Charge Photogeneration and Recombination Dynamics in PTQ10:Y6 Solar Cells

 ,
, {kind=link}
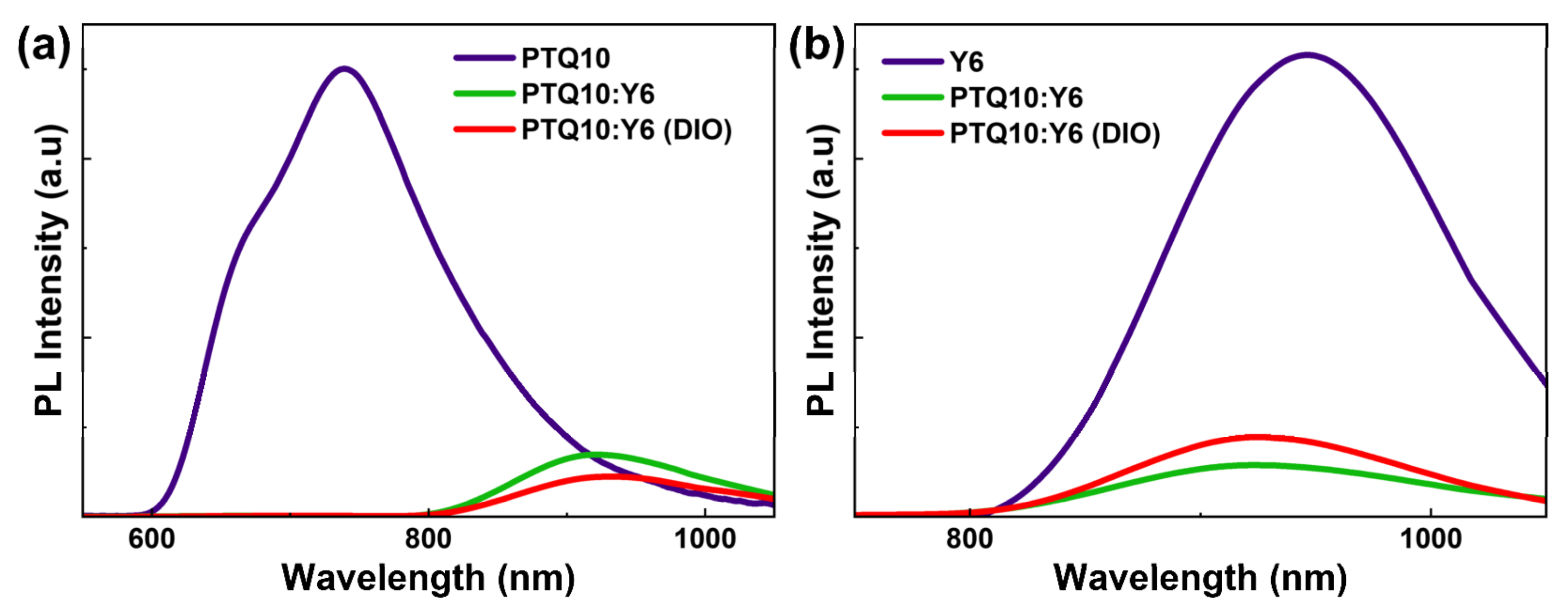{kind=link}
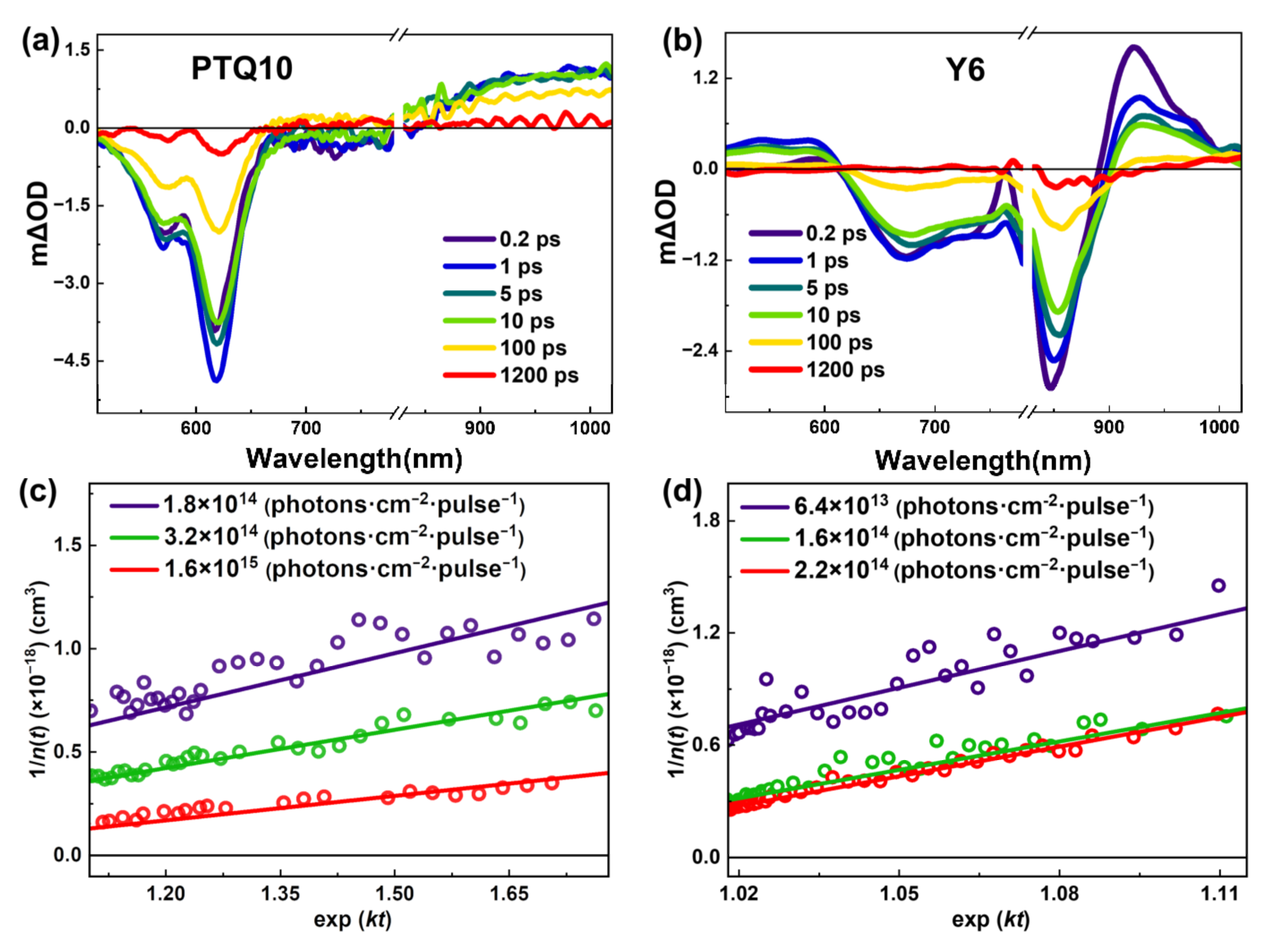{kind=link}
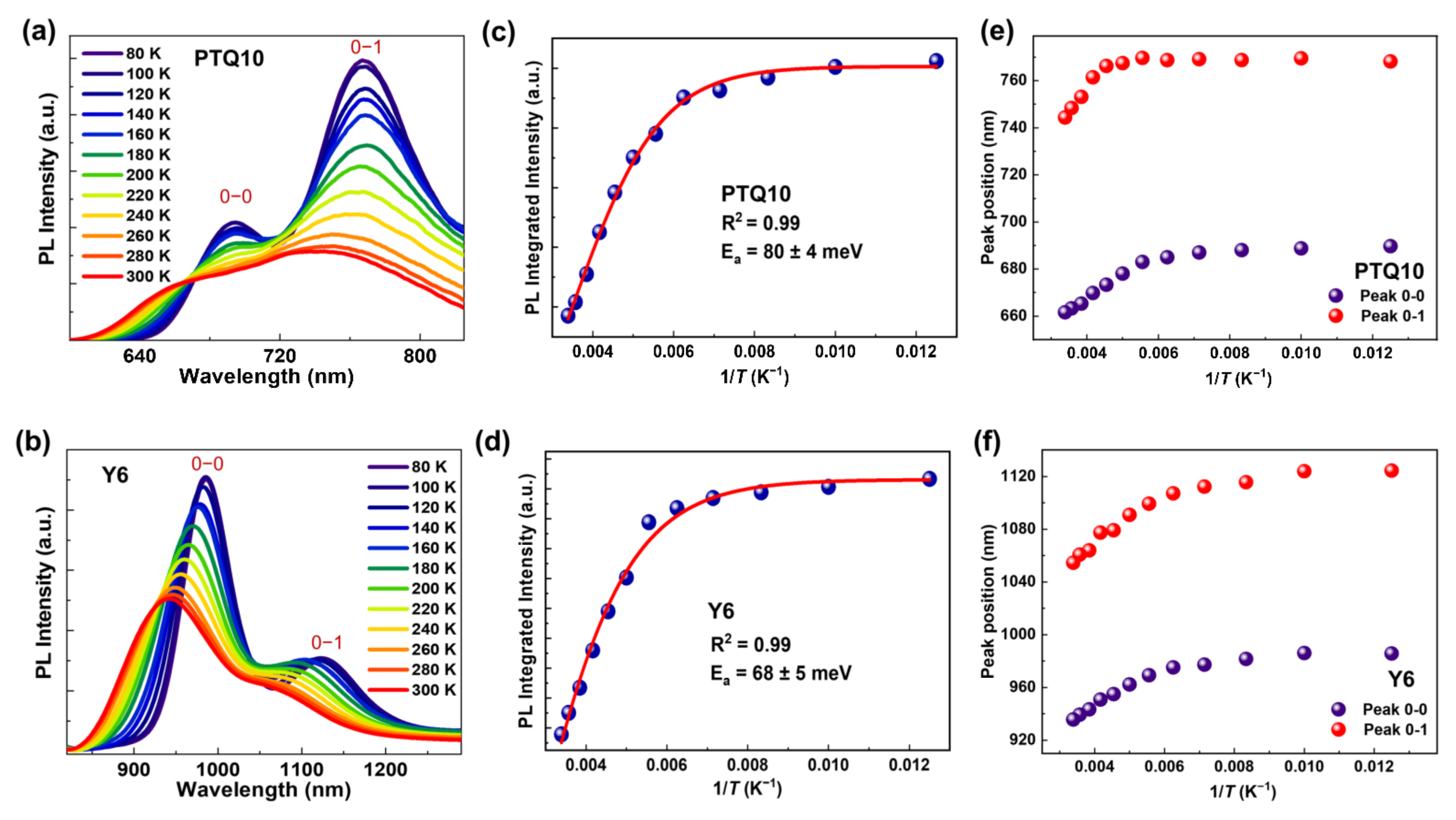{kind=link}
{kind=link}
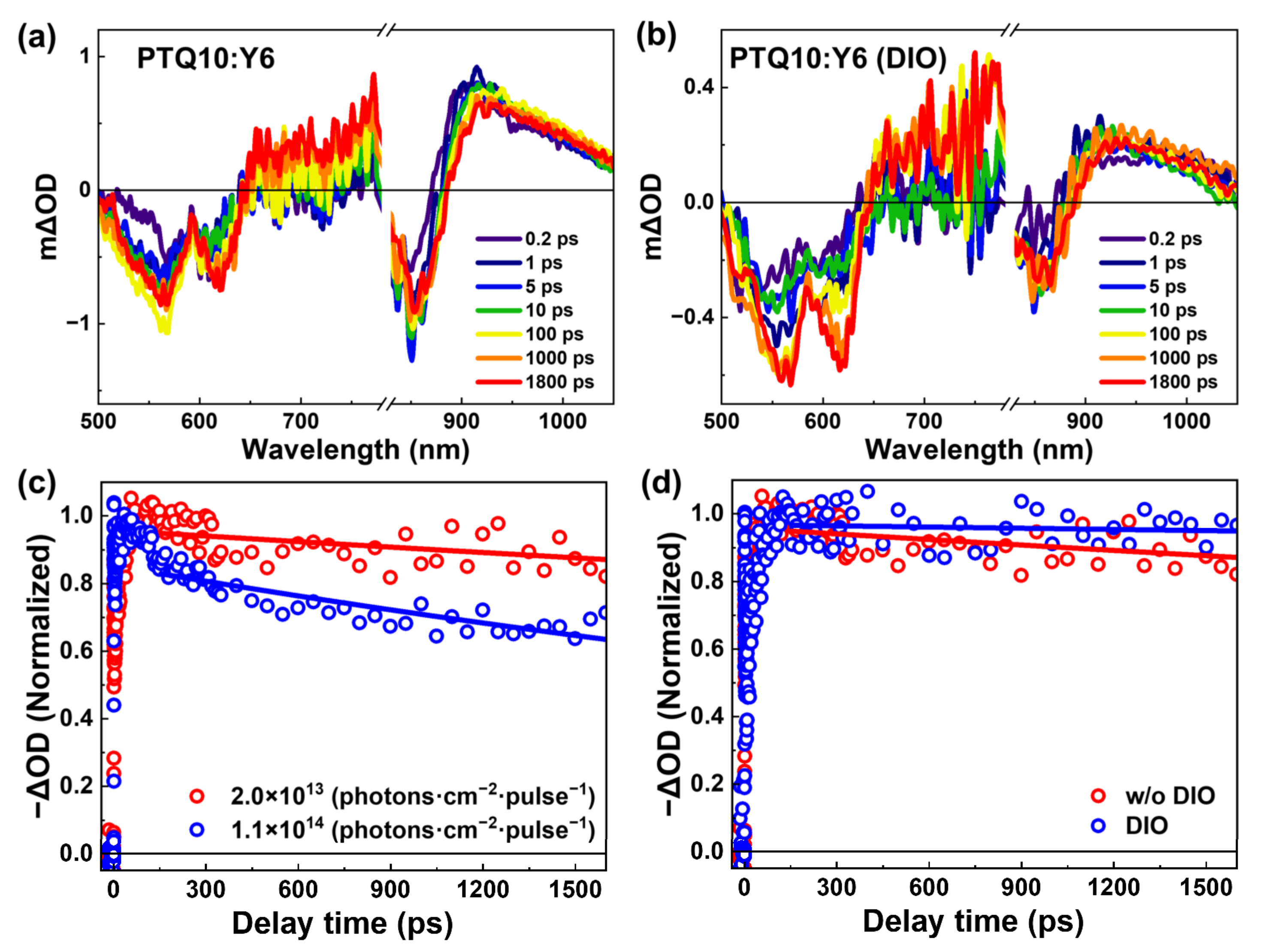{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Information and Film Preparation
2.2. Steady-State Spectra Measurements
2.3. Transient Absorption Measurements
3. Results and Discussion
3.1. Molecular Structures and Steady-State Spectra
3.2. Exciton Properties of PTQ10 and Y6 Neat Films
3.3. Temperature-Dependent PL Properties of PTQ10 and Y6 Film
3.4. Charge Photogeneration and Recombination Processes in PTQ10:Y6 Blend Films
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gillett, A.J.; Privitera, A.; Dilmurat, R.; Karki, A.; Qian, D.; Pershin, A.; Londi, G.; Myers, W.K.; Lee, J.; Yuan, J.; et al. The Role of Charge Recombination to Triplet Excitons in Organic Solar Cells. Nature 2021, 597, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.W. Two-layer Organic Photovoltaic Cell. Appl. Phys. Lett. 1986, 48, 183–185. [Google Scholar] [CrossRef]
- Wadsworth, A.; Moser, M.; Marks, A.; Little, M.S.; Gasparini, N.; Brabec, C.J.; Baran, D.; McCulloch, I. Critical Review of the Molecular Design Progress in Non-Fullerene Electron Acceptors towards Commercially Viable Organic Solar Cells. Chem. Soc. Rev. 2019, 48, 1596–1625. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Inganäs, O.; Friend, R.H.; Gao, F. Organic Solar Cells Based on Non-Fullerene Acceptors. Nat. Mater. 2018, 17, 119–128. [Google Scholar] [CrossRef]
- Zhang, G.; Zhao, J.; Chow, P.C.Y.; Jiang, K.; Zhang, J.; Zhu, Z.; Zhang, J.; Huang, F.; Yan, H. Nonfullerene Acceptor Molecules for Bulk Heterojunction Organic Solar Cells. Chem. Rev. 2018, 118, 3447–3507. [Google Scholar] [CrossRef]
- Wang, G.; Melkonyan, F.S.; Facchetti, A.; Marks, T.J. All-Polymer Solar Cells: Recent Progress, Challenges, and Prospects. Angew. Chem. Int. Ed. 2019, 58, 4129–4142. [Google Scholar] [CrossRef]
- Yuan, J.; Zhang, Y.; Zhou, L.; Zhang, G.; Yip, H.-L.; Lau, T.-K.; Lu, X.; Zhu, C.; Peng, H.; Johnson, P.A.; et al. Single-Junction Organic Solar Cell with over 15% Efficiency Using Fused-Ring Acceptor with Electron-Deficient Core. Joule 2019, 3, 1140–1151. [Google Scholar] [CrossRef]
- Zhu, L.; Zhang, M.; Xu, J.; Li, C.; Yan, J.; Zhou, G.; Zhong, W.; Hao, T.; Song, J.; Xue, X.; et al. Single-Junction Organic Solar Cells with over 19% Efficiency Enabled by a Refined Double-Fibril Network Morphology. Nat. Mater. 2022, 21, 656–663. [Google Scholar] [CrossRef]
- Ma, R.; Yan, C.; Fong, P.W.-K.; Yu, J.; Liu, H.; Yin, J.; Huang, J.; Lu, X.; Yan, H.; Li, G. In Situ and Ex Situ Investigations on Ternary Strategy and Co-Solvent Effects towards High-Efficiency Organic Solar Cells. Energy Environ. Sci. 2022, 15, 2479–2488. [Google Scholar] [CrossRef]
- He, Q.; Sheng, W.; Zhang, M.; Xu, G.; Zhu, P.; Zhang, H.; Yao, Z.; Gao, F.; Liu, F.; Liao, X.; et al. Revealing Morphology Evolution in Highly Efficient Bulk Heterojunction and Pseudo-Planar Heterojunction Solar Cells by Additives Treatment. Adv. Energy Mater. 2021, 11, 2003390. [Google Scholar] [CrossRef]
- Song, J.; Zhu, L.; Li, C.; Xu, J.; Wu, H.; Zhang, X.; Zhang, Y.; Tang, Z.; Liu, F.; Sun, Y. High-Efficiency Organic Solar Cells with Low Voltage Loss Induced by Solvent Additive Strategy. Matter 2021, 4, 2542–2552. [Google Scholar] [CrossRef]
- Su, X.; Hu, R.; Wen, G.; Zou, X.; Qing, M.; Peng, J.; He, X.; Zhang, W. Understanding of Photophysical Processes in DIO Additive-Treated PTB7:PC71BM Solar Cells. Crystals 2021, 11, 1139. [Google Scholar] [CrossRef]
- Zhao, L.; Zhao, S.; Xu, Z.; Qiao, B.; Huang, D.; Xu, X. Two Effects of 1,8-Diiodooctane on PTB7-Th:PC71BM Polymer Solar Cells. Org. Electron. 2016, 34, 188–192. [Google Scholar] [CrossRef]
- Zusan, A.; Gieseking, B.; Zerson, M.; Dyakonov, V.; Magerle, R.; Deibel, C. The Effect of Diiodooctane on the Charge Carrier Generation in Organic Solar Cells Based on the Copolymer PBDTTT-C. Sci. Rep. 2015, 5, 8286. [Google Scholar] [CrossRef]
- Wang, J.; Jiao, C.; Huang, H.; Zhang, F. The Effect of DIO Additive on Performance Improvement of Polymer Solar Cells. Chin. Sci. Bull. 2014, 59, 3227–3231. [Google Scholar] [CrossRef]
- Huo, M.-M.; Hu, R.; Zhang, Q.-S.; Chen, S.; Gao, X.; Zhang, Y.; Yan, W.; Wang, Y. Morphology and Carrier Non-Geminate Recombination Dynamics Regulated by Solvent Additive in Polymer/Fullerene Solar Cells. RSC Adv. 2020, 10, 23128–23135. [Google Scholar] [CrossRef]
- Ding, Y.; Zhang, X.; Feng, H.; Ke, X.; Meng, L.; Sun, Y.; Guo, Z.; Cai, Y.; Jiao, C.; Wan, X.; et al. Subtle Morphology Control with Binary Additives for High-Efficiency Non-Fullerene Acceptor Organic Solar Cells. ACS Appl. Mater. Interfaces 2020, 12, 27425–27432. [Google Scholar] [CrossRef]
- Choi, J.Y.; Han, Y.W.; Jeon, S.J.; Ko, E.J.; Moon, D.K. Introduction of Co-Additives to Form Well Dispersed Photoactive Layer to Improve Performance and Stability of Organic Solar Cells. Sol. Energy 2019, 185, 1–12. [Google Scholar] [CrossRef]
- Sun, C.; Pan, F.; Bin, H.; Zhang, J.; Xue, L.; Qiu, B.; Wei, Z.; Zhang, Z.-G.; Li, Y. A Low Cost and High Performance Polymer Donor Material for Polymer Solar Cells. Nat. Commun. 2018, 9, 743. [Google Scholar] [CrossRef]
- Fan, H.; Yang, H.; Wu, Y.; Yildiz, O.; Zhu, X.; Marszalek, T.; Blom, P.W.M.; Cui, C.; Li, Y. Anthracene-Assisted Morphology Optimization in Photoactive Layer for High-Efficiency Polymer Solar Cells. Adv. Funct. Mater. 2021, 31, 2103944. [Google Scholar] [CrossRef]
- Sun, C.; Pan, F.; Chen, S.; Wang, R.; Sun, R.; Shang, Z.; Qiu, B.; Min, J.; Lv, M.; Meng, L.; et al. Achieving Fast Charge Separation and Low Nonradiative Recombination Loss by Rational Fluorination for High-Efficiency Polymer Solar Cells. Adv. Mater. 2019, 31, 1905480. [Google Scholar] [CrossRef] [PubMed]
- Spano, F.C. The Spectral Signatures of Frenkel Polarons in H- and J-Aggregates. Acc. Chem. Res. 2010, 43, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Wen, G.; Hu, R.; Dong, G.; Zhang, C.; Zhang, W.; Huang, H.; Dang, W. An Insight into the Excitation States of Small Molecular Semiconductor Y6. Molecules 2020, 25, 4118. [Google Scholar] [CrossRef]
- Cha, H.; Zheng, Y.; Dong, Y.; Lee, H.H.; Wu, J.; Bristow, H.; Zhang, J.; Lee, H.K.H.; Tsoi, W.C.; Bakulin, A.A.; et al. Exciton and Charge Carrier Dynamics in Highly Crystalline PTQ10:IDIC Organic Solar Cells. Adv. Energy Mater. 2020, 10, 2001149. [Google Scholar] [CrossRef]
- Shin, H.-Y.; Woo, J.H.; Gwon, M.J.; Barthelemy, M.; Vomir, M.; Muto, T.; Takaishi, K.; Uchiyama, M.; Hashizume, D.; Aoyama, T.; et al. Exciton Diffusion in Near-Infrared Absorbing Solution-Processed Organic Thin Films. Phys. Chem. Chem. Phys. 2013, 15, 2867–2872. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.J.; Ruseckas, A.; Gaudin, O.P.M.; Webster, G.R.; Burn, P.L.; Samuel, I.D.W. Singlet Exciton Diffusion in MEH-PPV Films Studied by Exciton–Exciton Annihilation. Org. Electron. 2006, 7, 452–456. [Google Scholar] [CrossRef]
- Shaw, P.E.; Ruseckas, A.; Samuel, I.D.W. Exciton Diffusion Measurements in Poly(3-Hexylthiophene). Adv. Mater. 2008, 20, 3516–3520. [Google Scholar] [CrossRef]
- Hedley, G.J.; Ruseckas, A.; Samuel, I.D.W. Light Harvesting for Organic Photovoltaics. Chem. Rev. 2017, 117, 796–837. [Google Scholar] [CrossRef]
- Wen, G.; Zou, X.; Hu, R.; Peng, J.; Chen, Z.; He, X.; Dong, G.; Zhang, W. Ground- and Excited-State Characteristics in Photovoltaic Polymer N2200. RSC Adv. 2021, 11, 20191–20199. [Google Scholar] [CrossRef]
- Long, Y.; Hedley, G.J.; Ruseckas, A.; Chowdhury, M.; Roland, T.; Serrano, L.A.; Cooke, G.; Samuel, I.D.W. Effect of Annealing on Exciton Diffusion in a High Performance Small Molecule Organic Photovoltaic Material. ACS Appl. Mater. Interfaces 2017, 9, 14945–14952. [Google Scholar] [CrossRef]
- Riley, D.B.; Sandberg, O.J.; Li, W.; Meredith, P.; Armin, A. Quasi-Steady-State Measurement of Exciton Diffusion Lengths in Organic Semiconductors. Phys. Rev. Appl. 2022, 17, 024076. [Google Scholar] [CrossRef]
- Sajjad, M.T.; Ruseckas, A.; Jagadamma, L.K.; Zhang, Y.; Samuel, I.D.W. Long-Range Exciton Diffusion in Non-Fullerene Acceptors and Coarse Bulk Heterojunctions Enable Highly Efficient Organic Photovoltaics. J. Mater. Chem. A 2020, 8, 15687–15694. [Google Scholar] [CrossRef]
- Chandrabose, S.; Chen, K.; Barker, A.J.; Sutton, J.J.; Prasad, S.K.K.; Zhu, J.; Zhou, J.; Gordon, K.C.; Xie, Z.; Zhan, X.; et al. High Exciton Diffusion Coefficients in Fused Ring Electron Acceptor Films. J. Am. Chem. Soc. 2019, 141, 6922–6929. [Google Scholar] [CrossRef]
- Zhu, L.; Zhang, J.; Guo, Y.; Yang, C.; Yi, Y.; Wei, Z. Small Exciton Binding Energies Enabling Direct Charge Photogeneration Towards Low-Driving-Force Organic Solar Cells. Angew. Chem. Int. Ed. 2021, 60, 15348–15353. [Google Scholar] [CrossRef]
- Lim, S.-H.; Bjorklund, T.G.; Bardeen, C.J. Temperature-Dependent Exciton Dynamics in Poly(p-Phenylene Vinylene) Measured by Femtosecond Transient Spectroscopy. Chem. Phys. Lett. 2001, 342, 555–562. [Google Scholar] [CrossRef]
- Guha, S.; Rice, J.D.; Yau, Y.T.; Martin, C.M.; Chandrasekhar, M.; Chandrasekhar, H.R.; Guentner, R.; Scanduicci de Freitas, P.; Scherf, U. Temperature-Dependent Photoluminescence of Organic Semiconductors with Varying Backbone Conformation. Phys. Rev. B 2003, 67, 125204. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, L.; Xu, C.; Jin, P.; Huang, M.; Li, Y.; Wang, H.; Yi, Y.; Zhang, C.; Yang, Y.; et al. Single Photovoltaic Material Solar Cells with Enhanced Exciton Dissociation and Extended Electron Diffusion. Cell Rep. Phys. Sci. 2022, 3, 100895. [Google Scholar] [CrossRef]
- Chen, Z.; Zhu, H. Photoinduced Charge Transfer and Recombination Dynamics in Star Nonfullerene Organic Solar Cells. J. Phys. Chem. Lett. 2022, 13, 1123–1130. [Google Scholar] [CrossRef]
- Chong, K.; Xu, X.; Meng, H.; Xue, J.; Yu, L.; Ma, W.; Peng, Q. Realizing 19.05% Efficiency Polymer Solar Cells by Progressively Improving Charge Extraction and Suppressing Charge Recombination. Adv. Mater. 2022, 34, 2109516. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, W.; Zhou, G.; Yi, Y.; Xu, S.; Liu, F.; Zhu, H.; Zhu, X. Accurate Determination of the Minimum HOMO Offset for Efficient Charge Generation Using Organic Semiconducting Alloys. Adv. Energy Mater. 2020, 10, 1903298. [Google Scholar] [CrossRef]
- Ruderer, M.A.; Guo, S.; Meier, R.; Chiang, H.-Y.; Körstgens, V.; Wiedersich, J.; Perlich, J.; Roth, S.V.; Müller-Buschbaum, P. Solvent-Induced Morphology in Polymer-Based Systems for Organic Photovoltaics. Adv. Funct. Mater. 2011, 21, 3382–3391. [Google Scholar] [CrossRef]
- Zhang, W.; Hu, R.; Li, D.; Huo, M.-M.; Ai, X.-C.; Zhang, J.-P. Primary Dynamics of Exciton and Charge Photogeneration in Solvent Vapor Annealed P3HT/PCBM Films. J. Phys. Chem. C 2012, 116, 4298–4310. [Google Scholar] [CrossRef]
- Zhang, W.; Zhao, N.-J.; Huo, M.-M.; Fu, L.-M.; Ai, X.-C.; Zhang, J.-P. Subnanosecond Charge Recombination Dynamics in P3HT/PC61BM Films. Molecules 2012, 17, 13923–13936. [Google Scholar] [CrossRef] [PubMed]
- Clarke, T.M.; Durrant, J.R. Charge Photogeneration in Organic Solar Cells. Chem. Rev. 2010, 110, 6736–6767. [Google Scholar] [CrossRef] [PubMed]
- Kyaw, A.K.K.; Wang, D.H.; Luo, C.; Cao, Y.; Nguyen, T.-Q.; Bazan, G.C.; Heeger, A.J. Effects of Solvent Additives on Morphology, Charge Generation, Transport, and Recombination in Solution-Processed Small-Molecule Solar Cells. Adv. Energy Mater. 2014, 4, 1301469. [Google Scholar] [CrossRef]
- Peet, J.; Brocker, E.; Xu, Y.; Bazan, G.C. Controlledβ-Phase Formation in Poly(9,9-Di-n-Octylfluorene) by Processing with Alkyl Additives. Adv. Mater. 2008, 20, 1882–1885. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.; Wen, G.; Xiao, Z.; Peng, J.; Hu, R.; Chen, Z.; Zhang, C.; Zhang, W. Charge Photogeneration and Recombination Dynamics in PTQ10:Y6 Solar Cells. Photonics 2022, 9, 892. https://doi.org/10.3390/photonics9120892
Chen C, Wen G, Xiao Z, Peng J, Hu R, Chen Z, Zhang C, Zhang W. Charge Photogeneration and Recombination Dynamics in PTQ10:Y6 Solar Cells. Photonics. 2022; 9(12):892. https://doi.org/10.3390/photonics9120892
Chicago/Turabian StyleChen, Chuan, Guanzhao Wen, Zijie Xiao, Jun Peng, Rong Hu, Zhifeng Chen, Chengyun Zhang, and Wei Zhang. 2022. "Charge Photogeneration and Recombination Dynamics in PTQ10:Y6 Solar Cells" Photonics 9, no. 12: 892. https://doi.org/10.3390/photonics9120892
APA StyleChen, C., Wen, G., Xiao, Z., Peng, J., Hu, R., Chen, Z., Zhang, C., & Zhang, W. (2022). Charge Photogeneration and Recombination Dynamics in PTQ10:Y6 Solar Cells. Photonics, 9(12), 892. https://doi.org/10.3390/photonics9120892