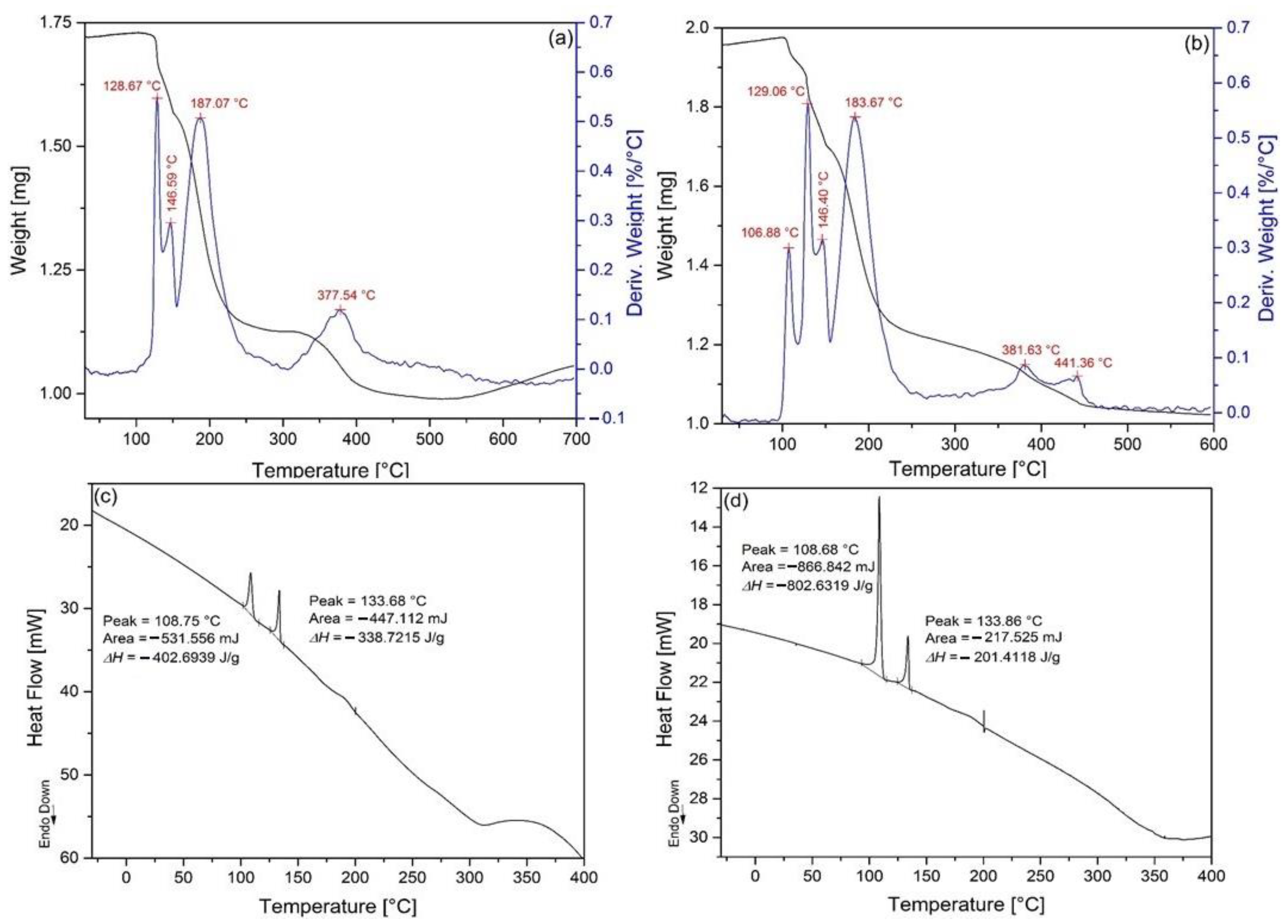
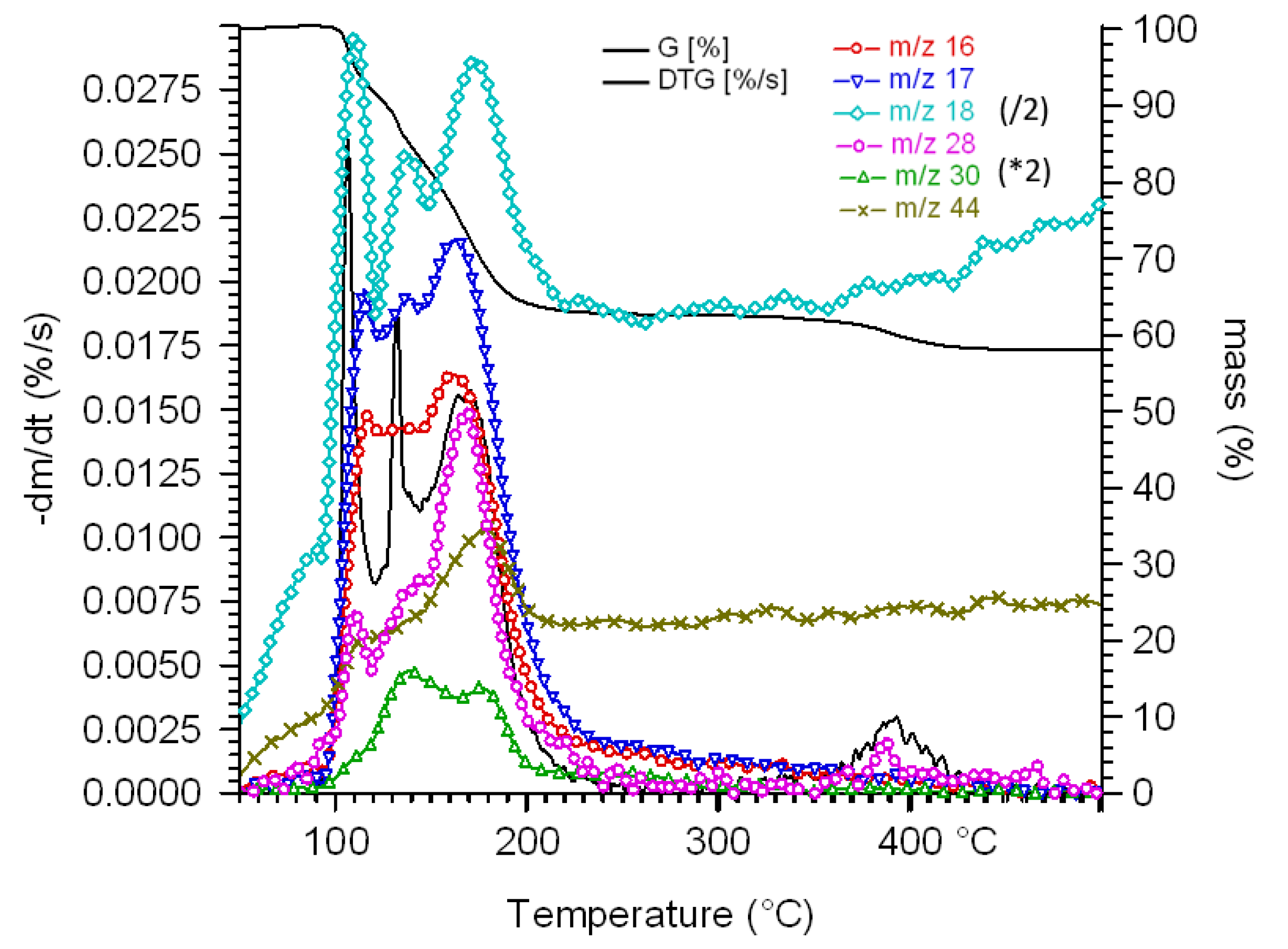
2.5. Non-Isothermal Thermal Decomposition of Compound 1
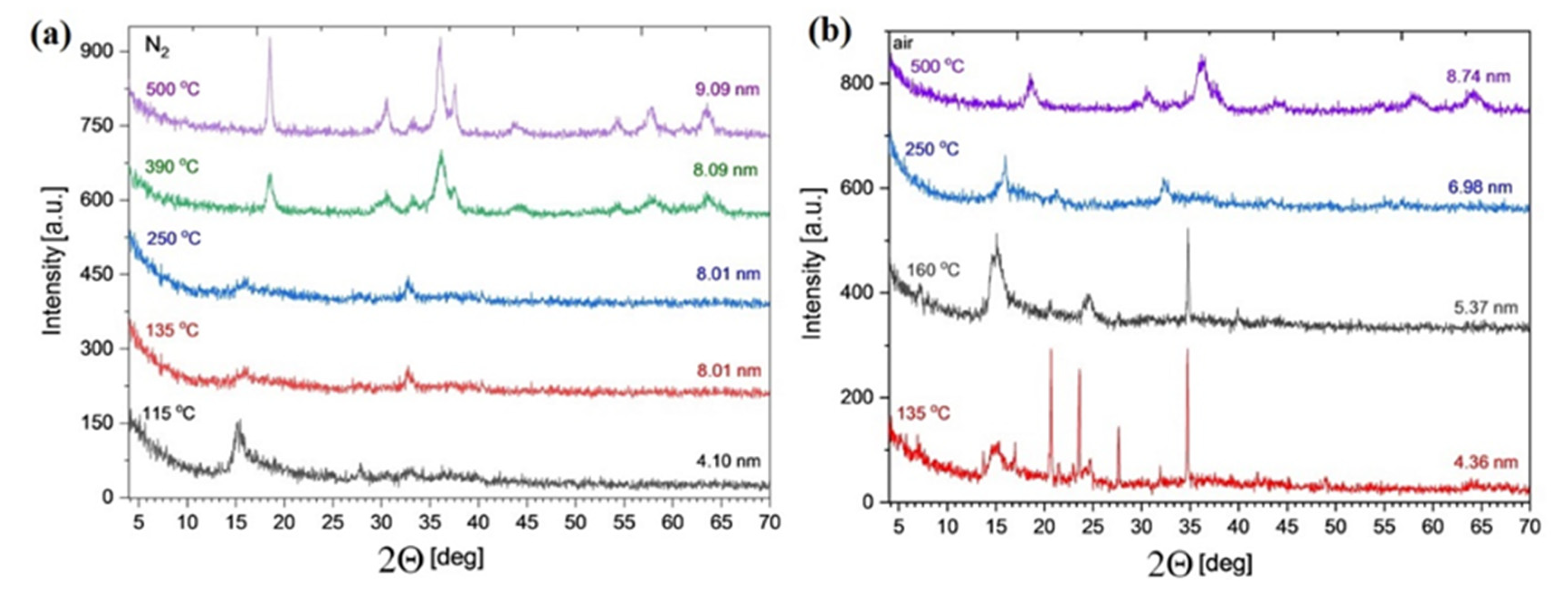
2.6. Isothermal Heat Treatments of Compound 1
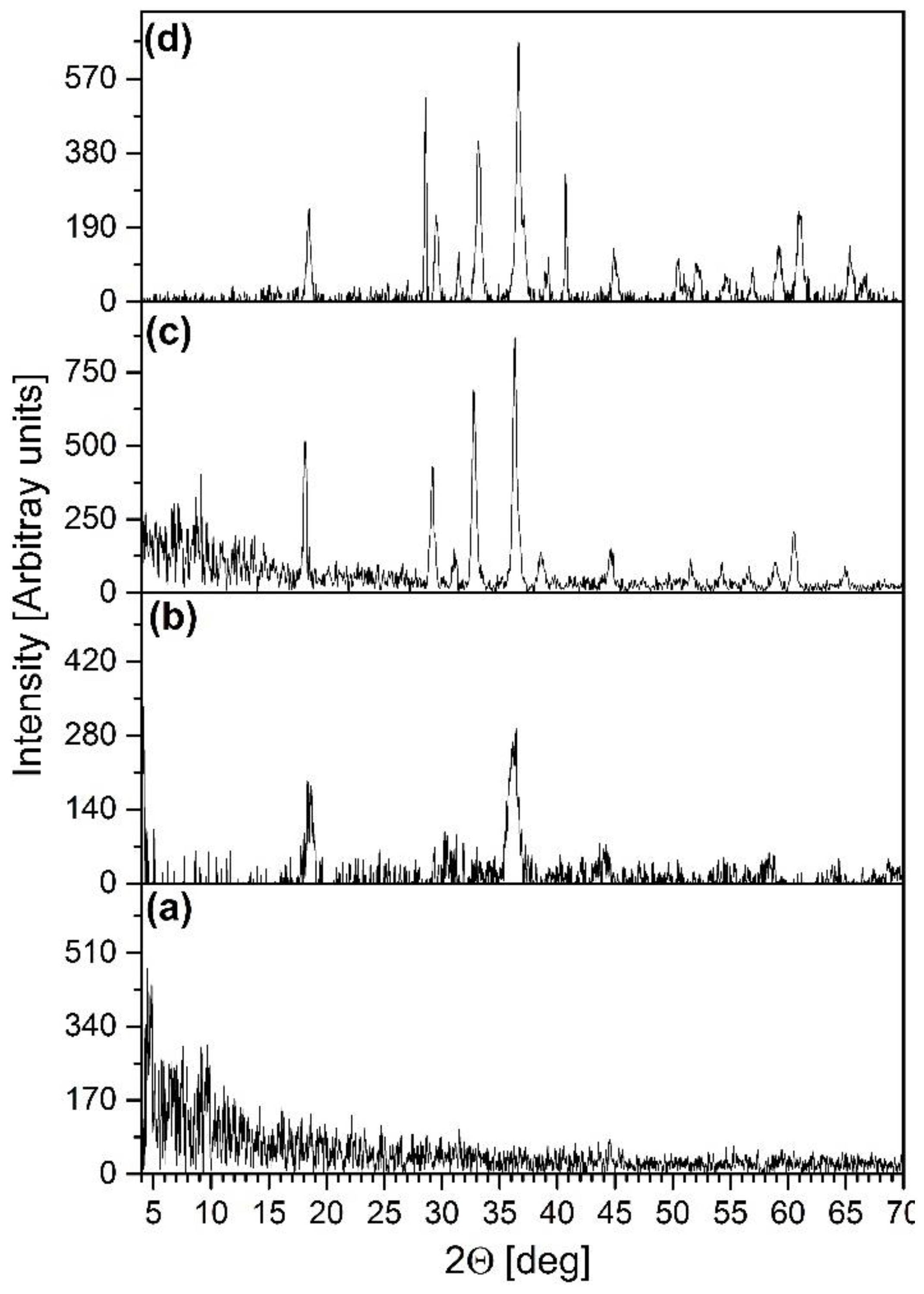
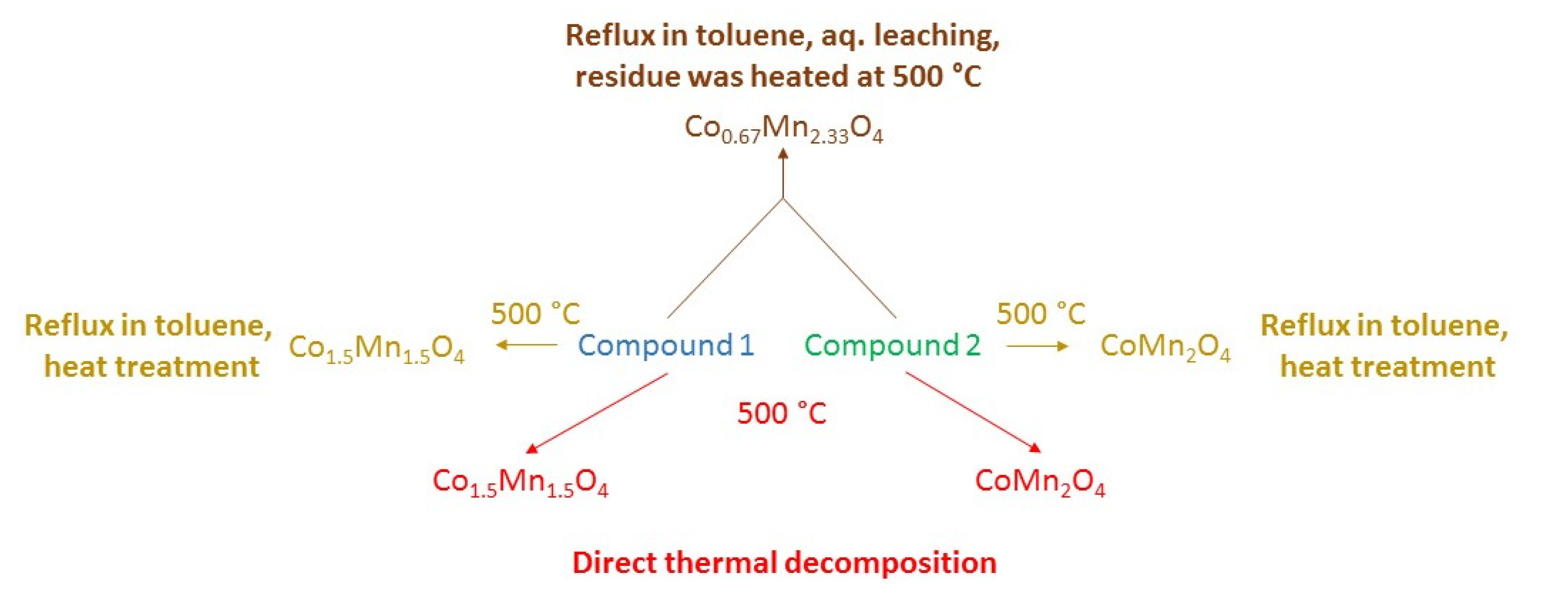
2.7. Isothermal Decomposition of Compound 1 in Boiling Toluene
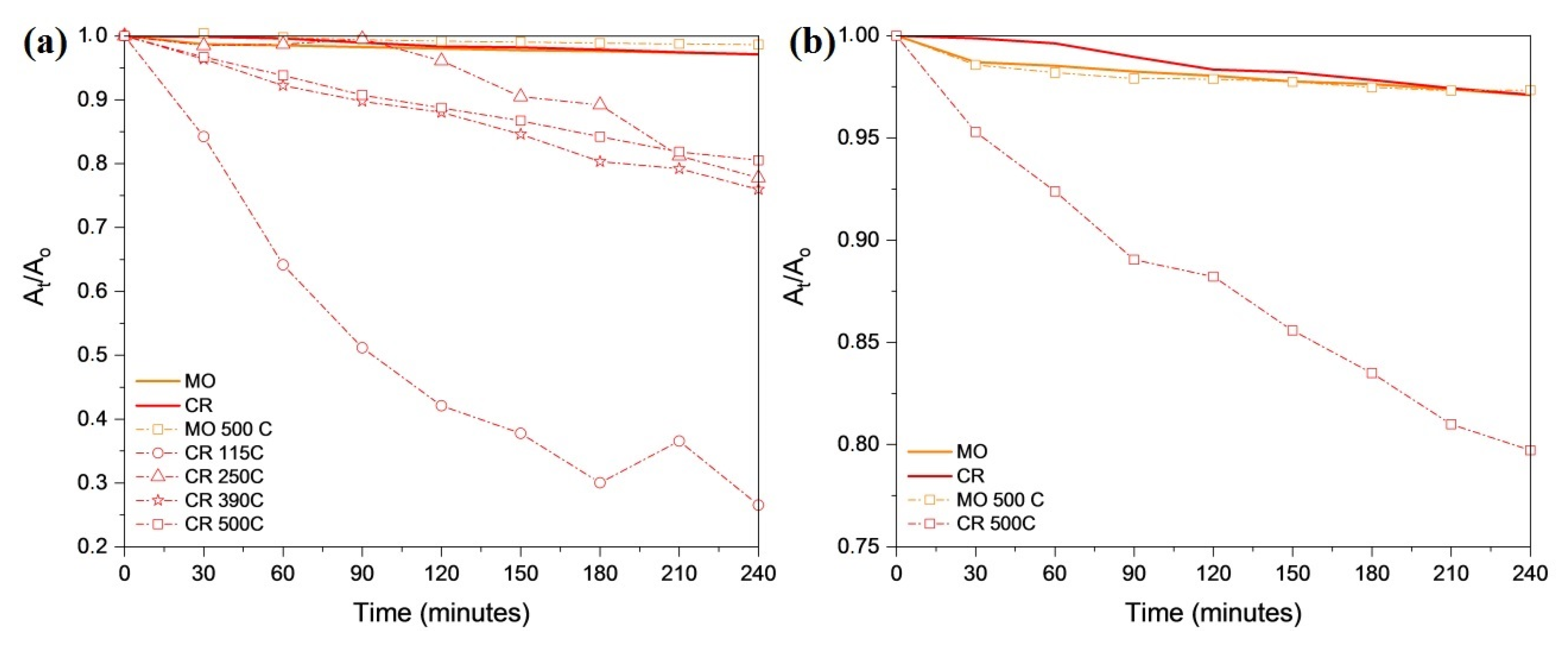
2.8. Surface Characterization of the Decomposition Products Form from Compound 1 and Their Photocatalytic Activity in the Degradation of Organic Dyes
3. Materials and Methods
3.1. Synthesis of Compound 1
3.2. Preparation of Co(NH3)6Cl3 (Compound 4)
3.3. Preparation of [Co(NH3)6](MnO4)3 (Compound 3)
3.4. Analytical Methods
3.5. Elemental Analysis
3.6. Vibrational Spectroscopy
3.7. UV−Vis Spectroscopy
3.8. Scanning Electron Microscopy
3.9. Powder X-ray Diffractometry
3.10. Single-Crystal X-ray Diffraction
3.11. Thermal Studies
3.12. Measurement of Magnetic Susceptibility
3.13. Photocatalytic Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Béres, K.A.; Homonnay, Z.; Kvitek, L.; Dürvanger, Z.; Kubikova, M.; Harmat, V.; Szilágyi, F.; Czégény, Z.; Németh, P.; Bereczki, L.; et al. Thermally Induced Solid-Phase Quasi-Intramolecular Redox Reactions of [Hexakis(urea−O)iron(III)] Permanganate: An Easy Reaction Route to Prepare Potential (Fe,Mn)Ox Catalysts for CO2 Hydrogenation. Inorg. Chem. 2022, 61, 14403–14418. [Google Scholar] [CrossRef] [PubMed]
- Bereczki, L.; Fogaca, L.A.; Durvanger, Z.; Harmat, V.; Kamarás, K.; Németh, G.; Holló, B.B.; Petruševski, V.; Bódis, E.; Farkas, A.; et al. Dynamic disorder in the high-temperature polymorph of bis[iamminesilver(I)] sulfate–reasons and consequences of simultaneous ammonia release from two different polymorphs. J. Coord. Chem. 2021, 74, 2144–2162. [Google Scholar] [CrossRef]
- Petruševski, V.M.; Béres, K.A.; Bombicz, P.; Farkas, A.; Kótai, L.; Bereczki, L. Structural and Raman Spectroscopic Characterization of Tetrapyridinesilver(I) Perrhenate, [Agpy4]ReO4. Maced. J. Chem. Chem. Eng. 2022, 41, 37–46. [Google Scholar] [CrossRef]
- Holló, B.B.; Petruševski, V.M.; Kovács, G.B.; Franguelli, F.P.; Farkas, A.; Menyhárd, A.; Lendvay, G.; Sajó, I.E.; Bereczki, L.; Pawar, R.P.; et al. Thermal and spectroscopic studies on a double-salt-type pyridine–silver perchlorate complex having κ1−O coordinated perchlorate ions. J. Therm. Anal. Calorim. 2019, 138, 1193–1205. [Google Scholar] [CrossRef]
- Béres, K.A.; Petruševski, V.; Holló, B.B.; Németh, P.; Fogaça, L.; Franguelli, F.P.; Farkas, A.; Menyhárd, A.; Szilágyi, I.M.; Kótai, L. AgNO3⋅NH4NO3 – an enigmatic double-salt type “decomposition intermediate” of diamminesilver(I) permanganate. Z. Anorg. Allg. Chem. 2021, 647, 1166. [Google Scholar] [CrossRef]
- Franguelli, F.P.; Barta Holló, B.; Petruševski, V.M.; Sajó, I.E.; Klébert, S.; Farkas, A.; Bódis, E.; Szilágyi, I.M.; Pawar, R.P.; Kótai, L. Thermal decomposition and spectral characterization of di[carbonatotetraamminecobalt(III)] sulfate trihydrate and the nature of its thermal decomposition products. J. Therm. Anal. Calorim. 2021, 145, 2907–2923. [Google Scholar] [CrossRef]
- Béres, K.A.; Sajó, I.E.; Lendvay, G.; Trif, L.; Petruševski, V.M.; Barta-Holló, B.; Korecz, L.; Franguelli, F.P.; László, K.; Szilágyi, I.M.; et al. Solid-Phase “Self-Hydrolysis” of [Zn(NH3)4MoO4@2H2O] Involving Enclathrated Water—An Easy Route to a Layered Basic Ammonium Zinc Molybdate Coordination Polymer. Molecules 2021, 26, 4022. [Google Scholar] [CrossRef]
- Franguelli, F.P.; Kováts, É.; Czégény, Z.; Bereczki, L.; Petruševski, V.M.; Barta Holló, B.; Béres, K.A.; Farkas, A.; Szilágyi, I.M.; Kótai, L. Multi-Centered Solid-Phase Quasi-Intramolecular Redox Reactions of [(Chlorido)Pentaamminecobalt(III)] Permanganate—An Easy Route to Prepare Phase Pure CoMn2O4 Spinel. Inorganics 2022, 10, 18. [Google Scholar] [CrossRef]
- Kovács, G.B.; May, N.V.; Bombicz, P.A.; Klébert, S.; Németh, P.; Menyhárd, A.; Novodárszki, G.; Petruševski, V.; Franguelli, F.P.; Magyari, J.; et al. An unknown component of a selective and mild oxidant: Structure and oxidative ability of a double salt-type complex having κ1O−coordinated permanganate anions and three- and four-fold coordinated silver cations. RSC Adv. 2019, 9, 28387–38398. [Google Scholar] [CrossRef]
- Fogaca, L.A.; Kováts, É.; Németh, G.; Kamarás, K.; Berés, K.A.; Németh, P.; Petruševski, V.; Bereczki, L.; Holló, B.B.; Sajó, I.E.; et al. Solid-Phase Quasi-Intramolecular Redox Reaction of [Ag(NH3)2]MnO4: An Easy Way to Prepare Pure AgMnO2. Inorg. Chem. 2021, 60, 3749–3760. [Google Scholar] [CrossRef]
- Franguelli, F.P.; Béres, K.A.; Kótai, L. Pyridinesilver Tetraoxometallate Complexes: Overview of the Synthesis, Structure and Properties of Pyridine Complexed AgXO4 (X = Cl, Mn, Re) Compounds. Inorganics 2021, 9, 79. [Google Scholar] [CrossRef]
- Solt, H.E.; Németh, P.; Mohai, M.; Sajó, I.E.; Klébert, S.; Franguelli, F.P.; Fogaca, L.A.; Pawar, R.P.; Kótai, L. Temperature-Limited Synthesis of Copper Manganites along the Borderline of the Amorphous/Crystalline State and Their Catalytic Activity in CO Oxidation. ACS Omega 2021, 6, 1523–1533. [Google Scholar] [CrossRef] [PubMed]
- Domonov, D.P.; Pechenyuk, S.I.; Semushina, Y.P.; Yusenko, K.V. Solid-state transformations in inner coordination sphere of [Co(NH3)6][Fe(C2O4)3]·3H2O as a route to access catalytically active Co–Fe materials. Materials 2019, 12, 221. [Google Scholar] [CrossRef] [PubMed]
- Serebrennikova, P.S.; Komarov, V.Y.; Sukhikh, A.S.; Khranenko, S.P.; Zadesenets, A.V.; Gromilov, S.A.; Yusenko, K.V. [Nien3](MoO4)0.5(WO4)0.5 co-crystals as single-source precursors for ternary refractory Ni–Mo–W alloys. Nanomaterials 2021, 11, 3272. [Google Scholar] [CrossRef]
- Garkul’, I.A.; Zadesenets, A.V.; Plyusnin, P.E.; Filatov, E.Y.; Asanova, T.I.; Kozlov, D.V.; Korenev, S.V. Zinc(II) and Manganese(II) Oxalatopalladates as Precursors of Bimetallic Nanomaterials. Russ. J. Inorg. Chem. 2020, 65, 1571–1576. [Google Scholar] [CrossRef]
- Lagunova, V.I.; Filatov, E.Y.; Plyusnin, P.E.; Korenev, S.V. In Situ and Ex Situ Studies of Tetrammineplatinum(II) Chromate Thermolysis. Russ. J. Inorg. Chem. 2020, 65, 1566–1570. [Google Scholar] [CrossRef]
- Hetmańczyk, Ł.; Hetmańczyk, J. Phase transition, thermal dissociation and dynamics of NH3 ligands in [Cd(NH3)4](ReO4)2. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2016, 164, 24–32. [Google Scholar] [CrossRef]
- Yusenko, K.V.; Pechenyuk, S.I.; Vikulova, E.S.; Semushina, Y.P.; Baidina, I.A.; Filatov, E.Y. Isostructurality and Thermal Properties in the Series of Double Complex Salts [M1(NH3)6][M2(C2O4)3]·3H2O (M1=Co, Ir, M2 = Fe, Cr). J. Struct. Chem. 2019, 60, 1062–1071. [Google Scholar] [CrossRef]
- Farhadi, S.; Pourzare, K.; Bazgir, S. Co3O4 nanoplates: Synthesis, characterization and study of optical and magnetic properties. J. Alloys Compd. 2014, 587, 632–637. [Google Scholar] [CrossRef]
- Ilyushin, M.A.; Shugalei, I.V.; Tverjanovich, A.S.; Smirnov, A.V. Influence of the Mechanism of the Initial Stages of the Ligand Decomposition on the Initiating Ability of Cobalt(III) Ammine Tetrazolate Complexes. Russ. J. Inorg. Chem. 2020, 90, 640–647. [Google Scholar] [CrossRef]
- Müller, T.G.; Mogk, J.; Conrad, M.; Kraus, F. Octaammine EuII and YbII Azides and Their Thermal Decompositions to the Nitrides. Eur. J. Inorg. Chem. 2016, 26, 4162–4169. [Google Scholar] [CrossRef]
- Gosteva, A.; Kossoy, A.; Tsvetov, N.; Semushina, Y.; Kozerozhets, I. Dynamics of Thermal Decomposition of the Double Complex Salt [Cr(ur)6][Co(CN)6]⋅4H2O. Russ. J. Inorg. Chem. 2022, 67, 1257–1263. [Google Scholar] [CrossRef]
- Saloutin, V.I.; Edilova, Y.O.; Kudyakova, Y.S.; Burgart, Y.V.; Bazhin, D.N. Heterometallic Molecular Architectures Based on Fluorinated β−Diketone Ligands. Molecules 2022, 27, 7894. [Google Scholar] [CrossRef] [PubMed]
- Tao, F.F. Synthesis, catalysis, surface chemistry and structure of bimetallic nanocatalysts. Chem. Soc. Rev. 2012, 41, 7977–7979. [Google Scholar] [CrossRef]
- Kótai, L.; Banerji, K.K.; Sajó, I.; Kristof, J.; Sreedhar, B.; Holly, S.; Keresztury, G.; Rockenbauer, A. An unprecedented-type intramolecular redox reaction of solid tetraamminecopper(2+) bis(permanganate)—[Cu(NH3)4](MnO4)2−A low-temperature synthesis of copper dimanganese tetraoxide-type (CuMn2O4) nanocrystalline catalyst precursors. Helv. Chim. Acta 2002, 85, 2316–2327. [Google Scholar] [CrossRef]
- Sajó, I.E.; Kótai, L.; Keresztury, G.; Gács, I.; Pokol, G.; Kristóf, J.; Soptrayanov, B.; Petruševski, V.; Timpu, D.; Sharma, P.K. Studies on the Chemistry of Tetraamminezinc(II) Dipermanganate ([Zn(NH3)4](MnO4)2): Low-Temperature Synthesis of the Manganese Zinc Oxide (ZnMn2O4) Catalyst Precursor. Helv. Chim. Acta 2008, 91, 1646–1658. [Google Scholar] [CrossRef]
- Kótai, L.; Sajó, I.E.; Jakab, E.; Keresztury, G.; Németh, C.; Gács, I.; Menyhárd, A.; Kristóf, J.; Hajba, L.; Petruševski, V.; et al. Studies on the chemistry of [Cd(NH3)4](MnO4)2. A low-temperature synthesis route of the CdMn2O4+x type NOx and CH3SH sensor. Z. Anorg. All. Chem. 2012, 638, 177–186. [Google Scholar] [CrossRef]
- Mansouri, M.; Atashi, H.; Tabrizi, F.F.; Mirzaei, A.A.; Mansouri, G. Kinetics studies of nano-structured cobalt-manganese oxide catalysts in Fischer-Tropsch synthesis. J. Ind. Eng. Chem. 2013, 19, 1177–1183. [Google Scholar] [CrossRef]
- Mansouri, G.; Mansouri, M. Synthesis and characterization of Co–Mn nanocatalyst prepared by thermal decomposition for Fischer-Tropsch reaction. Iran. J. Chem. Eng. 2018, 37, 1–9. [Google Scholar]
- Fogaça, L.A.; Bereczki, L.; Petruševski, V.M.; Barta-Holló, B.; Franguelli, F.P.; Mohai, M.; Béres, K.A.; Sajó, I.E.; Szilágyi, I.M.; Kotai, L. A quasi-intramolecular solid-phase redox reaction of ammonia ligands and perchlorate anion in diamminesilver(I) perchlorate. Inorganics 2021, 9, 38. [Google Scholar] [CrossRef]
- Klobb, T. De quelques permanganates nouveaux. Bull. Soc. Chim. 1887, 47, 240–244. [Google Scholar]
- Kótai, L.; Gács, L.; Sajó, I.E.; Sharma, P.K.; Banerji, K.K. Beliefs and facts in permanganate chemistry–and overview on the synthesis and the reactivity of simple and complex permanganates. Trends. Inorg. Chem. 2011, 11, 25–104. [Google Scholar] [CrossRef]
- Kótai, L.; Sajó, I.E.; Gács, I.; Pradeep, K.S.; Banerji, K.K. Convenient routes for the preparation of barium permanganate and other permanganate salts. Z. Anorg. Allg. Chem. 2007, 633, 1257–1260. [Google Scholar] [CrossRef]
- Kótai, L.; Keszler, A.; Pató, J.; Holly, S.; Banerji, K.K. The reaction of barium manganate with acids and their precursors. Ind. J. Chem. A 1998, 38A, 966–968. [Google Scholar]
- Kótai, L.; Banerji, K.K. An improved method for the preparation of high-purity permanganate salts. Synth. React. Inorg. Met.-Org. Chem. 2001, 31, 491–495. [Google Scholar] [CrossRef]
- Klobb, T. Contribution à l’étude des sels lutéocobaltiques. Bull. Soc. Chim. Paris 1901, 25, 1022–1031. [Google Scholar]
- Alvisi, G.U. Ricerche sui Perclorati. Perclorati di Luteocobaltiammine ed osservazioni sui metalloammoni. Gazz. Chim. Ital. 1901, 31, 289–301. [Google Scholar]
- Sacconi, L.; Sabatini, A.; Cans, P. Infrared Spectra from of Some Metal-Ammine Complexes. Inorg. Chem. 1964, 3, 1772–1774. [Google Scholar] [CrossRef]
- Schmidt, K.H.; Müller, A. Vibrational Spectra and Force Constants of [Cr(NH3)6]3+, [Co(NH3)6]3+, [Cu(NH3)]42+, and [Pd(NH3]42+ with 50Cr/53Cr, 63Cu/65Cu, 104Pd/110Pd, and H/D isotopic substitution. J. Mol. Struct. 1974, 22, 343–352. [Google Scholar] [CrossRef]
- Chen, Y.; Christensen, D.H.; Sorensen, G.O.; Nielsen, O.F.; Pedersen, E.B. The skeletal vibrational spectra and metal-ligand force constants of cobalt(III) ammine complexes. J. Mol. Struct. 1993, 299, 61–72. [Google Scholar] [CrossRef]
- Block, H. A vibrational study of the hexaamminecobalt(III) ion. Trans. Faraday Soc. 1959, 55, 867–875. [Google Scholar] [CrossRef]
- Téllez, C. Análisis de coordenadas normales para [Cr(NH3)6]3+ Y [Co(NH3)6]3+ con substitución isotópica 50Cr/53Cr, 14N/15N y H/D. Semin. Exact Technol. Sci. 1982, 3, 185–188. [Google Scholar] [CrossRef]
- Kiefer, W.; Bernstein, H.J. The resonance Raman effect of the permanganate and chromate ions. Mol. Phys. 1972, 23, 835–851. [Google Scholar] [CrossRef]
- Grinberg, A.A.; Varshavskii, Y.S. The frequency of coordinated ammonia deformation mode and its relationship with the chemical properties of transition metal ammonia complexes. Primen. Molekul. Spektr. Khim. 1966, 104–107. [Google Scholar]
- Baran, E.J.; Aymonino, P.J. Hexaamminecobalt(III) permanganate. Z. Anorg. Allg. Chem. 1968, 362, 215–219. [Google Scholar] [CrossRef]
- Hendry, P.; Ludi, A. Structure, Reactivity, Spectra, and Redox Properties of Cobalt(III) Hexaamines. Adv. Inorg. Chem. 1990, 35, 117–198. [Google Scholar]
- Mitsutsuka, Y.; Kondo, I.; Nakahara, M. Splitting parameters in Ligand field absorption bands of cobalt(III) ammine complexes with chloro or aqua ligands. Bull. Chem. Soc. Jpn. 1986, 59, 2767–2771. [Google Scholar] [CrossRef]
- Sakiyama, H.; Ishiyama, Y.; Sugawara, H. Trigonallycompressed octahedral geometry of hexaamminecobalt(III) complex cation in aqueous solution investigated from the electronic spectra. Spectrosc. Lett. 2017, 50, 111–114. [Google Scholar] [CrossRef]
- Wilson, R.B.; Solomon, E.I. Spectroscopic Studies of the Photoactive 4T2g Excited State of Hexaamminechromium(lII). Inorg. Chem. 1978, 17, 1729–1736. [Google Scholar] [CrossRef]
- Benedix, R.; Hennig, H.; Nieke, C. NDDO/CI Calculations of the Electronic Spectra of Cobalt(III) Ammine Complexes. Inorg. Chim. Acta 1990, 172, 109–112. [Google Scholar] [CrossRef]
- Wilson, R.B.; Solomon, E.I. Spectroscopic Studies of Photochemically Important Transition Metal Excited States. 2. The 1T1g, 3T1g, and 5T2g Excited States of Hexaamminecobalt(III). J. Am. Chem. Soc. 1980, 102, 4085–4095. [Google Scholar] [CrossRef]
- Kocsis, T.; Magyari, J.; Sajó, I.E.; Pasinszki, T.; Homonnay, Z.; Szilagyi, I.M.; Farkas, A.; May, Z.; Effenberger, H.; Szakall, S.; et al. Evidence of quasi-intramolecular redox reactions during thermal decomposition of ammonium hydroxodisulfitoferriate(III), (NH4)2[Fe(OH)(SO3)2]H2O. J. Therm. Anal. Calorim. 2018, 132, 493–502. [Google Scholar] [CrossRef]
- Habibi, M.H.; Bagheri, P. Spinel cobalt manganese oxide nano-composites grown hydrothermally on nanosheets for enhanced photocatalytic mineralization of Acid Black 1 textile dye: XRD, FTIR, FESEM, EDS and TOC studies. J. Iran. Chem. Soc. 2017, 14, 1643–1649. [Google Scholar] [CrossRef]
- Naresh, B.; Kuchi, C.; Rajasekhar, D.; Reddy, P.S. Solvothermal synthesis of MnCo2O4 microspheres for high-performance electrochemical supercapacitors. Colloids Surf. A: Physicochem. Eng. Asp. 2022, 640, 128443. [Google Scholar] [CrossRef]
- Bordeneuve, H.; Guillemet-Fritsch, S.; Rousset, A.; Schuurman, S.; Poulain, V. Structure and electrical properties of single-phase cobalt manganese oxide spinels Mn3−xCoxO4 sintered classically and by spark plasma sintering(SPS). J. Solid State Chem. 2009, 182, 396–401. [Google Scholar] [CrossRef]
- Wickham, D.G.; Croft, W.J. Crystallographic and magnetic properties of several spinels containing trivalent manganese. J. Phys. Chem. Solids 1958, 7, 351–360. [Google Scholar] [CrossRef]
- Boucher, B.; Buhl, R.; Di Bella, R.; Perrin, M. Etude par des mesures de diffraction de neutrons et de magnétisme des propriétés cristallines et magnétiques de composés cubiques spinelles Co3−xMnxO4(0,6 ≤ x ≤ 1,2). J. Phys. Fr. 1970, 31, 113–119. [Google Scholar] [CrossRef]
- Kolomiets, B.T.; Sheftel, J.; Kurlina, E. Electrical Properties of Some Complex Oxide Semiconductors. Sov. Phys. Tech. Phys. 1957, 2, 40–58. [Google Scholar]
- Blasse, G. Superexchange in the spinel structure. Some magnetic properties of oxides M2+ Co2O4 and M2+ Rh2O4 with spinel structure. Philips Res. Rep. 1963, 18, 383–392. [Google Scholar]
- Gautier, J.L.; Barbato, S.; Brenet, J. Ionic repartition in the manganese cobaltite. C. R. Acad. Sci. 1982, 294, 427–430. [Google Scholar]
- Jabry, E.; Rousset, A.; Lagrange, A. Preparation and characterization of manganese and cobalt based semiconducting ceramics. Phase Transit. 1988, 13, 63–71. [Google Scholar] [CrossRef]
- Aoki, I. Tetragonal Distortion of the Oxide Spinels Containing Cobalt and Manganese. J. Phys. Soc. Jpn. 1962, 17, 53–61. [Google Scholar] [CrossRef]
- Krebs, B.; Müller, A.; Roesky, H.W. Kraftkonstanten tetraedrischer Oxoanionen des Mangans (MnO4−, MnO42−, MnO43−) und Rutheniums (RuO4, RuO4−, RuO42−). Mol. Phys. 1967, 12, 469–474. [Google Scholar] [CrossRef]
- Vratny, F. Infrared Spectra of Metal Nitrates. Appl. Spectrosc. 1959, 13, 59–70. [Google Scholar] [CrossRef]
- Kótai, L.; Argay, G.; Holly, S.; Keszler, Á.; Pukánszky, B.; Baneerji, K.K. Study on the Existence of Hydrogen Bonds in Ammonium Permanganate. Z. Anorg. Allg. Chem. 2001, 627, 114–118. [Google Scholar] [CrossRef]
- Wendland, W.W.; Smith, J.P. The thermal decomposition of metal complexes–VII a thermomagnetic study of the Co(III)~Co(II) reduction in cobalt(III) ammine complexes. J. Inorg. Nucl. Chem. 1963, 25, 1267–1272. [Google Scholar] [CrossRef]
- Tanaka, Y.; Nanjo, M. The mechanism of the thermal decomposition of hexaamminecobalt(III) chloride. Bull. Soc. Chem. Jpn. 1964, 37, 1330–1332. [Google Scholar] [CrossRef]
- Wendlandt, W.W.; Smith, J.P. Thermal decomposition of metal complexes—VI some ammine cobalt (II) complexes. J. Inorg. Nucl. Chem. 1963, 25, 985–993. [Google Scholar] [CrossRef]
- Potter, R.M.; Rossman, G.R. The tetravalent manganese oxides: Identification, hydration and structural relationships by infrared spectroscopy. Am. Miner. 1979, 64, 1199–1218. [Google Scholar]
- Turner, S.; Buseck, P.R. Todorokites: A new family of naturally occurring manganese oxides. Science 1981, 212, 1024–1027. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Sachdeva, S.; Papadaki, M.I.; Mannan, M.S. Ammonium nitrate thermal decomposition with additives. J. Loss Prev. Process Ind. 2015, 35, 307–315. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Dave, P.N. Review on thermal decomposition of ammonium nitrate. J. Energ. Mater. 2013, 31, 1–26. [Google Scholar] [CrossRef]
- Jörgensen, S.M., VIII. Ueber die Luteokobaltsalze. J. Prakt. Chem. 1887, 35, 417–448. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Label |
|---|---|
| [Co(NH3)6]Cl2(MnO4) | 1 |
| [Co(NH3)5Cl](MnO4)2 | 2 |
| [Co(NH3)6](MnO4)3 | 3 |
| [Co(NH3)6]Cl3 | 4 |
| [Co(NH3)6]Cl2(ClO4) | 5 |
| [Co(NH3)6](ClO4)3 | 6 |
| Empirical Formula | [Co(NH3)6]Cl2(MnO4) |
|---|---|
| Formula weight | 350.97 g·mol−1 |
| Crystal system | Monoclinic |
| Space group | P21/c |
| Unit cell dimensions, Å | a = 13.6133 (7) b = 7.3658 (5) c = 12.3682 (6); |
| β = 108.547 (8)° | |
| Z | 4 |
| Density (calcd.) (g·cm−3) | 1.983 |
| Temperature (K) | 163 |
| Volume (Å3) | 1175.78 (13) |
| R factor (%) | 4.22 |
| CSD deposition number | 2,220,607 |
| Temperature, °C | 115 | 135 | 160 | 250 | 390 | 500 | |
|---|---|---|---|---|---|---|---|
| Specific surface area, m2·g−1 | Under N2 | 6 | 17 | 16 | 7 | 27 | 8 |
| Under air | − | 6 | 4 | 4 | − | 35 | |
| Dyes (2∙10−5 M) and Catalysts Parameters | pH | Kapp/10−4, min−1 | R2 |
|---|---|---|---|
| Congo red, without catalyst | 5.7 | 1.0 | 0.98 |
| Congo red, catalyst made at 115 °C in N2 | 5.7 | 54.0 | 0.93 |
| Congo red, catalyst made at 250 °C in N2 | 5.7 | 12.0 | 0.87 |
| Congo red, catalyst made at 390 °C in N2 | 5.7 | 11.0 | 0.99 |
| Congo red, catalyst made at 500 °C in N2 | 5.7 | 9.0 | 0.99 |
| Congo red, catalyst made at 500 °C in air | 5.7 | 9.0 | 0.98 |
| Methyl orange, without catalyst | 5.6 | 0.8 | 0.99 |
| Methyl orange, catalyst made at 500 °C in N2 | 5.6 | 0.6 | 0.97 |
| Methyl orange, catalyst made at 500 °C in air | 5.6 | 0.6 | 0.94 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bereczki, L.; Petruševski, V.M.; Franguelli, F.P.; Béres, K.A.; Farkas, A.; Holló, B.B.; Czégény, Z.; Szilágyi, I.M.; Kótai, L. [Hexaamminecobalt(III)] Dichloride Permanganate—Structural Features and Heat-Induced Transformations into (CoII,MnII)(CoIII,MnIII)2O4 Spinels. Inorganics 2022, 10, 252. https://doi.org/10.3390/inorganics10120252
Bereczki L, Petruševski VM, Franguelli FP, Béres KA, Farkas A, Holló BB, Czégény Z, Szilágyi IM, Kótai L. [Hexaamminecobalt(III)] Dichloride Permanganate—Structural Features and Heat-Induced Transformations into (CoII,MnII)(CoIII,MnIII)2O4 Spinels. Inorganics. 2022; 10(12):252. https://doi.org/10.3390/inorganics10120252
Chicago/Turabian StyleBereczki, Laura, Vladimir M. Petruševski, Fernanda Paiva Franguelli, Kende Attila Béres, Attila Farkas, Berta Barta Holló, Zsuzsanna Czégény, Imre Miklós Szilágyi, and László Kótai. 2022. "[Hexaamminecobalt(III)] Dichloride Permanganate—Structural Features and Heat-Induced Transformations into (CoII,MnII)(CoIII,MnIII)2O4 Spinels" Inorganics 10, no. 12: 252. https://doi.org/10.3390/inorganics10120252
APA StyleBereczki, L., Petruševski, V. M., Franguelli, F. P., Béres, K. A., Farkas, A., Holló, B. B., Czégény, Z., Szilágyi, I. M., & Kótai, L. (2022). [Hexaamminecobalt(III)] Dichloride Permanganate—Structural Features and Heat-Induced Transformations into (CoII,MnII)(CoIII,MnIII)2O4 Spinels. Inorganics, 10(12), 252. https://doi.org/10.3390/inorganics10120252