Conformational Dynamics of Biopolymers in the Course of Their Interaction: Multifaceted Approaches to the Analysis by the Stopped-Flow Technique with Fluorescence Detection


Abstract
1. Introduction
2. General Principles of Investigation into Conformational Dynamics in Real Time
3. Conformational Changes in DNA Glycosylases of the HhH-GPD Structural Family and in DNA during Their Interaction
3.1. Human 8-Oxoguanine DNA Glycosylase hOGG1
3.2. Other Members of the HhH-GPD Structural Family: MutY, MBD4, and Nth
3.3. The General Model of Lesion Recognition by Enzymes of Structural Family HhH-GPD
4. Conformational Alterations in DNA Glycosylases of Structural Family H2tH and in DNA during Their Interaction
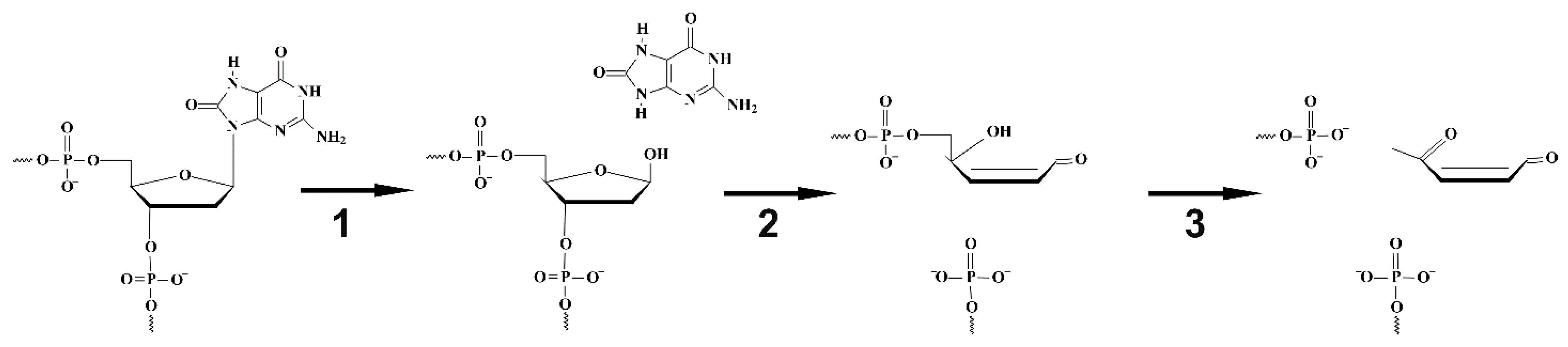
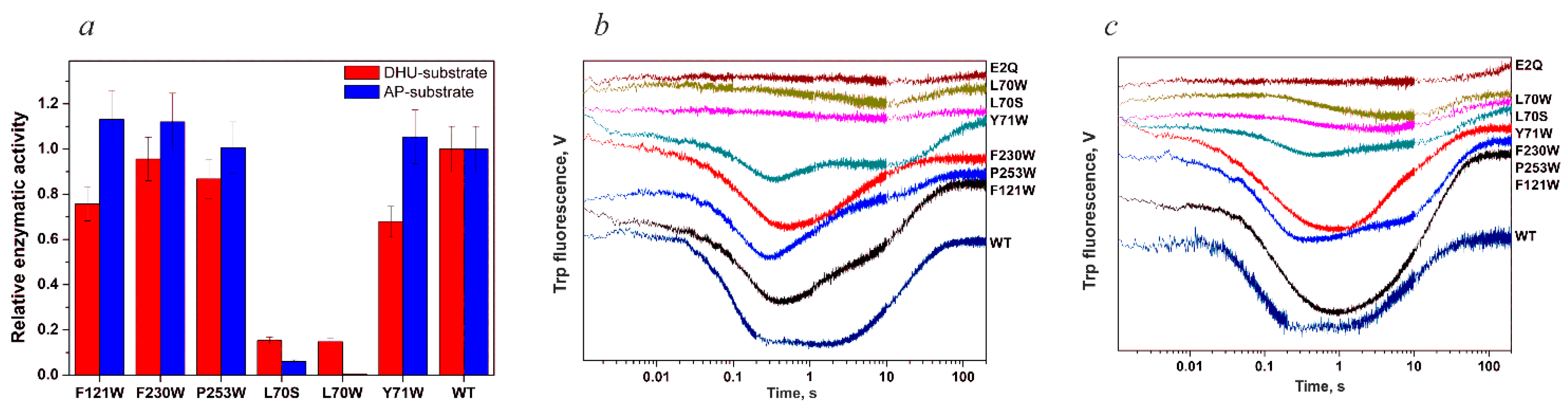
4.1. Endonuclease VIII Nei
4.2. Other Members of Structural Family H2tH: Fpg and NEIL1
4.3. A General Model for the Recognition of DNA Damage by Enzymes of Structural Family H2tH
5. Human AP Endonuclease APE1
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
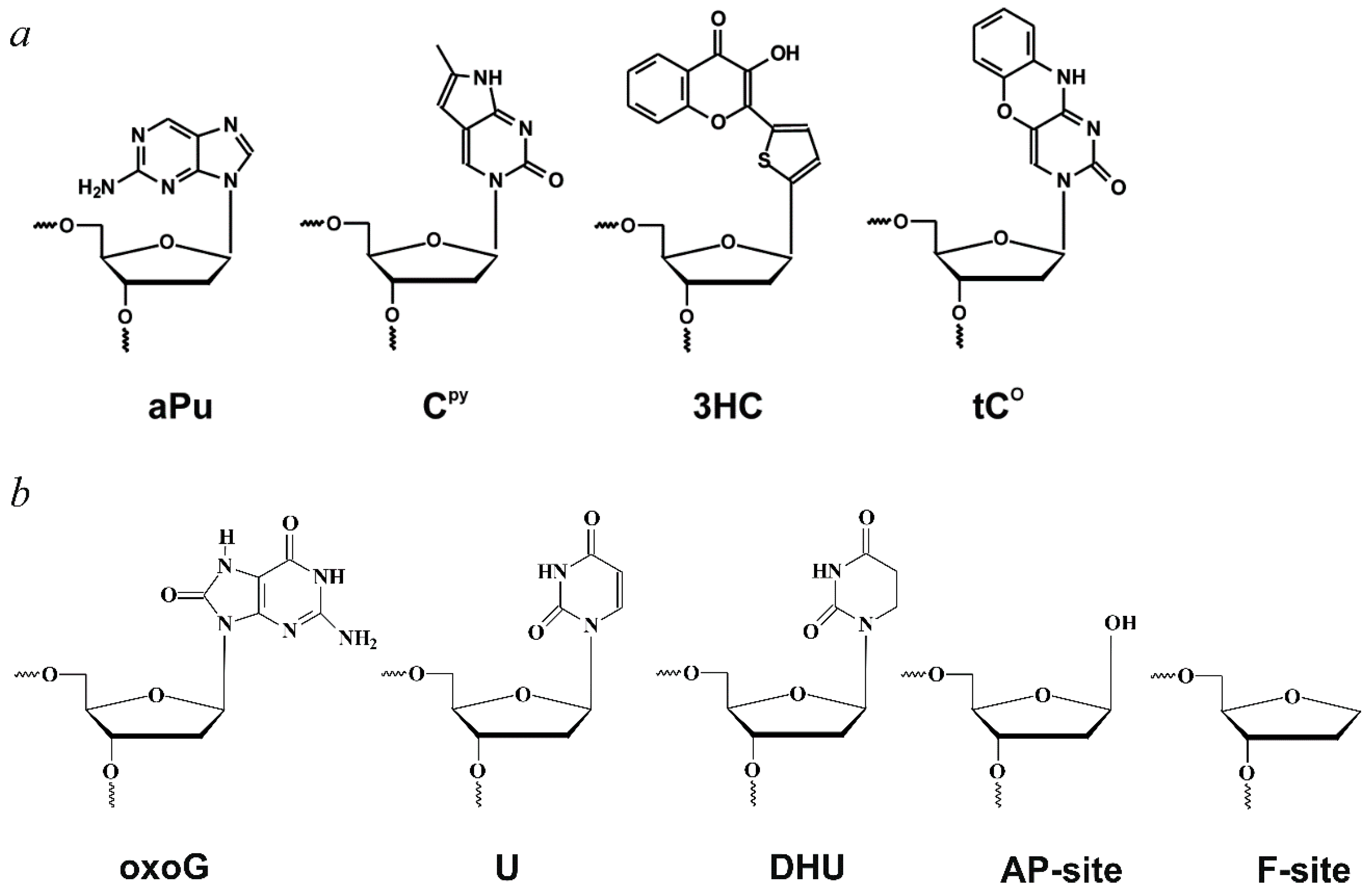
Glossary
| aPu | 2-aminopurine; |
| AP-site | apurinic/apyrimidinic site; |
| APE1 | human apurinic/apyrimidinic endonuclease 1; |
| BER | base excision repair; |
| BHQ1 | black hole quencher 1; |
| CPy | pyrrolocytosine; |
| DHU | 5,6-dihydrouracil; |
| F-site | (2R,3S)−2-(hydroxymethyl)−3-hydroxytetrahydrofuran residue (AP-site analogue); |
| FAM | 6-carboxyfluorescein; |
| Fpg | formamidopyrimidine DNA glycosylase from Escherichia coli; |
| FRET | Forster resonance energy transfer; |
| HhH-GPD | structural family of DNA glycosylases containing the helix-hairpin-helix motif and a region enriched with glycine and proline residues (Gly/Pro loop) and the aspartate residue (D); |
| H2tH | structural family of DNA glycosylases containing helix-two turn-helix motif; |
| hOGG1 | human 8-oxoguanine DNA glycosylase; |
| MutY | adenine-DNA glycosylase from Escherichia coli; |
| MBD4 | human methyl-CpG-binding domain 4; |
| Nth | endonuclease III from Escherichia coli; |
| Nei | endonuclease VIII from Escherichia coli; |
| NEIL1 | human endonuclease VIII; |
| oxoG | 8-oxoguanine; |
| PDB ID | Protein Data Bank Identification number; |
| tCO | 1,3-diaza-2-oxophenoxazine; |
| WT | wild type enzyme; |
| PAGE | polyacrylamide gel electrophoresis; |
| 3HC | 3-hydroxychromone. |
References
- Friedberg, E.C.; Walker, G.C.; Siede, W.; Wood, R.D.; Schultz, R.A.; Ellenberger, T. DNA Repair and Mutagenesis; ASM Press: Washington, DC, USA, 2006. [Google Scholar]
- Fromme, J.C.; Verdine, G.L. Base Excision Repair. Adv. Protein Chem. 2004, 69, 1–41. [Google Scholar] [CrossRef]
- David, S.S.; O’Shea, V.L.; Kundu, S. Base-Excision Repair of Oxidative DNA Damage. Nature 2007, 447, 941–950. [Google Scholar] [CrossRef]
- Wallace, S.S. Base Excision Repair: A Critical Player in Many Games. DNA Repair 2014, 19, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Zharkov, D.O. Base Excision DNA Repair. Cell. Mol. Life Sci. 2008, 65, 1544–1565. [Google Scholar] [CrossRef] [PubMed]
- Kladova, O.A.; Kuznetsov, N.A.; Fedorova, O.S. Initial Stages of DNA Base Excision Repair in Nucleosomes. Mol. Biol. 2021, 55, 167–181. [Google Scholar] [CrossRef]
- Krokan, H.E.; Bjørås, M. Base Excision Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef]
- Robertson, A.B.; Klungland, A.; Rognes, T.; Leiros, I. DNA Repair in Mammalian Cells: Base Excision Repair: The Long and Short of It. Cell. Mol. Life Sci. 2009, 66, 981–993. [Google Scholar] [CrossRef]
- Gros, L.; Saparbaev, M.K.; Laval, J. Enzymology of the Repair of Free Radicals-Induced DNA Damage. Oncogene 2002, 21, 8905–8925. [Google Scholar] [CrossRef]
- Tudek, B. Base Excision Repair Modulation as a Risk Factor for Human Cancers. Mol. Asp. Med. 2007, 28, 258–275. [Google Scholar] [CrossRef]
- Wallace, S.S.; Murphy, D.L.; Sweasy, J.B. Base Excision Repair and Cancer. Cancer Lett. 2012, 327, 73–89. [Google Scholar] [CrossRef]
- Marsden, C.G.; Dragon, J.A.; Wallace, S.S.; Sweasy, J.B. Base Excision Repair Variants in Cancer. Methods Enzym. 2017, 591, 119–157. [Google Scholar] [CrossRef]
- Chan, K.K.L.; Zhang, Q.M.; Dianov, G.L. Base Excision Repair Fidelity in Normal and Cancer Cells. Mutagenesis 2006, 21, 173–178. [Google Scholar] [CrossRef]
- Maynard, S.; Schurman, S.H.; Harboe, C.; de Souza-Pinto, N.C.; Bohr, V.A. Base Excision Repair of Oxidative DNA Damage and Association with Cancer and Aging. Carcinogenesis 2009, 30, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Kladova, O.A.; Fedorova, O.S.; Kuznetsov, N.A. The Role of Natural Polymorphic Variants of DNA Polymerase β in DNA Repair. Int. J. Mol. Sci. 2022, 23, 2390. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Eshtad, S.; Nik-Zainal, S. Mechanisms Underlying Mutational Signatures in Human Cancers. Nat. Rev. Genet. 2014, 15, 585–598. [Google Scholar] [CrossRef]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA Repair Pathways as Targets for Cancer Therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: New York, NY, USA, 2006. [Google Scholar]
- Carpenter, M.L.; Oliver, A.W.; Kneale, G.G. Analysis of DNA-Protein Interactions by Intrinsic Fluorescence. Methods Mol. Biol. 2001, 148, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Wong, I.; Lundquist, A.J.; Bernards, A.S.; Mosbaugh, D.W. Presteady-State Analysis of a Single Catalytic Turnover by Escherichia coli Uracil-DNA Glycosylase Reveals a “Pinch-Pull-Push” Mechanism. J. Biol. Chem. 2002, 277, 19424–19432. [Google Scholar] [CrossRef] [PubMed]
- Dunlap, C.A.; Tsai, M.D. Use of 2-Aminopurine and Tryptophan Fluorescence as Probes in Kinetic Analyses of DNA Polymerase β. Biochemistry 2002, 41, 11226–11235. [Google Scholar] [CrossRef]
- Cherepanov, A.V.; de Vries, S. Binding of Nucleotides by T4 DNA Ligase and T4 RNA Ligase: Optical Absorbance and Fluorescence Studies. Biophys. J. 2001, 81, 3545–3559. [Google Scholar] [CrossRef]
- Wilhelmsson, L.M. Fluorescent Nucleic Acid Base Analogues. Q. Rev. Biophys. 2010, 43, 159–183. [Google Scholar] [CrossRef] [PubMed]
- Sinkeldam, R.W.; Greco, N.J.; Tor, Y. Fluorescent Analogs of Biomolecular Building Blocks: Design, Properties, and Applications. Chem. Rev. 2010, 110, 2579–2619. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.T.; Kim, H.W.; Moon, D.; Rhee, Y.M.; Kim, B.H. (DNS)C: A Fluorescent, Environmentally Sensitive Cytidine Derivative for the Direct Detection of GGG Triad Sequences. Org. Biomol. Chem. 2013, 11, 5605–5614. [Google Scholar] [CrossRef]
- Suzuki, A.; Takahashi, N.; Okada, Y.; Saito, I.; Nemoto, N.; Saito, Y. Naphthalene-Based Environmentally Sensitive Fluorescent 8-Substituted 2′-Deoxyadenosines: Application to DNA Detection. Bioorg. Med. Chem. Lett. 2013, 23, 886–892. [Google Scholar] [CrossRef] [PubMed]
- Pawar, M.G.; Nuthanakanti, A.; Srivatsan, S.G. Heavy Atom Containing Fluorescent Ribonucleoside Analog Probe for the Fluorescence Detection of RNA-Ligand Binding. Bioconjug. Chem. 2013, 24, 1367–1377. [Google Scholar] [CrossRef]
- Pawar, M.G.; Srivatsan, S.G. Environment-Responsive Fluorescent Nucleoside Analogue Probe for Studying Oligonucleotide Dynamics in a Model Cell-like Compartment. J. Phys. Chem. B 2013, 117, 14273–14282. [Google Scholar] [CrossRef]
- Purohit, V.; Grindley, N.D.F.; Joyce, C.M. Use of 2-Aminopurine Fluorescence to Examine Conformational Changes during Nucleotide Incorporation by DNA Polymerase I (Klenow Fragment). Biochemistry 2003, 42, 10200–10211. [Google Scholar] [CrossRef]
- Mandal, S.S.; Fidalgo da Silva, E.; Reha-Krantz, L.J. Using 2-Aminopurine Fluorescence to Detect Base Unstacking in the Template Strand during Nucleotide Incorporation by the Bacteriophage T4 DNA Polymerase. Biochemistry 2002, 41, 4399–4406. [Google Scholar] [CrossRef]
- Kuznetsov, N.A.; Koval, V.V.; Zharkov, D.O.; Vorobjev, Y.N.; Nevinsky, G.A.; Douglas, K.T.; Fedorova, O.S. Pre-Steady-State Kinetic Study of Substrate Specificity of Escherichia coli Formamidopyrimidine-DNA Glycosylase. Biochemistry 2007, 46, 424–435. [Google Scholar] [CrossRef]
- Kuznetsova, A.A.; Fedorova, O.S.; Kuznetsov, N.A. Kinetic Features of 3′-5′ Exonuclease Activity of Human AP-Endonuclease APE1. Molecules 2018, 23, 2101. [Google Scholar] [CrossRef]
- Davletgildeeva, A.T.; Kuznetsova, A.A.; Fedorova, O.S.; Kuznetsov, N.A. Activity of Human Apurinic/Apyrimidinic Endonuclease APE1 toward Damaged DNA and Native RNA with Non-Canonical Structures. Front. Cell Dev. Biol. 2020, 8, 590848. [Google Scholar] [CrossRef]
- Zang, H.; Fang, Q.; Pegg, A.E.; Guengerich, F.P. Kinetic Analysis of Steps in the Repair of Damaged DNA by Human O6-Alkylguanine-DNA Alkyltransferase. J. Biol. Chem. 2005, 280, 30873–30881. [Google Scholar] [CrossRef]
- Kuznetsova, A.A.; Kuznetsov, N.A.; Vorobjev, Y.N.; Barthes, N.P.F.; Michel, B.Y.; Burger, A.; Fedorova, O.S. New Environment-Sensitive Multichannel DNA Fluorescent Label for Investigation of the Protein-DNA Interactions. PLoS ONE 2014, 9, e100007. [Google Scholar] [CrossRef]
- Kuznetsov, N.A.N.A.; Vorobjev, Y.N.Y.N.; Krasnoperov, L.N.L.N.; Fedorova, O.S.O.S. Thermodynamics of the Multi-Stage DNA Lesion Recognition and Repair by Formamidopyrimidine-DNA Glycosylase Using Pyrrolocytosine Fluorescence—Stopped-Flow Pre-Steady-State Kinetics. Nucleic Acids Res. 2012, 40, 7384–7392. [Google Scholar] [CrossRef]
- Yang, K.; Stanley, R.J. The Extent of DNA Deformation in DNA Photolyase-Substrate Complexes: A Solution State Fluorescence Study. Photochem. Photobiol. 2008, 84, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Sandin, P.; Borjesson, K.; Li, H.; Martensson, J.; Brown, T.; Wilhelmsson, L.M.; Albinsson, B. Characterization and Use of an Unprecedentedly Bright and Structurally Non-Perturbing Fluorescent DNA Base Analogue. Nucleic Acids Res. 2008, 36, 157–167. [Google Scholar] [CrossRef]
- Borjesson, K.; Sandin, P.; Wilhelmsson, L.M. Nucleic Acid Structure and Sequence Probing Using Fluorescent Base Analogue TC(O). Biophys. Chem. 2009, 139, 24–28. [Google Scholar] [CrossRef]
- Stengel, G.; Purse, B.W.; Wilhelmsson, L.M.; Urban, M.; Kuchta, R.D. Ambivalent Incorporation of the Fluorescent Cytosine Analogues TC and TCo by Human DNA Polymerase Alpha and Klenow Fragment. Biochemistry 2009, 48, 7547–7555. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stengel, G.; Urban, M.; Purse, B.W.; Kuchta, R.D. High Density Labeling of Polymerase Chain Reaction Products with the Fluorescent Base Analogue TCo. Anal. Chem. 2009, 81, 9079–9085. [Google Scholar] [CrossRef][Green Version]
- Rodgers, B.J.; Elsharif, N.A.; Vashisht, N.; Mingus, M.M.; Mulvahill, M.A.; Stengel, G.; Kuchta, R.D.; Purse, B.W. Functionalized Tricyclic Cytosine Analogues Provide Nucleoside Fluorophores with Improved Photophysical Properties and a Range of Solvent Sensitivities. Chemistry 2014, 20, 2010–2015. [Google Scholar] [CrossRef]
- Sandin, P.; Stengel, G.; Ljungdahl, T.; Borjesson, K.; Macao, B.; Wilhelmsson, L.M. Highly Efficient Incorporation of the Fluorescent Nucleotide Analogs TC and TCO by Klenow Fragment. Nucleic Acids Res. 2009, 37, 3924–3933. [Google Scholar] [CrossRef]
- Kuznetsov, N.A.; Kladova, O.A.; Kuznetsova, A.A.; Ishchenko, A.A.; Saparbaev, M.K.; Zharkov, D.O.; Fedorova, O.S. Conformational Dynamics of DNA Repair by Escherichia coli Endonuclease III. J. Biol. Chem. 2015, 290, 14338–14349. [Google Scholar] [CrossRef] [PubMed]
- Rist, M.J.; Marino, J.P. Fluorescent Nucleotide Base Analogs as Probes of Nucleic Acid Structure, Dynamics and Interactions. Curr. Org. Chem. 2002, 6, 775–793. [Google Scholar] [CrossRef]
- Berry, D.A.; Jung, K.Y.; Wise, D.S.; Sercel, A.D.; Pearson, W.H.; Mackie, H.; Randolph, J.B.; Somers, R.L. Pyrrolo-DC and Pyrrolo-C: Fluorescent Analogs of Cytidine and 2′-Deoxycytidine for the Study of Oligonucleotides. Tetrahedron Lett. 2004, 45, 2457–2461. [Google Scholar] [CrossRef]
- Vasilyeva, S.V.; Kuznetsov, N.A.; Kuznetsova, A.S.; Khalyavina, J.G.; Tropina, D.A.; Lavrikova, T.I.; Kargina, O.I.; Gornostaev, L.M. DNA Fluorescent Labeling with Naphtho[1,2,3-Cd]Indol-6(2H)-One for Investigation of Protein-DNA Interactions. Bioorg. Chem. 2017, 72, 268–272. [Google Scholar] [CrossRef]
- Dziuba, D.; Postupalenko, V.Y.; Spadafora, M.; Klymchenko, A.S.; Guerineau, V.; Mely, Y.; Benhida, R.; Burger, A. A Universal Nucleoside with Strong Two-Band Switchable Fluorescence and Sensitivity to the Environment for Investigating DNA Interactions. J. Am. Chem. Soc. 2012, 134, 10209–10213. [Google Scholar] [CrossRef]
- Kuznetsova, A.A.; Kladova, O.A.; Barthes, N.P.F.; Michel, B.Y.; Burger, A.; Fedorova, O.S.; Kuznetsov, N.A. Comparative Analysis of Nucleotide Fluorescent Analogs for Registration of DNA Conformational Changes Induced by Interaction with Formamidopyrimidine-DNA Glycosylase Fpg. Russ. J. Bioorg. Chem. 2019, 45, 591–598. [Google Scholar] [CrossRef]
- Kladova, O.A.; Kuznetsova, A.A.; Barthes, N.P.F.; Michel, B.Y.; Burger, A.; Fedorova, O.S.; Kuznetsov, N.A. New Fluorescent Analogs of Nucleotides Based on 3-Hydroxychromone for Recording Conformational Changes of DNA. Russ. J. Bioorg. Chem. 2019, 45, 598–606. [Google Scholar] [CrossRef]
- Hopkins, B.B.; Reich, N.O. Simultaneous DNA Binding, Bending, and Base Flipping Evidence for a Novel M.EcoRI Methyltransferase-DNA Complex. J. Biol. Chem. 2004, 279, 37049–37060. [Google Scholar] [CrossRef] [PubMed]
- Koval, V.V.; Kuznetsov, N.A.; Ishchenko, A.A.; Saparbaev, M.K.; Fedorova, O.S. Real-Time Studies of Conformational Dynamics of the Repair Enzyme E. Coli Formamidopyrimidine-DNA Glycosylase and Its DNA Complexes during Catalytic Cycle. Mutat. Res. 2010, 685, 3–10. [Google Scholar] [CrossRef]
- Kuznetsov, N.A.; Koval, V.V.; Fedorova, O.S. Mechanism of Recognition and Repair of Damaged DNA by Human 8-Oxoguanine DNA Glycosylase HOGG1. Biochemistry 2011, 76, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Davletgildeeva, A.T.; Ishchenko, A.A.; Saparbaev, M.; Fedorova, O.S.; Kuznetsov, N.A. The Enigma of Substrate Recognition and Catalytic Efficiency of APE1-Like Enzymes. Front. Cell Dev. Biol. 2021, 9, 617161. [Google Scholar] [CrossRef] [PubMed]
- Bakman, A.S.; Kuznetsova, A.A.; Yanshole, L.V.; Ishchenko, A.A.; Saparbaev, M.; Fedorova, O.S.; Kuznetsov, N.A. Fluorescently Labeled Human Apurinic/Apyrimidinic Endonuclease APE1 Reveals Effects of DNA Polymerase β on the APE1–DNA Interaction. DNA Repair 2023, 123, 103450. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, A.A.; Matveeva, A.G.; Milov, A.D.; Vorobjev, Y.N.; Dzuba, S.A.; Fedorova, O.S.; Kuznetsov, N.A. Substrate Specificity of Human Apurinic/Apyrimidinic Endonuclease APE1 in the Nucleotide Incision Repair Pathway. Nucleic Acids Res. 2018, 46, 11454–11465. [Google Scholar] [CrossRef]
- Nevinsky, G.A. Structural, Thermodynamic, and Kinetic Basis for the Activities of Some Nucleic Acid Repair Enzymes. J. Mol. Recognit. 2011, 24, 656–677. [Google Scholar] [CrossRef]
- David, S.S.; Williams, S.D. Chemistry of Glycosylases and Endonucleases Involved in Base-Excision Repair. Chem. Rev. 1998, 98, 1221–1261. [Google Scholar] [CrossRef]
- Bjoras, M.; Luna, L.; Johnsen, B.; Hoff, E.; Haug, T.; Rognes, T.; Seeberg, E. Opposite Base-Dependent Reactions of a Human Base Excision Repair Enzyme on DNA Containing 7,8-Dihydro-8-Oxoguanine and Abasic Sites. EMBO J. 1997, 16, 6314–6322. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, N.A.; Koval, V.V.; Nevinsky, G.A.; Douglas, K.T.; Zharkov, D.O.; Fedorova, O.S. Kinetic Conformational Analysis of Human 8-Oxoguanine-DNA Glycosylase. J. Biol. Chem. 2007, 282, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Roldán-Arjona, T.; Wei, Y.F.; Carter, K.C.; Klungland, A.; Anselmino, C.; Wang, R.P.; Augustus, M.; Lindahl, T. Molecular Cloning and Functional Expression of a Human CDNA Encoding the Antimutator Enzyme 8-Hydroxyguanine-DNA Glycosylase. Proc. Natl. Acad. Sci. USA 1997, 94, 8016–8020. [Google Scholar] [CrossRef]
- Radicella, J.P.P.; Dherin, C.; Desmaze, C.; Fox, M.S.S.; Boiteux, S. Cloning and Characterization of HOGG1, a Human Homolog of the OGG1 Gene of Saccharomyces Cerevisiae. Proc. Natl. Acad. Sci. USA 1997, 94, 8010–8015. [Google Scholar] [CrossRef]
- Girard, P.M.; Guibourt, N.; Boiteux, S. The Ogg1 Protein of Saccharomyces Cerevisiae: A 7,8-Dihydro-8-Oxoguanine DNA Glycosylase/AP Lyase Whose Lysine 241 Is a Critical Residue for Catalytic Activity. Nucleic Acids Res. 1997, 25, 3404–3411. [Google Scholar] [CrossRef]
- Kuznetsov, N.A.; Koval, V.V.; Zharkov, D.O.; Nevinsky, G.A.; Douglas, K.T.; Fedorova, O.S. Kinetics of Substrate Recognition and Cleavage by Human 8-Oxoguanine-DNA Glycosylase. Nucleic Acids Res. 2005, 33, 3919–3931. [Google Scholar] [CrossRef] [PubMed]
- Zharkov, D.O.; Rosenquist, T.A.; Gerchman, S.E.; Grollman, A.P. Substrate Specificity and Reaction Mechanism of Murine 8-Oxoguanine-DNA Glycosylase. J. Biol. Chem. 2000, 275, 28607–28617. [Google Scholar] [CrossRef]
- Bruner, S.D.D.; Norman, D.P.G.P.; Verdine, G.L.L. Structural Basis for Recognition and Repair of the Endogenous Mutagen 8-Oxoguanine in DNA. Nature 2000, 403, 859–866. [Google Scholar] [CrossRef]
- Crenshaw, C.M.; Nam, K.; Oo, K.; Kutchukian, P.S.; Bowman, B.R.; Karplus, M.; Verdine, G.L. Enforced Presentation of an Extrahelical Guanine to the Lesion Recognition Pocket of Human 8-Oxoguanine Glycosylase, HOGG1. J. Biol. Chem. 2012, 287, 24916–24928. [Google Scholar] [CrossRef]
- Kuznetsova, A.A.; Kuznetsov, N.A.; Ishchenko, A.A.; Saparbaev, M.K.; Fedorova, O.S. Step-by-Step Mechanism of DNA Damage Recognition by Human 8-Oxoguanine DNA Glycosylase. Biochim. Biophys. Acta 2014, 1840, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Lukina, M.V.; Kuznetsova, A.A.; Kuznetsov, N.A.; Fedorova, O.S. The Kinetic Analysis of Recognition of the Damaged Nucleotides by Mutant Forms of the 8-Oxoguanine DNA Glycosylase HOGG1. Russ. J. Bioorg. Chem. 2017, 43, 1–12. [Google Scholar] [CrossRef]
- Tyugashev, T.E.; Vorobjev, Y.N.; Kuznetsova, A.A.; Lukina, M.V.; Kuznetsov, N.A.; Fedorova, O.S. Roles of Active-Site Amino Acid Residues in Specific Recognition of DNA Lesions by Human 8-Oxoguanine-DNA Glycosylase (OGG1). J. Phys. Chem. B 2019, 123, 4878–4887. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, A.A.; Kuznetsov, N.A.; Ishchenko, A.A.; Saparbaev, M.K.; Fedorova, O.S. Pre-Steady-State Fluorescence Analysis of Damaged DNA Transfer from Human DNA Glycosylases to AP Endonuclease APE1. Biochim. Biophys. Acta 2014, 1840, 3042–3051. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, N.A.; Kuznetsova, A.A.; Vorobjev, Y.N.; Krasnoperov, L.N.; Fedorova, O.S. Thermodynamics of the DNA Damage Repair Steps of Human 8-Oxoguanine DNA Glycosylase. PLoS ONE 2014, 9, e98495. [Google Scholar] [CrossRef]
- Brooks, S.C.; Adhikary, S.; Rubinson, E.H.; Eichman, B.F. Recent Advances in the Structural Mechanisms of DNA Glycosylases. Biochim. Biophys. Acta 2013, 1834, 247–271. [Google Scholar] [CrossRef] [PubMed]
- Au, K.G.; Cabrera, M.; Miller, J.H.; Modrich, P. Escherichia-Coli Muty Gene-Product Is Required for Specific a-G-]C.G Mismatch Correction. Proc. Natl. Acad. Sci. USA 1988, 85, 9163–9166. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.-L.; Tsai-Wu, J.-J.; Cillo, J. DNA Determinants and Substrate Specificities of Escherichia coli MutY. J. Biol. Chem. 1995, 270, 23582–23588. [Google Scholar] [CrossRef]
- Bulychev, N.V.; Varaprasad, C.V.; Dorman, G.; Miller, J.H.; Eisenberg, M.; Grollman, A.P.; Johnson, F. Substrate Specificity of Escherichia coli MutY Protein. Biochemistry 1996, 35, 13147–13156. [Google Scholar] [CrossRef] [PubMed]
- Tyugashev, T.E.; Kuznetsova, A.A.; Kuznetsov, N.A.; Fedorova, O.S. Interaction Features of Adenine DNA Glycosylase MutY from E. coli with DNA Substrates. Russ. J. Bioorg. Chem. 2017, 43, 13–22. [Google Scholar] [CrossRef]
- Kuznetsov, N.A.; Kiryutin, A.S.; Kuznetsova, A.A.; Panov, M.S.; Barsukova, M.O.; Yurkovskaya, A.V.; Fedorova, O.S. The Formation of Catalytically Competent Enzyme-Substrate Complex Is Not a Bottleneck in Lesion Excision by Human Alkyladenine DNA Glycosylase. J. Biomol. Struct. Dyn. 2017, 35, 950–967. [Google Scholar] [CrossRef]
- Petronzelli, F.; Riccio, A.; Markham, G.D.; Seeholzer, S.H.; Genuardi, M.; Karbowski, M.; Yeung, A.T.; Matsumoto, Y.; Bellacosa, A. Investigation of the Substrate Spectrum of the Human Mismatch-Specific DNA N-Glycosylase MED1 (MBD4): Fundamental Role of the Catalytic Domain. J. Cell. Physiol. 2000, 185, 473–480. [Google Scholar] [CrossRef]
- Cortellino, S.; Turner, D.; Masciullo, V.; Schepis, F.; Albino, D.; Daniel, R.; Skalka, A.M.; Meropol, N.J.; Alberti, C.; Larue, L.; et al. The Base Excision Repair Enzyme MED1 Mediates DNA Damage Response to Antitumor Drugs and Is Associated with Mismatch Repair System Integrity. Proc. Natl. Acad. Sci. USA 2003, 100, 15071–15076. [Google Scholar] [CrossRef]
- Yakovlev, D.A.; Kuznetsova, A.A.; Fedorova, O.S.; Kuznetsov, N.A. Search for Modified DNA Sites with the Human Methyl-CpG-Binding Enzyme MBD4. Acta Nat. 2017, 9, 88–98. [Google Scholar] [CrossRef]
- Denver, D.R.; Swenson, S.L.; Lynch, M. An Evolutionary Analysis of the Helix-Hairpin-Helix Superfamily of DNA Repair Glycosylases. Mol. Biol. Evol. 2003, 20, 1603–1611. [Google Scholar] [CrossRef]
- Kladova, O.A.; Krasnoperov, L.N.; Kuznetsov, N.A.; Fedorova, O.S. Kinetics and Thermodynamics of DNA Processing by Wild Type DNA-Glycosylase Endo III and Its Catalytically Inactive Mutant Forms. Genes 2018, 9, 190. [Google Scholar] [CrossRef] [PubMed]
- Thayer, M.M.; Ahern, H.; Xing, D.; Cunningham, R.P.; Tainer, J.A. Novel DNA Binding Motifs in the DNA Repair Enzyme Endonuclease III Crystal Structure. EMBO J. 1995, 14, 4108–4120. [Google Scholar] [CrossRef]
- Fromme, J.C.; Verdine, G.L. Structure of a Trapped Endonuclease III-DNA Covalent Intermediate. EMBO J. 2003, 22, 3461–3471. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, N.A.; Fedorova, O.S. Kinetic Milestones of Damage Recognition by DNA Glycosylases of the Helix-Hairpin-Helix Structural Superfamily. Adv. Exp. Biol. Med. 2020, 1241, 1–18. [Google Scholar]
- Wallace, S.S.; Bandaru, V.; Kathe, S.D.; Bond, J.P. The Enigma of Endonuclease VIII. DNA Repair 2003, 2, 441–453. [Google Scholar] [CrossRef]
- Jiang, D.; Hatahet, Z.; Melamede, R.J.; Kow, Y.W.; Wallace, S.S. Characterization of Escherichia coli Endonuclease VIII. J. Biol. Chem. 1997, 272, 32230–32239. [Google Scholar] [CrossRef]
- Zharkov, D.O.; Golan, G.; Gilboa, R.; Fernandes, A.S.; Gerchman, S.E.; Kycia, J.H.; Rieger, R.A.; Grollman, A.P.; Shoham, G. Structural Analysis of an Escherichia coli Endonuclease VIII Covalent Reaction Intermediate. EMBO J. 2002, 21, 789–800. [Google Scholar] [CrossRef]
- Zharkov, D.O.; Shoham, G.; Grollman, A.P. Structural Characterization of the Fpg Family of DNA Glycosylases. DNA Repair 2003, 2, 839–862. [Google Scholar] [CrossRef]
- Kuznetsov, N.A.; Koval, V.V.; Zharkov, D.O.; Fedorova, O.S. Conformational Dynamics of the Interaction of Escherichia coli Endonuclease VIII with DNA Substrates. DNA Repair 2012, 11, 884–891. [Google Scholar] [CrossRef]
- Kladova, O.A.; Kuznetsov, N.A.; Fedorova, O.S. Thermodynamic Parameters of Endonuclease VIII Interactions with Damaged DNA. Acta Nat. 2019, 11, 29–37. [Google Scholar] [CrossRef]
- Kladova, O.A.; Kuznetsova, A.A.; Fedorova, O.S.; Kuznetsov, N.A. Mutational and Kinetic Analysis of Lesion Recognition by Escherichia coli Endonuclease VIII. Genes 2017, 8, 140. [Google Scholar] [CrossRef]
- Tchou, J.; Kasai, H.; Shibutani, S.; Chung, M.H.; Laval, J.; Grollman, A.P.; Nishimura, S. 8-Oxoguanine (8-Hydroxyguanine) DNA Glycosylase and Its Substrate Specificity. Proc. Natl. Acad. Sci. USA 1991, 88, 4690–4694. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.H.; Kasai, H.; Jones, D.S.; Inoue, H.; Ishikawa, H.; Ohtsuka, E.; Nishimura, S. An Endonuclease Activity of Escherichia-coli That Specifically Removes 8-Hydroxyguanine Residues from DNA. Mutat. Res. 1991, 254, 1–12. [Google Scholar] [CrossRef]
- Gilboa, R.; Zharkov, D.O.; Golan, G.; Fernandes, A.S.; Gerchman, S.E.; Matz, E.; Kycia, J.H.; Grollman, A.P.; Shoham, G. Structure of Formamidopyrimidine-DNA Glycosylase Covalently Complexed to DNA. J. Biol. Chem. 2002, 277, 19811–19816. [Google Scholar] [CrossRef]
- Kuznetsov, N.A.; Zharkov, D.O.; Koval, V.V.; Buckle, M.; Fedorova, O.S. Reversible Chemical Step and Rate-Limiting Enzyme Regeneration in the Reaction Catalyzed by Formamidopyrimidine-DNA Glycosylase. Biochemistry 2009, 48, 11335–11343. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, N.A.; Milov, A.D.; Koval, V.V.; Samoilova, R.I.; Grishin, Y.A.; Knorre, D.G.; Tsvetkov, Y.D.; Fedorova, O.S.; Dzuba, S.A. PELDOR Study of Conformations of Double-Spin-Labeled Single- and Double-Stranded DNA with Non-Nucleotide Inserts. Phys. Chem. Chem. Phys. 2009, 11, 6826–6832. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, N.A.; Milov, A.D.; Isaev, N.P.; Vorobjev, Y.N.; Koval, V.V.; Dzuba, S.A.; Fedorova, O.S.; Tsvetkov, Y.D. PELDOR Analysis of Enzyme-Induced Structural Changes in Damaged DNA Duplexes. Mol. Biosyst. 2011, 7, 2670–2680. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; Doublie, S.; Wallace, S.S. The Fpg/Nei Family of DNA Glycosylases: Substrates, Structures, and Search for Damage. Prog. Mol. Biol. Transl. Sci. 2012, 110, 71–91. [Google Scholar] [CrossRef]
- Hazra, T.K.; Mitra, S. Purification and Characterization of NEIL1 and NEIL2, Members of a Distinct Family of Mammalian DNA Glycosylases for Repair of Oxidized Bases. DNA Repair Pt. A 2006, 408, 33–48. [Google Scholar] [CrossRef]
- Doublie, S.; Bandaru, V.; Bond, J.P.; Wallace, S.S. The Crystal Structure of Human Endonuclease VIII-like 1 (NEIL1) Reveals a Zincless Finger Motif Required for Glycosylase Activity. Proc. Natl. Acad. Sci. USA 2004, 101, 10284–10289. [Google Scholar] [CrossRef]
- Prakash, A.; Carroll, B.L.; Sweasy, J.B.; Wallace, S.S.; Doublie, S. Genome and Cancer Single Nucleotide Polymorphisms of the Human NEIL1 DNA Glycosylase: Activity, Structure, and the Effect of Editing. DNA Repair 2014, 14, 17–26. [Google Scholar] [CrossRef]
- Kladova, O.A.; Grin, I.R.; Fedorova, O.S.; Kuznetsov, N.A.; Zharkov, D.O. Conformational Dynamics of Damage Processing by Human DNA Glycosylase NEIL1. J. Mol. Biol. 2019, 431, 1098–1112. [Google Scholar] [CrossRef] [PubMed]
- Demple, B.; Sung, J.-S. Molecular and Biological Roles of Ape1 Protein in Mammalian Base Excision Repair. DNA Repair 2005, 4, 1442–1449. [Google Scholar] [CrossRef]
- Gros, L.; Ishchenko, A.A.; Ide, H.; Elder, R.H.; Saparbaev, M.K. The Major Human AP Endonuclease (Ape1) Is Involved in the Nucleotide Incision Repair Pathway. Nucleic Acids Res. 2004, 32, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wilson, D.M., 3rd. Human Apurinic/Apyrimidinic Endonuclease 1. Antioxid. Redox Signal 2014, 20, 678–707. [Google Scholar] [CrossRef]
- Dodson, M.L.; Lloyd, R.S. Mechanistic Comparison among Base Exision Repair Glycosylases. Free Radic. Biol. Med. 2002, 32, 678–682. [Google Scholar] [CrossRef] [PubMed]
- Burrows, C.J.; Muller, J.G. Oxidative Nucleobase Modifications Leading to Strand Scission. Chem. Rev. 1998, 98, 1109–1152. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, A.L.; Schar, P. DNA Glycosylases: In DNA Repair and Beyond. Chromosoma 2012, 121, 1–20. [Google Scholar] [CrossRef]
- Boiteux, S.; Guillet, M. Abasic Sites in DNA: Repair and Biological Consequences in Saccharomyces Cerevisiae. DNA Repair 2004, 3, 1–12. [Google Scholar] [CrossRef]
- Chen, D.S.; Herman, T.; Demple, B. Two Distinct Human DNA Diesterases That Hydrolyze 3′-Blocking Deoxyribose Fragments from Oxidized DNA. Nucleic Acids Res. 1991, 19, 5907–5914. [Google Scholar] [CrossRef]
- Golan, G.; Ishchenko, A.A.; Khassenov, B.; Shoham, G.; Saparbaev, M.K. Coupling of the Nucleotide Incision and 3′-5′ Exonuclease Activities in Escherichia coli Endonuclease IV: Structural and Genetic Evidences. Mutat. Res. 2010, 685, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Prorok, P.; Saint-Pierre, C.; Gasparutto, D.; Fedorova, O.S.; Ishchenko, A.A.; Leh, H.; Buckle, M.; Tudek, B.; Saparbaev, M. Highly Mutagenic Exocyclic DNA Adducts Are Substrates for the Human Nucleotide Incision Repair Pathway. PLoS ONE 2012, 7, e51776. [Google Scholar] [CrossRef] [PubMed]
- Christov, P.P.; Banerjee, S.; Stone, M.P.; Rizzo, C.J. Selective Incision of the Alpha-N-Methyl-Formamidopyrimidine Anomer by Escherichia coli Endonuclease IV. J. Nucleic Acids 2010, 2010, 850234. [Google Scholar] [CrossRef] [PubMed]
- Vrouwe, M.G.; Pines, A.; Overmeer, R.M.; Hanada, K.; Mullenders, L.H. UV-Induced Photolesions Elicit ATR-Kinase-Dependent Signaling in Non-Cycling Cells through Nucleotide Excision Repair-Dependent and -Independent Pathways. J. Cell Sci. 2011, 124, 435–446. [Google Scholar] [CrossRef]
- Guliaev, A.B.; Hang, B.; Singer, B. Structural Insights by Molecular Dynamics Simulations into Specificity of the Major Human AP Endonuclease toward the Benzene-Derived DNA Adduct, PBQ-C. Nucleic Acids Res. 2004, 32, 2844–2852. [Google Scholar] [CrossRef]
- Daviet, S.; Couve-Privat, S.; Gros, L.; Shinozuka, K.; Ide, H.; Saparbaev, M.; Ishchenko, A.A. Major Oxidative Products of Cytosine Are Substrates for the Nucleotide Incision Repair Pathway. DNA Repair 2007, 6, 8–18. [Google Scholar] [CrossRef]
- Prorok, P.; Alili, D.; Saint-Pierre, C.; Gasparutto, D.; Zharkov, D.O.; Ishchenko, A.A.; Tudek, B.; Saparbaev, M.K. Uracil in Duplex DNA Is a Substrate for the Nucleotide Incision Repair Pathway in Human Cells. Proc. Natl. Acad. Sci. USA 2013, 110, E3695–E3703. [Google Scholar] [CrossRef]
- Barzilay, G.; Walker, L.J.; Robson, C.N.; Hickson, I.D. Site-Directed Mutagenesis of the Human DNA Repair Enzyme HAP1: Identification of Residues Important for AP Endonuclease and RNase H Activity. Nucleic Acids Res. 1995, 23, 1544–1550. [Google Scholar] [CrossRef]
- Berquist, B.R.; McNeill, D.R.; Wilson, D.M., 3rd. Characterization of Abasic Endonuclease Activity of Human Ape1 on Alternative Substrates, as Well as Effects of ATP and Sequence Context on AP Site Incision. J. Mol. Biol. 2008, 379, 17–27. [Google Scholar] [CrossRef]
- Barnes, T.; Kim, W.C.; Mantha, A.K.; Kim, S.E.; Izumi, T.; Mitra, S.; Lee, C.H. Identification of Apurinic/Apyrimidinic Endonuclease 1 (APE1) as the Endoribonuclease That Cleaves c-Myc MRNA. Nucleic Acids Res. 2009, 37, 3946–3958. [Google Scholar] [CrossRef]
- Chou, K.M.; Cheng, Y.C. An Exonucleolytic Activity of Human Apurinic/Apyrimidinic Endonuclease on 3′ Mispaired DNA. Nature 2002, 415, 655–659. [Google Scholar] [CrossRef]
- Kerins, S.M.; Collins, R.; McCarthy, T.V. Characterization of an Endonuclease IV 3′-5′ Exonuclease Activity. J. Biol. Chem. 2003, 278, 3048–3054. [Google Scholar] [CrossRef]
- Schormann, N.; Ricciardi, R.; Chattopadhyay, D. Uracil-DNA Glycosylases-Structural and Functional Perspectives on an Essential Family of DNA Repair Enzymes. Protein Sci. 2014, 23, 1667–1685. [Google Scholar] [CrossRef] [PubMed]
- Huffman, J.L.; Sundheim, O.; Tainer, J.A. DNA Base Damage Recognition and Removal: New Twists and Grooves. Mutat. Res. 2005, 577, 55–76. [Google Scholar] [CrossRef] [PubMed]
- Mullins, E.A.; Shi, R.X.; Parsons, Z.D.; Yuen, P.K.; David, S.S.; Igarashi, Y.; Eichman, B.F. The DNA Glycosylase AlkD Uses a Non-Base-Flipping Mechanism to Excise Bulky Lesions. Nature 2015, 527, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Dunn, A.R.; Kad, N.M.; Nelson, S.R.; Warshaw, D.M.; Wallace, S.S. Single Qdot-Labeled Glycosylase Molecules Use a Wedge Amino Acid to Probe for Lesions While Scanning along DNA. Nucleic Acids Res. 2011, 39, 7487–7498. [Google Scholar] [CrossRef]
- Kuznetsova, A.A.; Iakovlev, D.A.; Misovets, I.V.; Ishchenko, A.A.; Saparbaev, M.K.; Kuznetsov, N.A.; Fedorova, O.S. Pre-Steady-State Kinetic Analysis of Damage Recognition by Human Single-Strand Selective Monofunctional Uracil-DNA Glycosylase SMUG1. Mol. Biosyst. 2017, 13, 2638–2649. [Google Scholar] [CrossRef]
- Nelson, S.R.; Dunn, A.R.; Kathe, S.D.; Warshaw, D.M.; Wallace, S.S. Two Glycosylase Families Diffusively Scan DNA Using a Wedge Residue to Probe for and Identify Oxidatively Damaged Bases. Proc. Natl. Acad. Sci. USA 2014, 111, E2091-9. [Google Scholar] [CrossRef]
- Kuznetsov, N.A.; Bergonzo, C.; Campbell, A.J.; Li, H.; Mechetin, G.V.; de los Santos, C.; Grollman, A.P.; Fedorova, O.S.; Zharkov, D.O.; Simmerling, C. Active Destabilization of Base Pairs by a DNA Glycosylase Wedge Initiates Damage Recognition. Nucleic Acids Res. 2015, 43, 272–281. [Google Scholar] [CrossRef]
- Lee, A.J.; Wallace, S.S. Hide and Seek: How Do DNA Glycosylases Locate Oxidatively Damaged DNA Bases amidst a Sea of Undamaged Bases? Free Radic. Biol. Med. 2017, 107, 170–178. [Google Scholar] [CrossRef]
- Mol, C.D.; Hosfield, D.J.; Tainer, J.A. Abasic Site Recognition by Two Apurinic/Apyrimidinic Endonuclease Families in DNA Base Excision Repair: The 3′ Ends Justify the Means. Mutat. Res. 2000, 460, 211–229. [Google Scholar] [CrossRef]
- Tsutakawa, S.E.; Shin, D.S.; Mol, C.D.; Izumi, T.; Arvai, A.S.; Mantha, A.K.; Szczesny, B.; Ivanov, I.N.; Hosfield, D.J.; Maiti, B.; et al. Conserved Structural Chemistry for Incision Activity in Structurally Non-Homologous Apurinic/Apyrimidinic Endonuclease APE1 and Endonuclease IV DNA Repair Enzymes. J. Biol. Chem. 2013, 288, 8445–8455. [Google Scholar] [CrossRef] [PubMed]
- Beernink, P.T.; Segelke, B.W.; Hadi, M.Z.; Erzberger, J.P.; Wilson, D.M., 3rd; Rupp, B. Two Divalent Metal Ions in the Active Site of a New Crystal Form of Human Apurinic/Apyrimidinic Endonuclease, Ape1: Implications for the Catalytic Mechanism. J. Mol. Biol. 2001, 307, 1023–1034. [Google Scholar] [CrossRef]
- Gorman, M.A.; Morera, S.; Rothwell, D.G.; de La Fortelle, E.; Mol, C.D.; Tainer, J.A.; Hickson, I.D.; Freemont, P.S. The Crystal Structure of the Human DNA Repair Endonuclease HAP1 Suggests the Recognition of Extra-Helical Deoxyribose at DNA Abasic Sites. EMBO J. 1997, 16, 6548–6558. [Google Scholar] [CrossRef] [PubMed]
- Manvilla, B.A.; Pozharski, E.; Toth, E.A.; Drohat, A.C. Structure of Human Apurinic/Apyrimidinic Endonuclease 1 with the Essential Mg2+ Cofactor. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 2555–2562. [Google Scholar] [CrossRef]
- Mol, C.D.; Izumi, T.; Mitra, S.; Tainer, J.A. DNA-Bound Structures and Mutants Reveal Abasic DNA Binding by APE1 and DNA Repair Coordination. Nature 2000, 403, 451–456. [Google Scholar] [CrossRef]
- Timofeyeva, N.A.; Koval, V.V.; Knorre, D.G.; Zharkov, D.O.; Saparbaev, M.K.; Ishchenko, A.A.; Fedorova, O.S. Conformational Dynamics of Human AP Endonuclease in Base Excision and Nucleotide Incision Repair Pathways. J. Biomol. Struct. Dyn. 2009, 26, 637–652. [Google Scholar] [CrossRef]
- Kanazhevskaya, L.Y.; Koval, V.V.; Zharkov, D.O.; Strauss, P.R.; Fedorova, O.S. Conformational Transitions in Human AP Endonuclease 1 and Its Active Site Mutant during Abasic Site Repair. Biochemistry 2010, 49, 6451–6461. [Google Scholar] [CrossRef]
- Kanazhevskaya, L.Y.; Koval, V.V.; Vorobjev, Y.N.; Fedorova, O.S. Conformational Dynamics of Abasic DNA upon Interactions with AP Endonuclease 1 Revealed by Stopped-Flow Fluorescence Analysis. Biochemistry 2012, 51, 1306–1321. [Google Scholar] [CrossRef]
- Bulygin, A.A.; Syryamina, V.N.; Kuznetsova, A.A.; Novopashina, D.S.; Dzuba, S.A.; Kuznetsov, N.A. Inner Amino Acid Contacts Are Key Factors of Multistage Structural Rearrangements of DNA and Affect Substrate Specificity of Apurinic/Apyrimidinic Endonuclease APE1. Int. J. Mol. Sci. 2023, 24, 11474. [Google Scholar] [CrossRef] [PubMed]
- Senchurova, S.I.; Syryamina, V.N.; Kuznetsova, A.A.; Novopashina, D.S.; Ishchenko, A.A.; Saparbaev, M.; Dzuba, S.A.; Fedorova, O.S.; Kuznetsov, N.A. The Mechanism of Damage Recognition by Apurinic/Apyrimidinic Endonuclease Nfo from Escherichia coli. BBA—Gen. Subj. 2022, 1866, 130216. [Google Scholar] [CrossRef] [PubMed]
- Bulygin, A.A.; Fedorova, O.S.; Kuznetsov, N. Insights into Mechanisms of Damage Recognition and Catalysis by APE1-like Enzymes. Int. J. Mol. Sci. 2022, 23, 4361. [Google Scholar] [CrossRef] [PubMed]
- Bulygin, A.A.; Kuznetsova, A.A.; Vorobjev, Y.N.; Fedorova, O.S.; Kuznetsov, N.A. The Role of Active-Site Plasticity in Damaged-Nucleotide Recognition by Human Apurinic/Apyrimidinic Endonuclease APE1. Molecules 2020, 25, 3940. [Google Scholar] [CrossRef] [PubMed]
- Alekseeva, I.V.; Kuznetsova, A.A.; Bakman, A.S.; Fedorova, O.S.; Kuznetsov, N.A. The Role of Active-Site Amino Acid Residues in the Cleavage of DNA and RNA Substrates by Human Apurinic/Apyrimidinic Endonuclease APE1. BBA—Gen. Subj. 2020, 1864, 129718. [Google Scholar] [CrossRef] [PubMed]
- Alekseeva, I.V.; Bakman, A.S.; Vorobjev, Y.N.; Fedorova, O.S.; Kuznetsov, N.A. Role of Ionizing Amino Acid Residues in the Process of DNA Binding by Human AP Endonuclease 1 and in Its Catalysis. J. Phys. Chem. B 2019, 123, 9546–9556. [Google Scholar] [CrossRef]
- Alekseeva, I.V.; Davletgildeeva, A.T.; Arkova, O.V.; Kuznetsov, N.A.; Fedorova, O.S. The Impact of Single-Nucleotide Polymorphisms of Human Apurinic/Apyrimidinic Endonuclease 1 on Specific DNA Binding and Catalysis. Biochimie 2019, 163, 73–83. [Google Scholar] [CrossRef]
- Bakman, A.S.; Boichenko, S.S.; Kuznetsova, A.A.; Ishchenko, A.A.; Saparbaev, M.; Kuznetsov, N.A. The Impact of Human DNA Glycosylases on the Activity of DNA Polymerase β toward Various Base Excision Repair Intermediates. Int. J. Mol. Sci. 2023, 24, 9594. [Google Scholar] [CrossRef]
- Davletgildeeva, A.T.; Kuznetsova, A.A.; Novopashina, D.S.; Ishchenko, A.A.; Saparbaev, M.; Fedorova, O.S.; Kuznetsov, N.A. Comparative Analysis of Exo-and Endonuclease Activities of APE1-like Enzymes. Int. J. Mol. Sci. 2022, 23, 2869. [Google Scholar] [CrossRef]
- Kuznetsova, A.A.; Gavrilova, A.A.; Novopashina, D.S.; Fedorova, O.S.; Kuznetsov, N.A. Mutational and Kinetic Analysis of APE1 Endoribonuclease Activity. Mol. Biol. 2021, 55, 211–224. [Google Scholar] [CrossRef]
- Miroshnikova, A.D.; Kuznetsova, A.A.; Vorobjev, Y.N.; Kuznetsov, N.A.; Fedorova, O.S. Effects of Mono- and Divalent Metal Ions on DNA Binding and Catalysis of Human Apurinic/Apyrimidinic Endonuclease 1. Mol. BioSyst. 2016, 12, 1527–1539. [Google Scholar] [CrossRef]
- Miroshnikova, A.D.; Kuznetsova, A.A.; Kuznetsov, N.A.; Fedorova, O.S. Thermodynamics of Damaged DNA Binding and Catalysis by Human AP Endonuclease 1. Acta Nat. 2016, 8, 103–110. [Google Scholar] [CrossRef]

























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
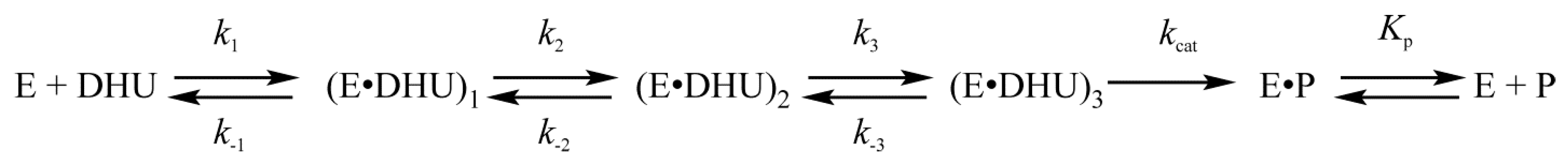{kind=link}
{kind=link}
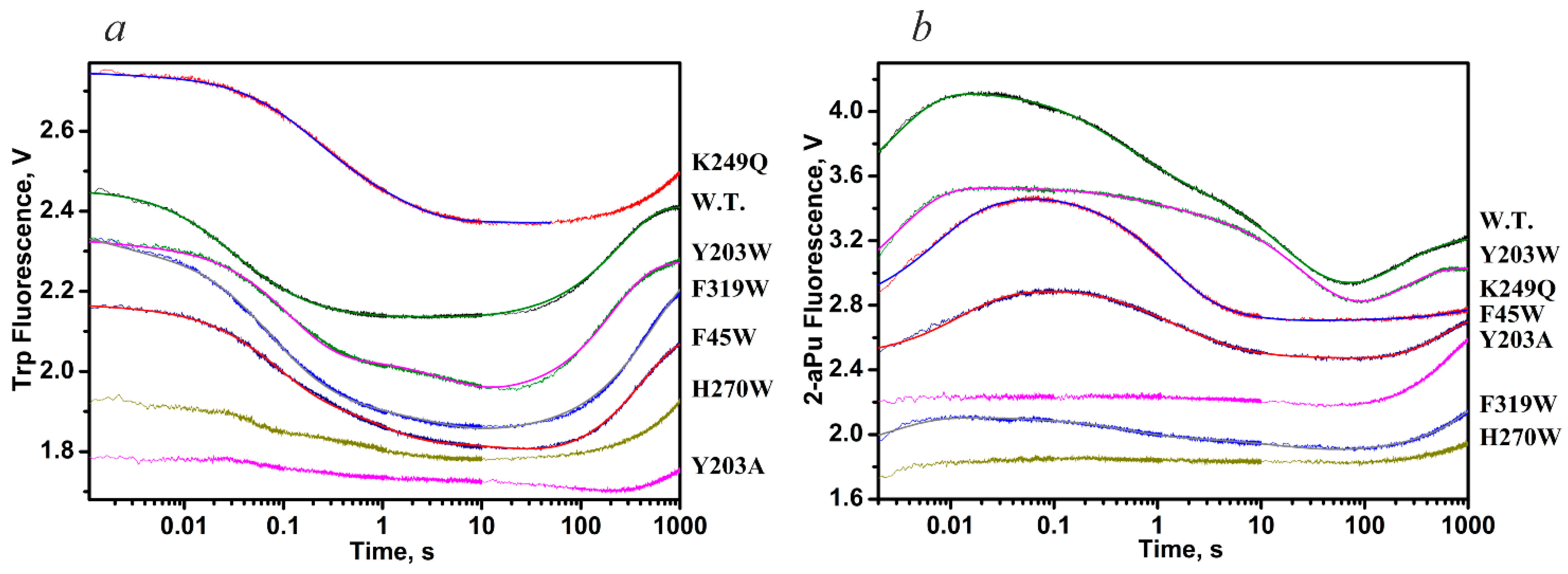
| Trp Detection | aPu Detection | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Constants | WT | K249Q | Y203W | F319W | F45W | WT | K249Q | Y203W | F319W | F45W |
| k1 × 10−8, M−1s−1 | 2.6 ± 0.1 | 1.7 ± 0.4 | 5.4 ± 1.0 | 2.2 ± 0.1 | 6.9 ± 1.8 | 1.2 ± 0.1 | 0.3 ± 0.1 | 0.66 ± 0.04 | 0.6 ± 0.2 | 0.5 ± 0.1 |
| k−1, s−1 | 130 ± 1 | 290 ± 60 | 520 ± 110 | 240 ± 10 | 500 ± 45 | 120 ± 10 | 62 ± 12 | 130 ± 7 | 260 ± 40 | 103 ± 9 |
| K1, M−1 | 2.0 × 106 | 0.6 × 106 | 1.0 × 106 | 0.9 × 106 | 1.4 × 106 | 1.0 × 106 | 0.5 × 106 | 0.5 × 106 | 0.2 × 106 | 0.5 × 106 |
| k2, s−1 | 13.3 ± 0.2 | 5.0 ± 0.5 | 7.4 ± 1.0 | 31.9 ± 1.2 | 12.1 ± 2.0 | 1.4 ± 0.1 | 1.4 ± 0.1 | 0.3 ± 0.1 | 4.1 ± 1.5 | 1.4 ± 0.1 |
| k−2, s−1 | 1.16 ± 0.02 | 2.8 ± 0.2 | 1.2 ± 0.2 | 0.5 ± 0.1 | 2.3 ± 0.6 | 1.5 ± 0.1 | 1.1 ± 0.2 | 1.3 ± 0.3 | 2.5 ± 0.8 | 1.4 ± 0.2 |
| K2 | 11.5 | 1.8 | 6.2 | 63.8 | 5.3 | 0.9 | 1.3 | 0.2 | 1.6 | 1.0 |
| k3, s−1 | 0.012 ± 0.001 | 0.26 ± 0.01 | 0.010 ± 0.001 | 0.08 ± 0.01 | 0.009 ± 0.001 | 0.10 ± 0.01 | 5.4 ± 1.1 | 0.28 ± 0.04 | 0.9 ± 0.2 | 0.48 ± 0.09 |
| k−3, s−1 | 0.07 ± 0.01 | 0.52 ± 0.01 | 0.12 ± 0.01 | 0.8 ± 0.1 | 0.4 ± 0.1 | 0.013 ± 0.002 | 0.8 ± 0.1 | 0.022 ± 0.008 | 0.5 ± 0.2 | 0.38 ± 0.04 |
| K3 | 0.17 | 0.5 | 0.08 | 0.1 | 0.02 | 7.7 | 6.7 | 12.7 | 1.8 | 1.3 |
| k4, s−1 | 0.06 ± 0.02 | - | 0.03 ± 0.01 | 0.018 ± 0.002 | 0.05 ± 0.01 | 0.029 ± 0.001 | - | 0.015 ± 0.004 | 0.015 ± 0.004 | 0.034 ± 0.002 |
| k5 × 103, s−1 | 6.4 ± 0.7 | - | 4.2 ± 0.9 | 1.8 ± 0.1 | 0.4 ± 0.1 | 3.6 ± 0.2 | - | 3.0 ± 0.7 | 1.9 ± 0.1 | 4.4 ± 0.1 |
| KEP, μM | 0.88 | - | 0.3 ± 0.1 | 1.1 ± 0.1 | 1.3 ± 0.4 | 7.0 ± 1.1 | - | 1.0 ± 0.2 | - | - |
| Kbind, M−1 | 2.9 × 107 | 2.2 × 106 | 8.0 × 106 | 6.5 × 107 | 8.8 × 106 | 9.2 × 106 | 5.3 × 106 | 2.1 × 106 | 1.3 × 106 | 1.6 × 106 |
| K1 × K2 × K3 | 3.9 × 106 | 0.54 × 106 | 0.50 × 106 | 6.1 × 106 | 0.15 × 106 | 6.9 × 106 | 4.3 × 106 | 1.3 × 106 | 0.58 × 106 | 0.65 × 106 |
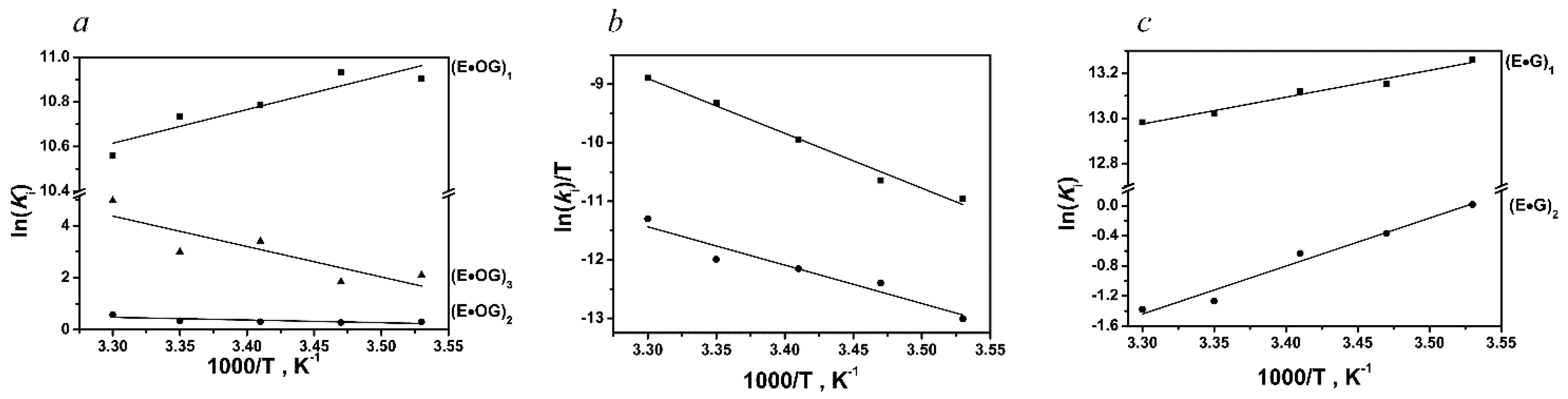
| DNA | Constants | Temperature | ||||
|---|---|---|---|---|---|---|
| 10 °C | 15 °C | 20 °C | 25 °C | 30 °C | ||
| oxoG-substrate | k1, M−1s−1 | (1.3 ± 0.7) × 107 | (2.2 ± 1.0) × 107 | (1.7 ± 0.8) × 107 | (1.9 ± 1.0) × 107 | (2.0 ± 1.0) × 107 |
| k−1, s−1 | 230 ± 80 | 390 ± 50 | 360 ± 20 | 410 ± 70 | 520 ± 120 | |
| k2, s−1 | 8.3 ± 3.8 | 15.6 ± 4.2 | 26.6 ± 6.0 | 53.5 ± 20.3 | 72.4 ± 31.4 | |
| k−2, s−1 | 6.1 ± 0.2 | 11.8 ± 0.2 | 19.6 ± 1.7 | 37.9 ± 11.1 | 40.5 ± 12.5 | |
| k3, s−1 | 0.087 ± 0.017 | 0.12 ± 0.03 | 0.19 ± 0.07 | 0.4 ± 0.1 | 0.3 ± 0.1 | |
| k−3, s−1 | 0.011 ± 0.001 | 0.018 ± 0.001 | 0.007 ± 0.002 | 0.02 ± 0.003 | 0.002 ± 0.001 | |
| k4, s−1 | 0.005 ± 0.001 | 0.007 ± 0.001 | 0.014 ± 0.002 | 0.026 ± 0.003 | 0.041 ± 0.007 | |
| k5, s−1 | 0.0006 ± 0.0001 | 0.0012 ± 0.0001 | 0.001 ± 0.0001 | 0.001 ± 0.001 | 0.0037 ± 0.007 | |
| undamaged DNA | k1, M−1s−1 | (5.6 ± 1.2) × 107 | (6.6 ± 1.1) × 107 | (5.9 ± 1.3) × 107 | (6.3 ± 0.3) × 107 | (7.4 ± 1.4) × 107 |
| k−1, s−1 | 97 ± 23 | 128 ± 21 | 119 ± 19 | 140 ± 14 | 170 ± 17 | |
| k2, s−1 | 12.6 ± 5.5 | 17.5 ± 2.2 | 20.7 ± 6.3 | 14.0 ± 1.5 | 18.2 ± 8.7 | |
| k−2, s−1 | 12.4 ± 0.5 | 25.2 ± 1.3 | 38.9 ± 1.9 | 49.8 ± 2.3 | 72.1 ± 9.9 | |
| DNA | Parameter | ∆G°i298, kcal/mol | ∆H°i, kcal/mol | ∆S°i, cal/(K·mol) | |
|---|---|---|---|---|---|
| Stage Number | |||||
| oxoG-substrate | 1 | −6.4 | −2.8 ± 0.7 | 11.2 ± 2.4 | |
| 2 | −0.2 | 2.1 ± 0.9 | 7.7 ± 3.3 | ||
| 3 | −1.8 | 23.2 ± 7.8 | 85.4 ± 26.6 | ||
| −8.4 | 22.5 ± 9.4 | 104.3 ± 32.3 | |||
| 4 | 19.6 | 18.6 ± 1.1 | −3.5 ± 3.9 | ||
| 5 | 21.0 | 13.0 ± 1.9 | −27.0 ± 6.7 | ||
| undamaged DNA | 1 | −7.7 | −2.4 ± 0.2 | 18.0 ± 0.7 | |
| 2 | 0.8 | −12.7 ± 1.2 | −44.7 ± 4.1 | ||
| −6.9 | −15.1 ± 1.4 | −26.7 ± 4.8 | |||
| hOGG1 | Nth (B. st.) | MBD4 | MutY (E. coli) | ||
|---|---|---|---|---|---|
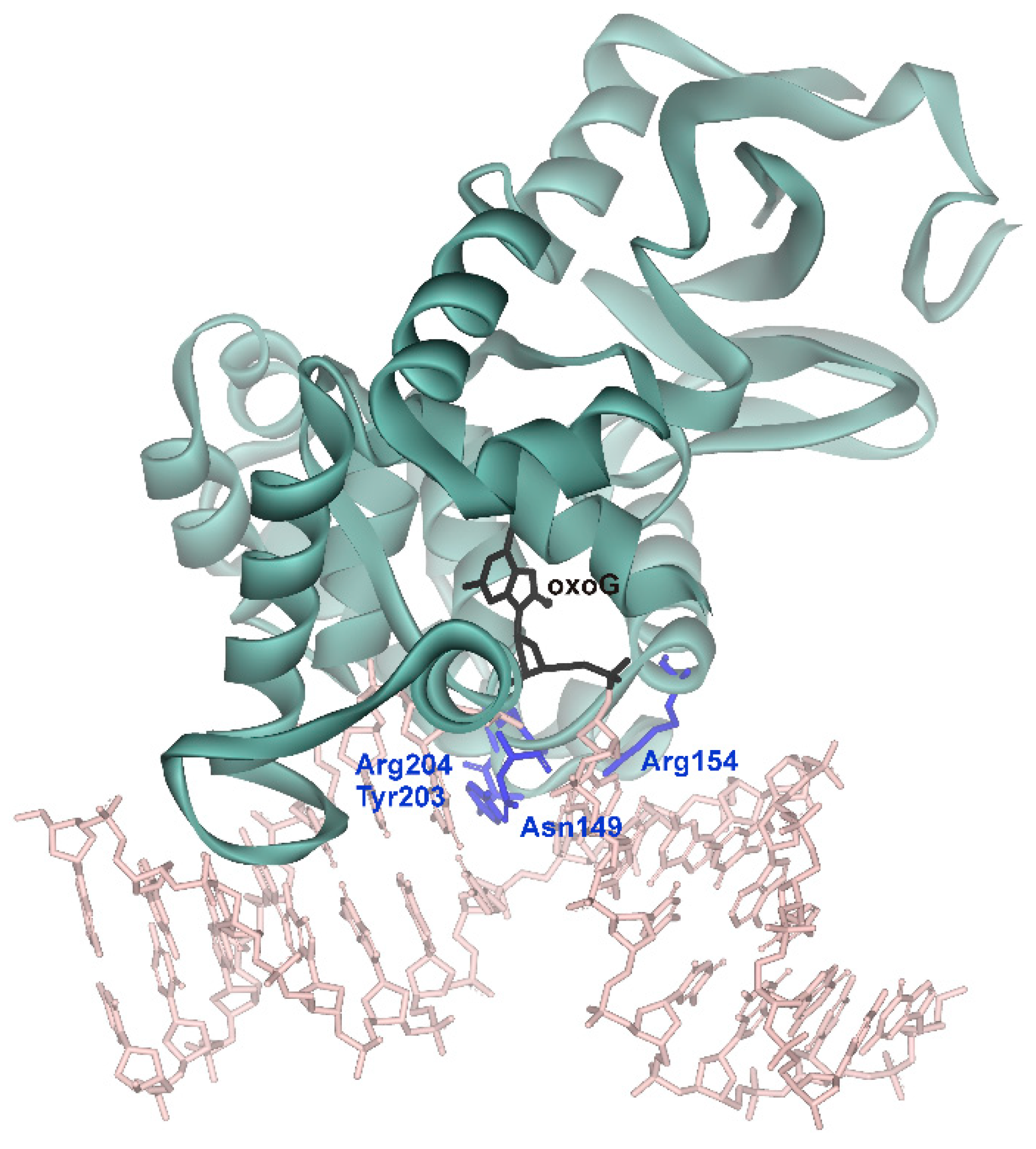
| Initial binding | Destabilization of double helix | Arg154, Arg204 | Arg78, Arg84 | Arg468 | Arg50, Arg91 |
| Intercalation of “sensor” | Tyr203 | Leu82 | Leu508 | Tyr88 | |
| Specific recognition | Complete eversion of damaged base | + | + | + | + |
| Intercalation of other amino acid residues | Asn149 | Gln42 | - | Gln48 |
| Constants | L70S | L70W | Y71W | F121W | F230W | P253W | WT |
|---|---|---|---|---|---|---|---|
| k1, M−1s−1 | (0.09 ± 0.02) × 106 | (0.06 ± 0.01) × 106 | (21 ± 3) × 106 | (39 ± 11) × 106 | (30 ± 2) × 106 | (27 ± 2) × 106 | (36 ± 7) × 106 |
| k−1, s−1 | 1 ± 0.1 | 0.4 ± 0.1 | 330 ± 30 | 120 ± 31 | 310 ± 45 | 310 ± 26 | 410 ± 20 |
| K1a, M−1 | 0.9 × 105 | 1.5 × 105 | 0.63 × 105 | 3 × 105 | 0.97 × 105 | 0.87 × 105 | 0.88 × 105 |
| k2, s−1 | 16 ± 3 | 17 ± 4 | 21 ± 3 | 23 ± 3 | 27 ± 2 | ||
| k−2, s−1 | 2.5 ± 0.1 | 0.55 ± 0.25 | 1.2 ± 0.1 | 0.8 ± 0.1 | 1.8 ± 0.3 | ||
| K2 | 6.4 | 31 | 17.5 | 28.7 | 15 | ||
| k3, s−1 | 0.4 ± 0.1 | 0.58 ± 0.15 | 0.82 ± 0.04 | 1.0 ± 0.03 | 1.6 ± 0.1 | ||
| k−3, s−1 | 1.1 ± 0.1 | 0.89 ± 0.14 | 0.66 ± 0.06 | 1.2 ± 0.1 | 1.5 ± 0.2 | ||
| K3 | 0.36 | 0.65 | 1.2 | 0.83 | 1.1 | ||
| Kassb, M−1 | 1.45 × 105 | 6 × 106 | 2 × 106 | 2 × 106 | 1.45 × 106 | ||
| kcat, s−1 | 0.09 ± 0.01 | 0.08 ± 0.03 | 0.14 ± 0.03 | 0.26 ± 0.09 | 0.38 ± 0.01 | 0.4 ± 0.03 | 0.35 ± 0.02 |
| Kp, M | (1.0 ± 0.2) × 10−6 | (0.7 ± 0.5) × 10−6 | (0.4 ± 0.1) × 10−6 | (0.6 ± 0.1) × 10−6 | (0.6 ± 0.2) × 10−6 | (0.63 ± 0.08) × 10−6 | (0.7 ± 0.1) × 10−6 |
| DNA | Parameter | ∆G°i298, kcal/mol | ∆H°i, kcal/mol | ∆S°i, cal/(K·mol) | Process | |
|---|---|---|---|---|---|---|
| Stage Number | ||||||
| G/CPy12 | 1 | −7.0 | −3.8 ± 0.9 | 10.9 ± 3.2 | Nonspecific binding, local “melting” of segment of DNA duplex, dehydration | |
| F/CPy12 | 1 | −7.2 | −4.0 ± 0.3 | 10.8 ± 1.0 | Nonspecific binding, local “melting” of segment of DNA duplex, dehydration | |
| 2 | 0.7 | 6.7 ± 0.3 | 20.3 ± 0.9 | Kinking of DNA and intercalation of Fpg amino acid residues into duplex | ||
| −6.5 | 2.7 ± 0.4 | 31.1 ± 1.3 | ||||
| oxoG/CPy12 | 1 | −7.0 | −3.2 ± 0.4 | 12.7 ± 1.5 | Nonspecific binding, local “melting” of segment of DNA duplex, dehydration | |
| 2 | 0.4 | 0.3 ± 0.8 | −0.3 ± 2.7 | Insertion of “wedge” into DNA (amino acid residue Phe110) for discrimination between damaged and intact segments of DNA | ||
| 3 | 0.8 | 6.3 ± 1.7 | 18.4 ± 5.8 | Kinking of DNA | ||
| 4 | −1.5 | −15.5 ± 3.9 | −46.9 ± 13.5 | Eversion of oxoG base into active site of Fpg concurrently with insertion of amino acid residues Arg108 and Met73 into resultant cavity in DNA helix | ||
| 5 | −1.9 | 31.2 ± 5.5 | 111.1 ± 18.6 | Final fine adjustment of active-site structure to attain catalytically competent state, dehydration of DNA grooves | ||
| −9.2 | 19.1 ± 12.3 | 95.0 ± 42.1 | ||||
| 6 | 19.6 | 6.0 ± 1.4 | −45.5 ± 4.7 | Transition state of catalytical chemical stage | ||
| 7 | −7.0 | −4.1 ± 0.3 | 9.6 ± 0.9 | Formation of enzyme–product complex | ||
| Nei (E. coli) | Fpg (E. coli) | NEIL1 | |
|---|---|---|---|
| Destabilization of double helix, local “melting” | + | + | + |
| Intercalation of “sensor” | Leu70 | Phe110 | Phe119 |
| Eversion of damaged base and duplex bending | + | + | + |
| Intercalation of other amino acid residues | Gln69, Tyr71 | Met78, Arg108 | Met80, Arg117 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuznetsov, N.A. Conformational Dynamics of Biopolymers in the Course of Their Interaction: Multifaceted Approaches to the Analysis by the Stopped-Flow Technique with Fluorescence Detection. Photonics 2023, 10, 1033. https://doi.org/10.3390/photonics10091033
Kuznetsov NA. Conformational Dynamics of Biopolymers in the Course of Their Interaction: Multifaceted Approaches to the Analysis by the Stopped-Flow Technique with Fluorescence Detection. Photonics. 2023; 10(9):1033. https://doi.org/10.3390/photonics10091033
Chicago/Turabian StyleKuznetsov, Nikita A. 2023. "Conformational Dynamics of Biopolymers in the Course of Their Interaction: Multifaceted Approaches to the Analysis by the Stopped-Flow Technique with Fluorescence Detection" Photonics 10, no. 9: 1033. https://doi.org/10.3390/photonics10091033
APA StyleKuznetsov, N. A. (2023). Conformational Dynamics of Biopolymers in the Course of Their Interaction: Multifaceted Approaches to the Analysis by the Stopped-Flow Technique with Fluorescence Detection. Photonics, 10(9), 1033. https://doi.org/10.3390/photonics10091033