1. Introduction
Super-resolved microscopy (SRM) is becoming an established tool for biomedical research that extends the ability to visualise structures and localise or colocalise proteins in cells beyond the Abbe (diffraction) limit of λ/(2NA). SRM can be realised utilising single molecule localisation microscopy (SMLM) techniques such as PALM [
1,
2] or STORM [
3,
4], using laser scanning point spread function (PSF) engineering approaches such as RESOLFT [
5] and STED [
6,
7], microscopy, or using structured illumination microscopy (SIM) approaches including wide-field linear [
8], and nonlinear [
9] or photo-switched [
10] approaches. While SMLM, RESOLFT, and STED can provide spatial resolution down to 10’s of nm, linear SIM is limited to a factor of two in improvement of the spatial resolution (region of support) compared to wide-field microscopy, which means lateral resolutions of >80 nm are achievable for visible radiation. However, this lateral resolution is accompanied by an improved axial resolution (<300 nm) compared to wide-field microscopy [
11] and SIM offers the significant advantages of working with many existing fluorophores and sample preparations (including live cells) and providing relatively high acquisition speeds at low intensities as is typical for wide-field microscopy. The experimental convenience and relatively low phototoxicity of SIM have led it to be widely applied for biological studies where superior resolution compared to confocal microscopy is required, e.g., [
12,
13].
A common goal in biological studies is to study protein interactions but no optical SRM technique is yet able to resolve on the sub-10 nm scale required for most protein interactions of interest in cell biology, although progress is being made using co-localisation techniques, e.g., [
14]. Rather, indirect spectroscopic readouts are employed that sense the close proximity of fluorophores and therefore of the proteins they label. Of these, Förster resonant energy transfer (FRET) is probably the most widely used approach to map protein binding and the readouts of genetically expressed biosensors that change their molecular conformation upon binding their analyte. Many techniques have been implemented to read out and quantify FRET [
15,
16] with the most widely used being spectral ratiometric imaging, where FRET is read out through the change in acceptor to donor intensity ratio, and fluorescence lifetime imaging (FLIM), where the increased de-excitation rate resulting from FRET results in a decrease in the donor fluorescence lifetime. While the former approach typically requires fewer detected photons per pixel, quantitative spectrally resolved FRET measurements require calibration of the spectral response of the system (including sample and instrument) and an independent measurement of the actual FRET efficiency is also required if the fractions of the FRETing donor and acceptor populations are needed [
17]. FLIM offers inherently ratiometric measurement of FRET in a single spectral channel (i.e., donor emission) that is insensitive to fluorophore concentration or the optical properties of sample or instrument [
18]. Furthermore, if sufficient photons are detected, the donor fluorescence can be fitted to a complex decay model to enable the FRET efficiency and the FRETing population fraction to be obtained from a single measurement. Using global analysis techniques, the number of photons required to fit to complex decay models can be reduced to the level required to fit monoexponential decay profiles, i.e., a few hundred photons per pixel [
19]. Besides FRET, FLIM can provide quantitative single channel readouts of other fluorescence-based sensors and is widely used to probe metabolic changes as reported by the lifetimes and populations fractions of free and bound NADH or flavoproteins, e.g., [
20].
For studies of biological function, it is interesting to combine the molecular readouts afforded by FLIM and FRET with the precision afforded by SRM techniques to image morphological changes. FLIM combined with SRM can also avoid issues associated with chromatic aberration in multi-label imaging if samples can be labelled with fluorophores of similar spectral properties but different fluorescence lifetimes. Super-resolved FLIM has been realised using laser scanning STED microscopy [
21,
22] where time-correlated single photon counting (TCSPC) was implemented in the fluorescence detection channel. While STED FLIM is a useful extension of STED, the requirement to detect sufficient photons for FLIM increases the corresponding light dose from both excitation and depletion beams, which can result in unwanted photobleaching, phototoxicity, and background fluorescence. At this time, FLIM has not been implemented with SMLM, to the best of our knowledge, and while it should be possible using single photon counting imaging detectors, e.g., [
23], this remains to be demonstrated and the acquisition times may be too long for live cell imaging. We demonstrate here, however, that the combination of SIM with wide-field FLIM to provide super-resolved images of cell morphology with colocalised fluorescence lifetime readouts is practical and straightforward since both techniques can be implemented using wide-field detection.
SIM essentially entails recording a sequence of fluorescence intensity images that contain the desired high spatial frequency content mixed with lower spatial frequency components—and typically contains out of focus contributions that are also recorded by the wide-field detector. The super-resolved (SIM) images are then calculated from the recorded image data. Wide-field FLIM can be implemented in the time-domain or frequency domains and, for both approaches, the fluorescence lifetime images are calculated from series of gated fluorescence intensity images acquired at different relative delays following excitation.
2. Experimental Approach
In principle, the camera used for wide-field detection in SIM could be replaced by a wide-field time-gated detection system, such as a gated optical image intensifier and readout camera configured to provide time-domain [
24] or frequency domain [
25] FLIM, or a modulated CMOS camera [
26]. However, as we established previously when we combined FLIM with the simpler form of structured illumination [
27] to realise optically sectioned FLIM [
28,
29], this approach suffers from lower signal to noise ratio of the recovered (optically sectioned) images (compared to wide-field fluorescence intensity images) that would limit the accuracy of fluorescence lifetime determination. It also significantly increases the data acquisition time—and therefore the potential for photobleaching or sample motion to cause artefacts in the reconstructed optically sectioned FLIM images. A single FLIM image based on 8 time gated image acquisitions with 1 s integration time typically requires ~8 s. If 3D SIM images are each calculated from 5 grating angles with 5 phase shifts, the total exposure time for a 3D SIM image with FLIM data acquired for every illumination condition would require ~200 s. Further artifacts could arise from any nonlinear processing steps in the calculation of the series of gated optically sectioned images. Thus, combining SIM and FLIM in this way would significantly degrade the performance of both techniques.
Accordingly, we have instead implemented an approach where we acquire optically sectioned time-gated intensity images and apply SIM to the same field of view. This approach maintains the advantages of both techniques and provides both molecular contrast from FLIM with super-resolved intensity imaging of subcellular structure. We illustrate this “SIM + FLIM” capability with two exemplar applications to map FRET readouts and correlate molecular function with the nanostructure presented by SIM.
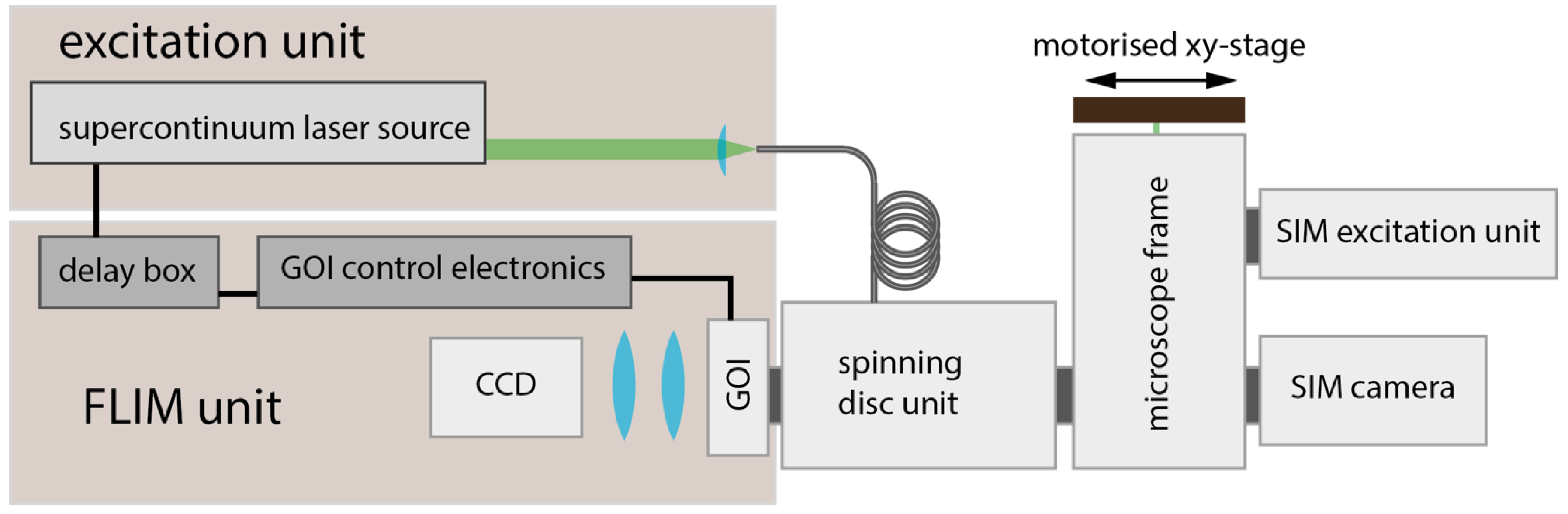
Figure 1 presents the configuration of the system, which is built around a commercial SIM microscope (Elyra S1, Carl Zeiss, Oberkochern, Germany), to which an optically sectioning FLIM system has been added. The performance of the SIM system is specified at down to 120 nm lateral and 300 nm axial resolution with excitation being provided by c.w. lasers at 405 nm, 488 nm, 561 nm, or 642 nm. SIM image data were acquired using a sCMOS camera (pco.edge 4.2, PCO GmbH, Kehlheim, Germany) mounted at the right-hand port of the microscope frame and the SIM images were reconstructed using the commercial SIM software (ZEN black edition, Carl Zeiss). Optically sectioned wide-field FLIM was implemented following our previous work where we demonstrated wide-field time-gated FLIM of fluorescent protein-labelled fixed or live cells at frame rates up to 10 Hz [
30]. Briefly, this FLIM system consists of a laser excitation unit, a spinning Nipkow disk confocal scanner, and a gated optical image intensifier (GOI) read out using a cooled scientific camera. The FLIM data acquisition was under the control of an in-house software plug-in for the open source µManager programme that controlled the camera, the electronic delay of the time-gating, the motorised excitation filter wheels, and the various functions of the motorised microscope (Elyra S1, Carl Zeiss), including z-focus and selection of dichroic beamsplitter cube, objectives or image port. A description of the data acquisition protocols is given in [
31], and the software can be downloaded from
https://github.com/imperial-photonics/openFLIM-HCA.git. The FLIM data analysis was undertaken using our open source software package, FLIMfit [
17] available at
http://flimfit.org/. FLIMfit [
17] can account for the temporal instrument response function of the system, background light, time varying background and a spatial response from the GOI. It can also provide image segmentation and global analysis of FLIM data.
The FLIM laser excitation unit is based on an ultrashort-pulsed fibre-laser-pumped supercontinuum source (WhiteLase, Fianium, Southhampton, UK), providing picosecond pulses at a repetition rate of 60 MHz, with motorised neutral density and spectral bandpass filter wheels to control the excitation intensity and wavelength. A single-mode polarisation-maintaining optical fiber conveys the pulsed excitation radiation to the spinning disk unit (CSU-X1, Yokogawa, Tokyo, Japan) and the GOI was triggered by an electronically delayed signal from the supercontinuum excitation source that is synchronous with the ultrashort optical pulse train. The GOI used (HRI-HL-S, Kentech Instruments Ltd, Wallingford, UK) provided time gates of 4 ns duration with a rising edge of ~50 ps and was read out using a sCMOS (Zyla-5.5, Andor, Belfast, UK) or a CCD (Retiga R1, QImaging, Inc., Surrey, BC, Canada) camera. For FLIM, sequences of 8 time-gated fluorescence intensity images were acquired. After fitting, FLIMfit provided maps of fluorescence lifetime, time-integrated fluorescence intensity, and an “intensity-weighted” fluorescence lifetime image for which the integrated fluorescence intensity is used to scale the intensity of each pixel of a fluorescence lifetime map. The latter is important since it gives a higher visual weighting to pixels where more photons are detected, which will provide more accurate values of fluorescence lifetime.
Figure S1a illustrates the calculation of an intensity-weighted fluorescence lifetime image. As illustrated below, these intensity-merged fluorescence lifetime images can be superimposed on SIM intensity images acquired immediately after the FLIM acquisition to enable variations in nanostructure and function to be correlated.
3. Results
An exemplar application of this approach is illustrated with the FLIM FRET readout of aggregation of the receptor tyrosine kinase known as discoidin domain receptor 1 (DDR1). It has previously been suggested that stimulating DDR1 with its ligand, collagen, causes aggregation [
32,
33,
34]. Here, we label DDR1 with a SNAP-tag that has been stochastically linked to Alexa488 or Alexa546. This labelling, which is illustrated in
Figure S1b, allows receptor aggregation to be measured as an increase in FRET between Alexa488 as the donor and Alexa546 as the acceptor, resulting in a decreased lifetime of the Alexa488 dye.
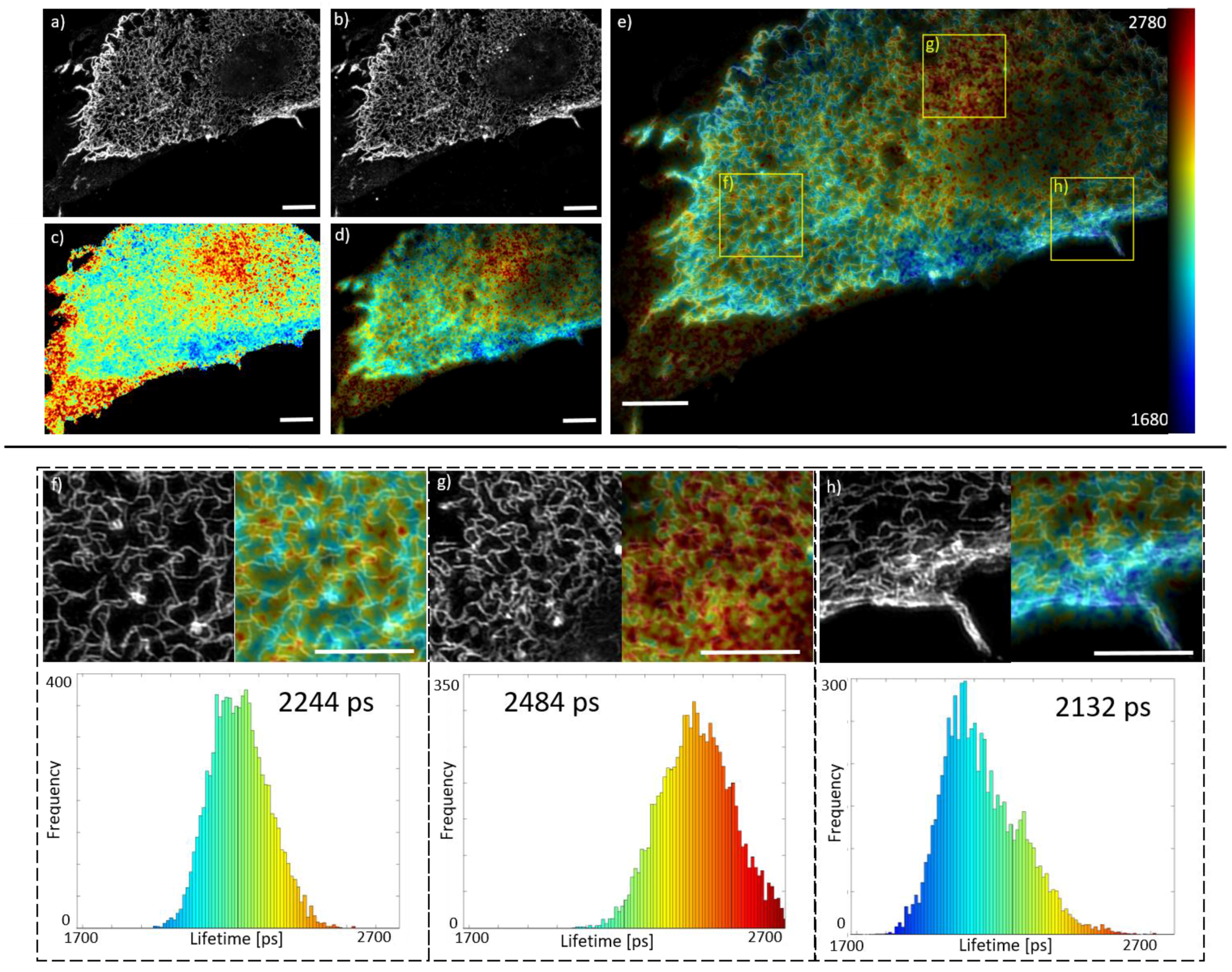
Figure 2 shows SIM and FLIM data of a Cos 7 cell expressing DDR1 labelled with AlexaFluor488 and AlexaFluor546 and stimulated with collagen for 60 min. The SIM images of
Figure 2a,b show that the donor and acceptor are well colocalised across the field of view, see
Figure S2 for an overlay of these two images. The SIM images reveal the receptor is aggregated to different extents across the cell.
The fluorescence lifetime maps of
Figure 2c,d show variation in donor fluorescence lifetime across the cell, indicating variation in FRET efficiency, which could be explained by variations in the extent of DDR1 aggregation, i.e., the number of DDR1 receptors clustering together and how close together the receptor molecules are.
Figure 2e and
Supplementary Video 1 show the superposition of the SIM and FLIM data, enabling variations in lifetime and nanostructure to be correlated.
Figure 2f–h show sub-regions of
Figure 2e as indicated, clearly showing the differences in Alexa488 lifetime. These differences are clearly seen in the corresponding lifetime histograms of
Figure 2. There appears to be a correlation between the state of aggregation seen in the SIM images and the amount of FRET occurring. For example,
Figure 2h shows high aggregation and high FRET, while
Figure 2g shows lower aggregation and lower FRET. However, in some areas of the cell the SIM and FRET images do not correlate. Thus, we believe the FRET map is reporting nanostructure variations that are not apparent in the SIM image of which the biological cause is yet to be determined. The images of donor and acceptor fluorescence intensity shown in
Figure S2 indicate that the observed lifetime changes are not simply resulting from the relative labelling densities of the donor and acceptor.
A second exemplar application illustrates how functional molecular readouts could provide information complementing observations of changes in nanostructure, e.g., during the cell cycle. SIM has previously been applied to study changes in nuclear structure, e.g., to study chromatin territories [
13], but many phenomena occur on spatial scales <<100 nm and remain elusive. One example is the compaction of chromatin in the cell nucleus. While SIM can provide sufficient resolution to indicate chromatin territories, DNA compaction can only be inferred, e.g., from the higher intensity of a DNA label such as DAPI. As reported in previous studies, e.g., [
35], FLIM FRET can be utilised to map the compaction of DNA that has been labelled with appropriate donor/acceptor fluorophores such that they come into closer proximity as the DNA folds more tightly. The increased FRET efficiency results in a lower donor fluorescence lifetime for more compacted chromatin.
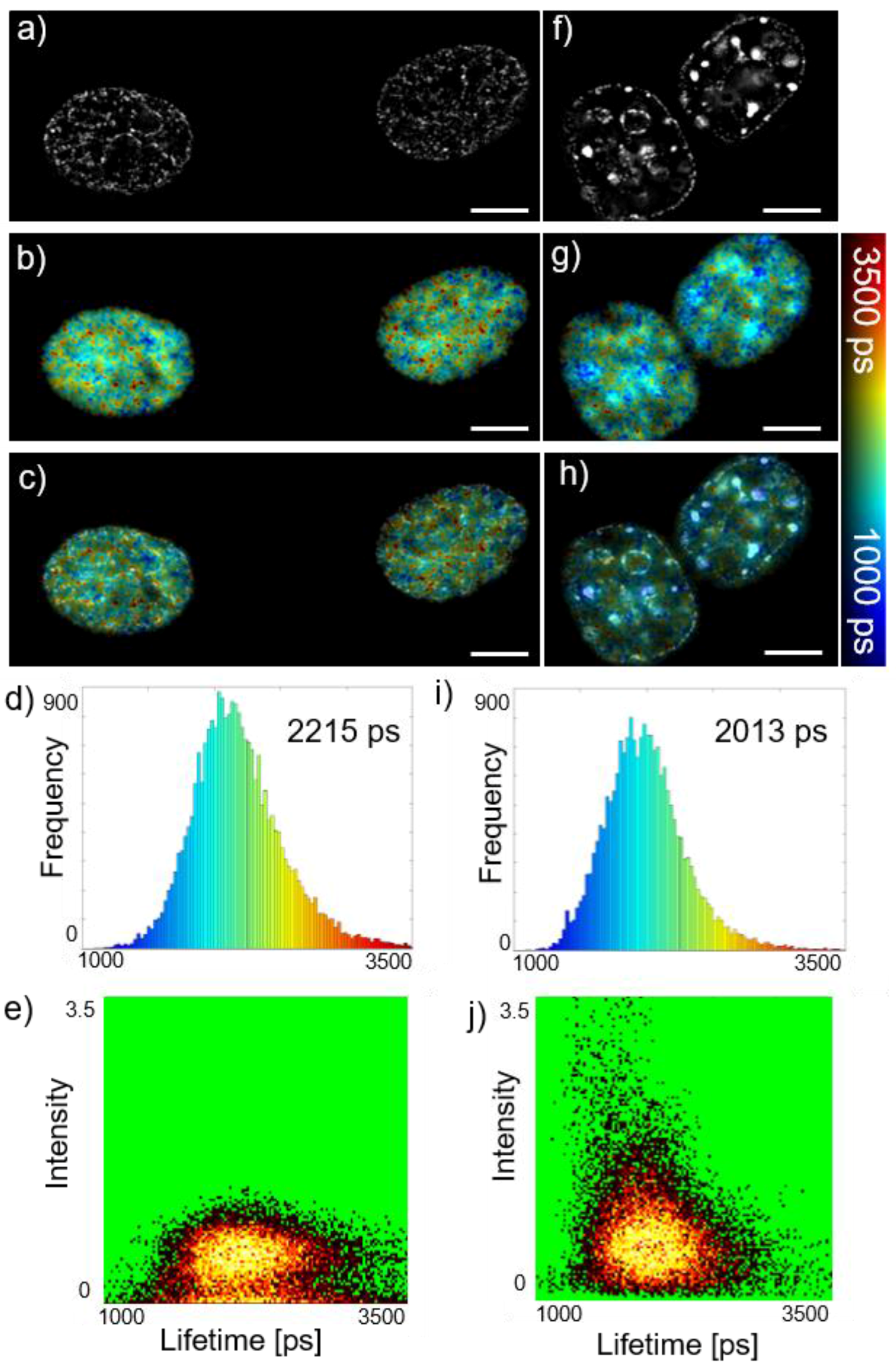
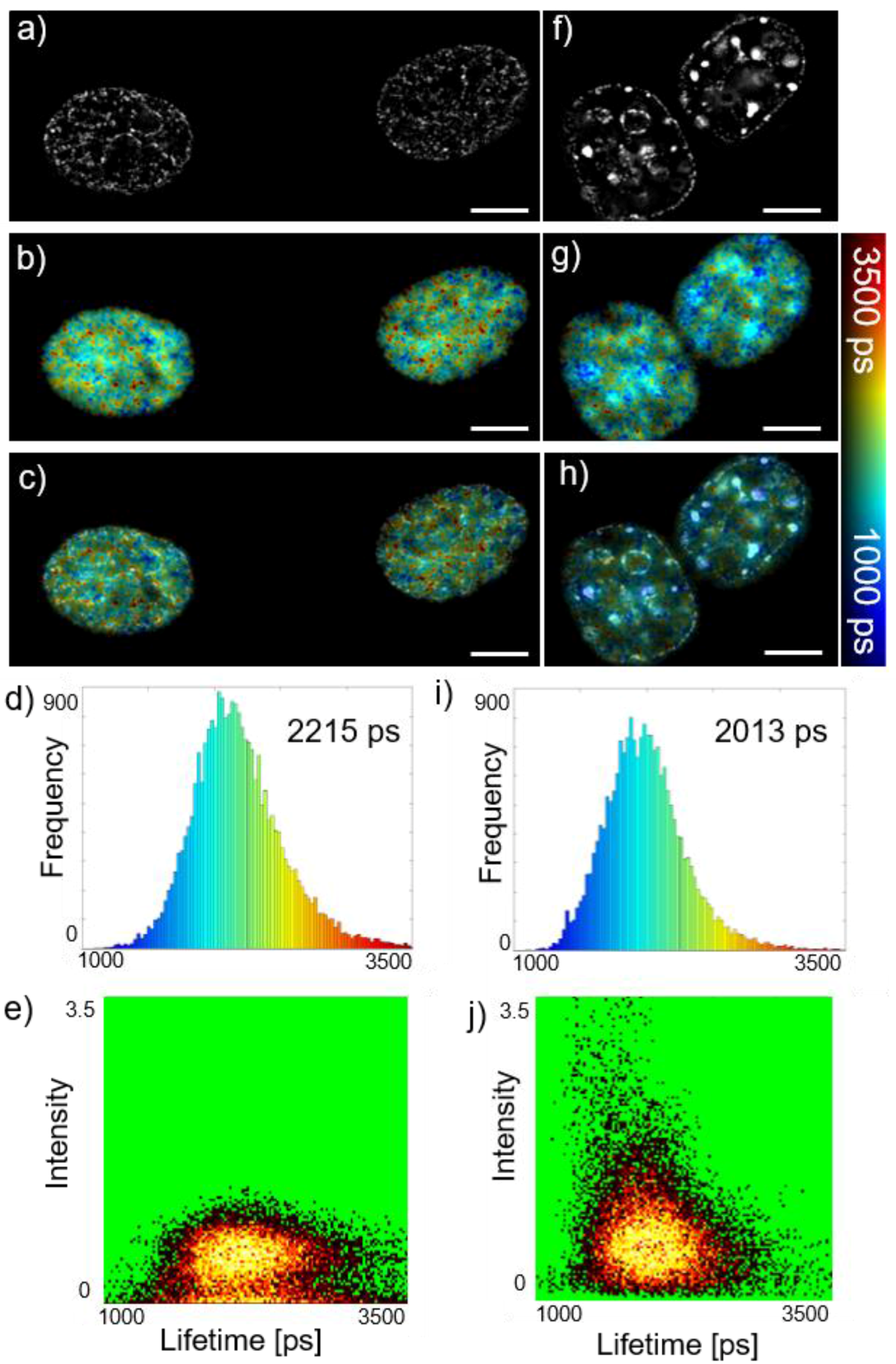
Here we have combined this FRET readout of DNA compaction with SIM of nucleotides in NIH3T3 cells as shown in
Figure 3, which presents SIM + FLIM applied to cells imaged in S-phase of the cell cycle with the DNA stochastically labelled with Alexa594 and Alexa647 using click chemistry via the synthetic nucleotide EdU. The cells corresponding to
Figure 3f–j present a more punctate distribution of DNA than those for
Figure 3a–e with the FLIM data indicating shorter fluorescence lifetimes and therefore more tightly packed nucleotides and corresponding higher intensities at the punctate features. The 2D intensity-lifetime histogram in
Figure 3j shows that there is a correlation between the lower donor lifetime and higher fluorescence intensity as would be expected if the DNA is concentrated in foci. This correlation is not present in
Figure 3e for the sample where the fluorescence distribution is less punctate. We believe the ability to quantitatively map DNA compaction and correlate this with SIM of nuclear nanostructure could be useful for elucidating nuclear processes. Other FLIM/FRET readouts could include biosensors for local nucleotide environments.
4. Materials and Methods
4.1. Sample Preparation
COS7 cells were seeded on a #1.5 thickness coverslip (VWR, Lutterworth, UK ) under standard cell culture conditions (DMEM supplemented 10% Fetal Bovine Serum, 5% CO2, 37 °C humidified atmosphere) for 24 h. The cells were then transiently transfected (using Lipofectamine 3000) with a construct encoding DDR1 with a SNAP-tag insertion in its extracellular region (NEB, pSNAPfVector) and incubated again for 24 h. To label the SNAP-tags the cells were incubated in complete DMEM containing both Alexa488 and Alexa546 SNAP reagents at 2.5 μM for 1 hour at 37 °C (NEB, SNAP-Surface®Alexa Fluor®488, SNAP-Surface®Alexa Fluor®546). To remove unbound reagent the cells were washed three times with DMEM. To prepare the DDR1 stimulation medium collagen I (from rat tail, C7661, Sigma-Aldrich, Gillingham, UK ) was diluted in DMEM to 20 μg/mL, from a 1 mg/mL stock solution in 0.1 M acetic acid. The stimulation medium was added to the cells and incubated at 37 °C for 60 min. After stimulation the cells were washed with PBS and fixed with 4% paraformaldehyde in phosphate buffered saline for 15 min at room temperature. ProLong Gold Antifade (Thermofisher Scientific, Paisley, UK) was used as mounting medium.
NIH3T3 cells were seeded in 6 well plates with a #1.5 coverslip (Ref 0117052 Marienfied, Würzburg, Germany) under standard cell culture conditions (DMEM supplemented with 10% Fetal Bovine Serum, 5% CO2, 37 °C humidified atmosphere). After achieving a 50% density cells were metabolically labelled with 10 μM of 5-ethynyl-2′-deoxyuridine (EdU), fixed in 4% paraformaldehyde in phosphate buffer saline for 10 min, and stochastically labelled with Alexa Fluor 594 and Alexa Fluor 647 Azides following manufacturer recommendations (Click-iT Edu, Thermofisher Scientific Paisley, UK). Slides were mounted in Tris/Glycerol Vectashield (12 mM Tris (pH 8.0), 67% Glycerol, 25% Vectashield) and sealed with nail polish. Samples were stored at 4 °C with light protection until documentation.
4.2. Image Acquisition Parameters
For the DDR1 FRET experiments presented in
Figure 2, 3D SIM images in two spectral channels were acquired with excitation laser lines at 488 nm and 561 nm using different dichroic beamsplitters (Carl Zeiss 405/488/642 nm and 561/642 nm). The integration time for raw images was 200 ms for both lines. The SIM image was calculated from 5 grating angles and 5 phase shifts, resulting in a total SIM acquisition time of 5 s per z-section. A z stack of 15 slices was taken over an axial distance of 1.65 µm. For the FLIM acquisition via the spinning disc scanner, a GFP like filter set was used (Yokogawa CSU-X 500/25 nm excitation filter, 488/561 nm dichroic beamsplitter, 542/27 nm emission filter). The GOI gate width was 4 ns, and the gain voltage 750 V. The sampling strategy was to set one gate at 3 ns before the peak fluorescence intensity, one at the peak intensity and 6 further gates spaced “logarithmically”, i.e. set to sample the fluorescence decay profile at approximately equal intensity intervals out to 12 ns after the intensity peak assuming a lifetime of 4 ns). The integration time was 3 s per gate, resulting in a total acquisition time of 24 s per fluorescence lifetime image. To read out the GOI, a sCMOS camera (Andor, Zyla-5.5) was used in this experiment.
For the DNA folding studies displayed in
Figure 3, 3D SIM images in two spectral channels were acquired with excitation laser lines at 561 nm and 642 nm using the same dichroic beamsplitter (561/642 nm). The acquisition time for each raw image was 200 ms for both excitation lines. For the calculation of the SIM images, 5 grating angles and 5 phase shifts were used. The FLIM experiment was performed with a 525/50 nm excitation filter, a dichroic (457/514/647 nm), and a 629/62 nm emission filter. The GOI gain voltage was 750 V, and the gate width was 4 ns. The same sampling strategy was used for the time gates and the integration time at each gate delay was 400 ms, resulting in a total acquisition time of <4 s per fluorescence lifetime image. The imaging camera of the FLIM port was from QImaging Inc. for this experiment. To read out the GOI, a sCMOS camera (QImaging Inc., Retiga R1) was used in this experiment.
4.3. FLIM Fitting and Post Processing of Images
For a full description of the fitting of lifetime data with
FLIMfit, please see
http://flimfit.org/. Briefly, for
Figure 2, the FLIM data were fitted pixelwise to a monoexponential decay model and the intensity-weighted mean lifetime was calculated for each pixel. The fitting took account of both static and time-varying backgrounds and a 3 × 3 smoothing was applied before fitting for better visualisation. A spatially varying instrument response function (IRF) measurement was employed to account for any spatial inhomogeneity in the time-gated imaging. To generate the FLIM + SIM images of
Figure 2e–h, a
z-projection of the SIM intensity images was used. Although the raw acquired SIM and FLIM images are reasonably well co-registered, they are not completely equivalent in image size or pixel size, and there can be some relative rotation and offset in their
x-
y positions. Using MATLAB, the FLIM image was scaled according to the size of the readout camera sensor and the FLIM image was rotated and translated in order to register it with the SIM image. For this image registration, a multispectral fluorescent bead sample was used. A similar procedure was applied for the data presented in
Figure 3.
Table 1 presents the mean and standard deviation of the intensity-weighted lifetimes calculated.
5. Conclusions
We believe that correlating spectroscopic readouts with super-resolved microscopy of nanostructure can provide useful approaches to study biological function. In particular, combining FRET with SRM can provide information concerning molecular function over a range of scales down to <10 nm. FLIM can provide robust readouts of FRET and of changes in tissue matrix components and cellular metabolic processes, which could be interesting to localise, e.g., to specific cellular compartments, with the precision of SRM. Unfortunately, however, the requirements of FLIM for specialised detectors and significantly increased number of detected photons present challenges when directly integrating FLIM with SRM. We therefore believe that parallel application of FLIM and SRM techniques represents a promising approach since both can be used without compromise—providing that the fluorophores have appropriate properties.
We have demonstrated the combination of super-resolved imaging using SIM with wide-field (optically sectioned) FLIM for the first time, here enabling images of cellular nanostructure to be correlated with FRET read out using FLIM. For both these wide-field imaging techniques, the excitation intensities and acquisition times required are compatible with live cell imaging. Our experience is that the FLIM results in significantly less photobleaching that the SIM, which we found to be challenging when imaging samples labelled with fluorescent proteins. The use of dye-based fluorophores, including in combination with genetically expressed tags such as SNAP-tags, provides superior performance for both FLIM and SIM.
We note that the FLIM data we present here is not super-resolved but is mapped onto the super-resolved (SIM) intensity images of nanostructure. Accordingly, in the SIM + FLIM image the false-colour encoding lifetime that is applied to a super-resolved structure is not strictly related to the structure itself, since it could be affected by the lifetimes of nearby structures that are within the point spread function of the FLIM image. However, it can provide (diffraction-limited) information about local molecular function that may not be available in the SIM image. This can permit changes in nanostructure to be correlated with variations in molecular function, for example, read out via a FRET sensor. Alternatively, the FLIM images could guide further investigation of the super-resolved nanostructure. For samples of “slowly spatially varying” photophysical changes that are constant in lifetime within the resolution of the confocal FLIM microscope, the true lifetime of the nanostructures would be reported. It could be possible to advance this approach by developing a more sophisticated image analysis approach to use the SIM intensity data with the FLIM data to reconstruct truly super-resolved lifetime maps reporting lifetime variations between pixels in the SIM image. This would be analogous to deconvolution—and would be subject to similar trade-offs, limitations, and pitfalls. Using fluorophores of known lifetimes and/or using global fitting approaches, e.g., for FRET, could simplify this process.




{kind=link}
{kind=link}
{kind=link}