Development of a Methodology to Adapt an Equilibrium Buffer/Wash Applied to the Purification of hGPN2 Protein Expressed in Escherichia coli Using an IMAC Immobilized Metal Affinity Chromatography System

 , , and
, , and
Abstract
:1. Introduction
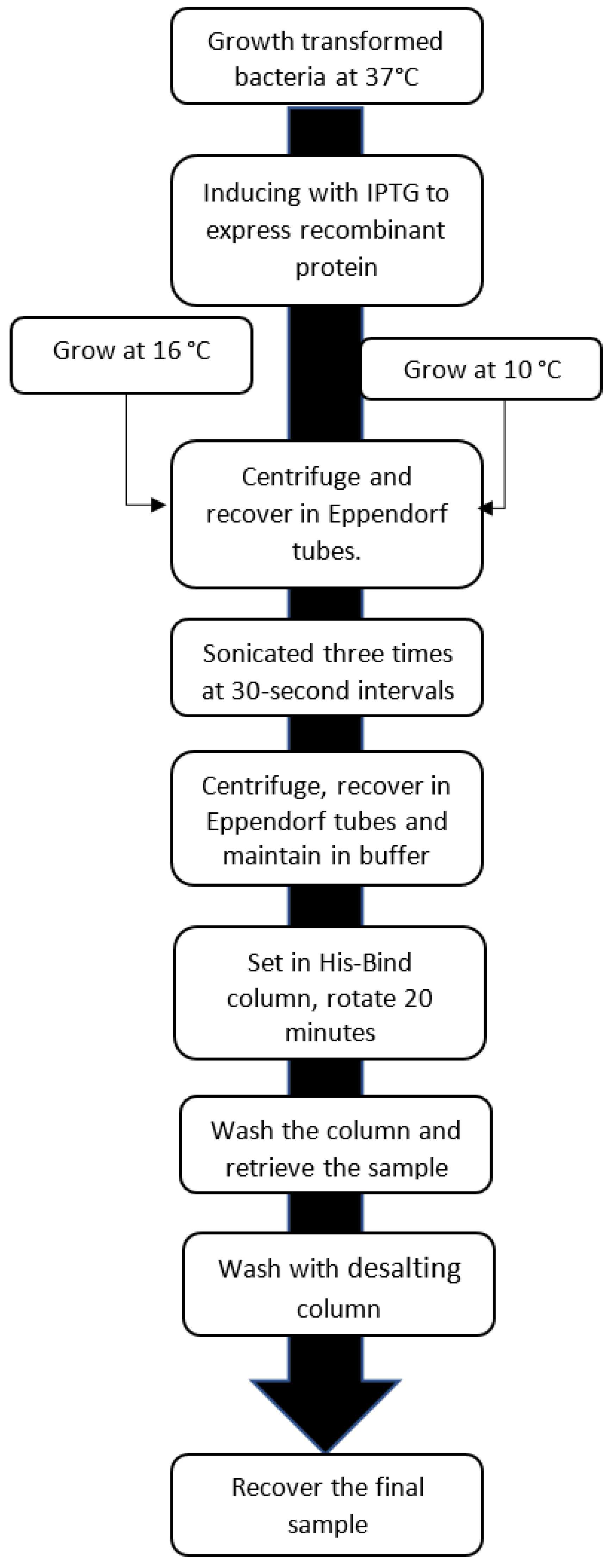
2. Materials and Methods
2.1. Construction of the Expression Vector for hGPN2
2.2. Preparation of the His-Bind Column
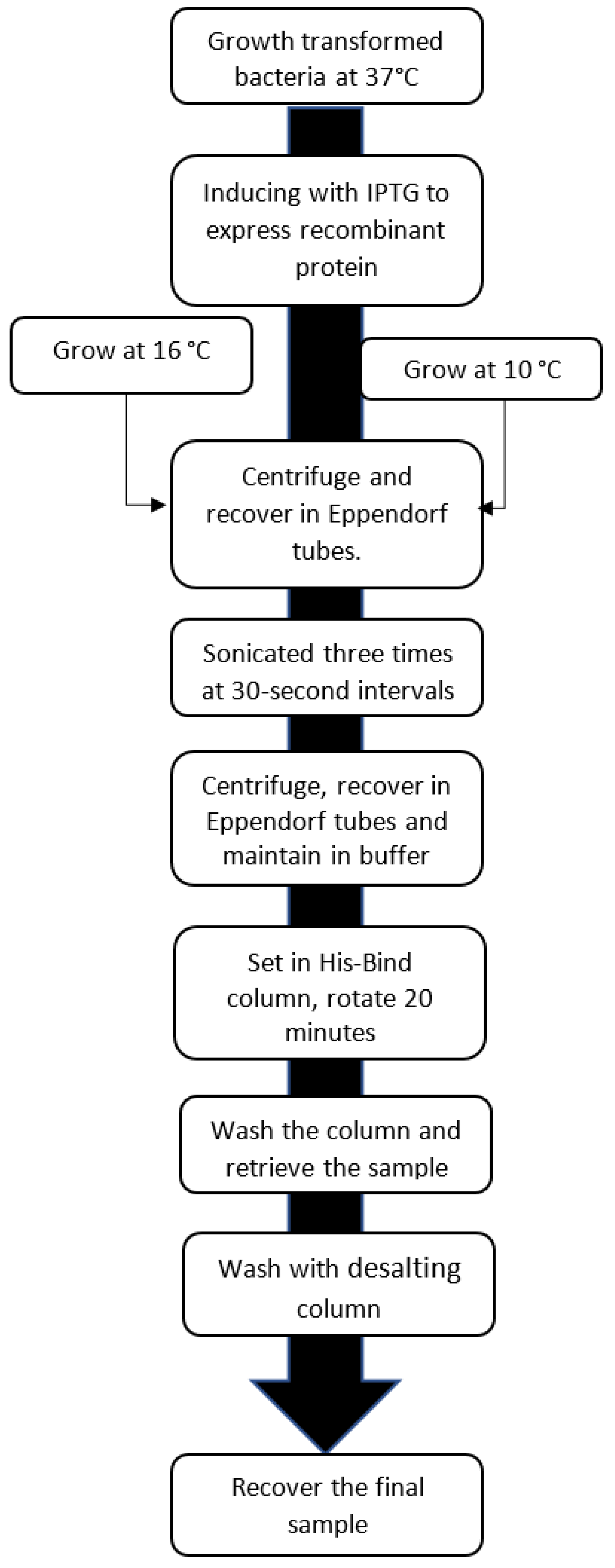
2.3. Expression of hGPN2 Protein and Growth at Different Temperatures
2.4. Protein Expression with NaCl
2.5. Protein Purification of Recombinant Protein with TRIS
2.6. Protein Purification of Recombinant Protein with EDTA
2.7. Protein Purification of Recombinant Protein with DTT
2.8. Protein Purification of Recombinant Protein with Amino Acids
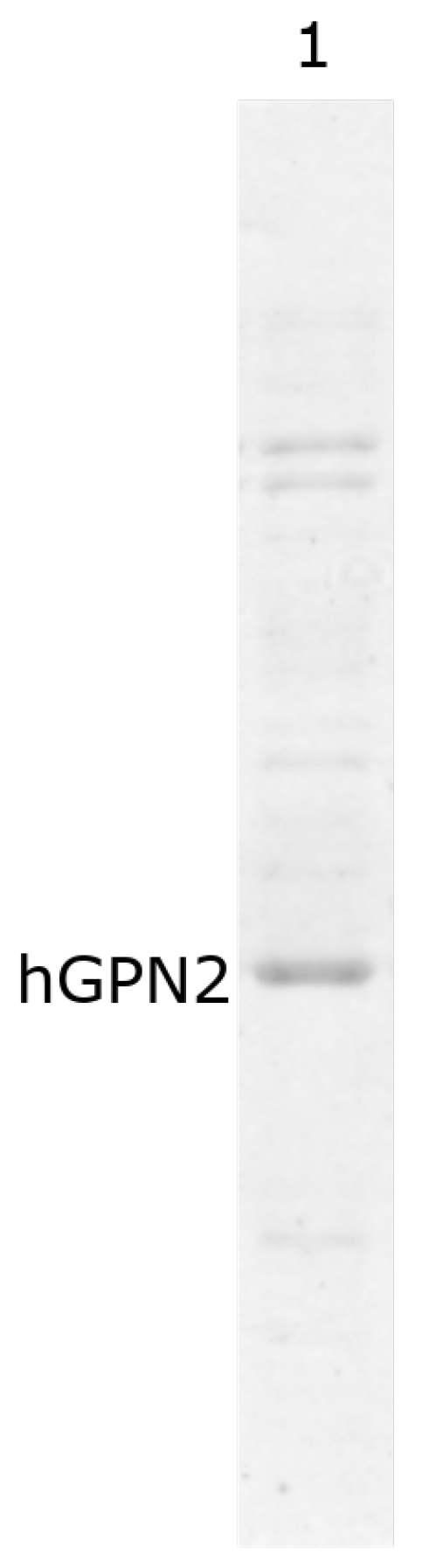
2.9. Purification with Final Buffer
2.10. Quantification of the hGPN2 Protein
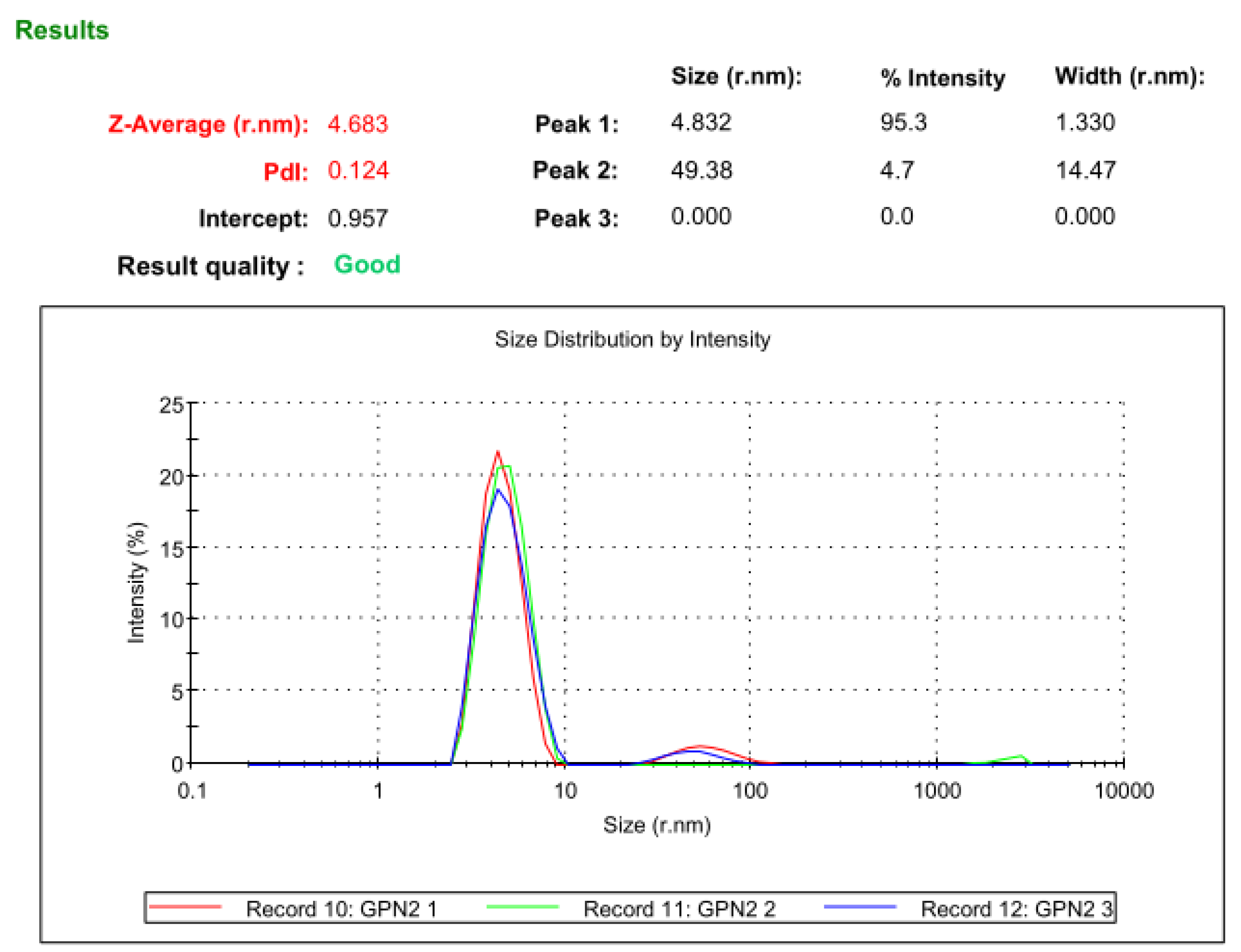
2.11. Dynamic light Scattering Analysis
3. Results
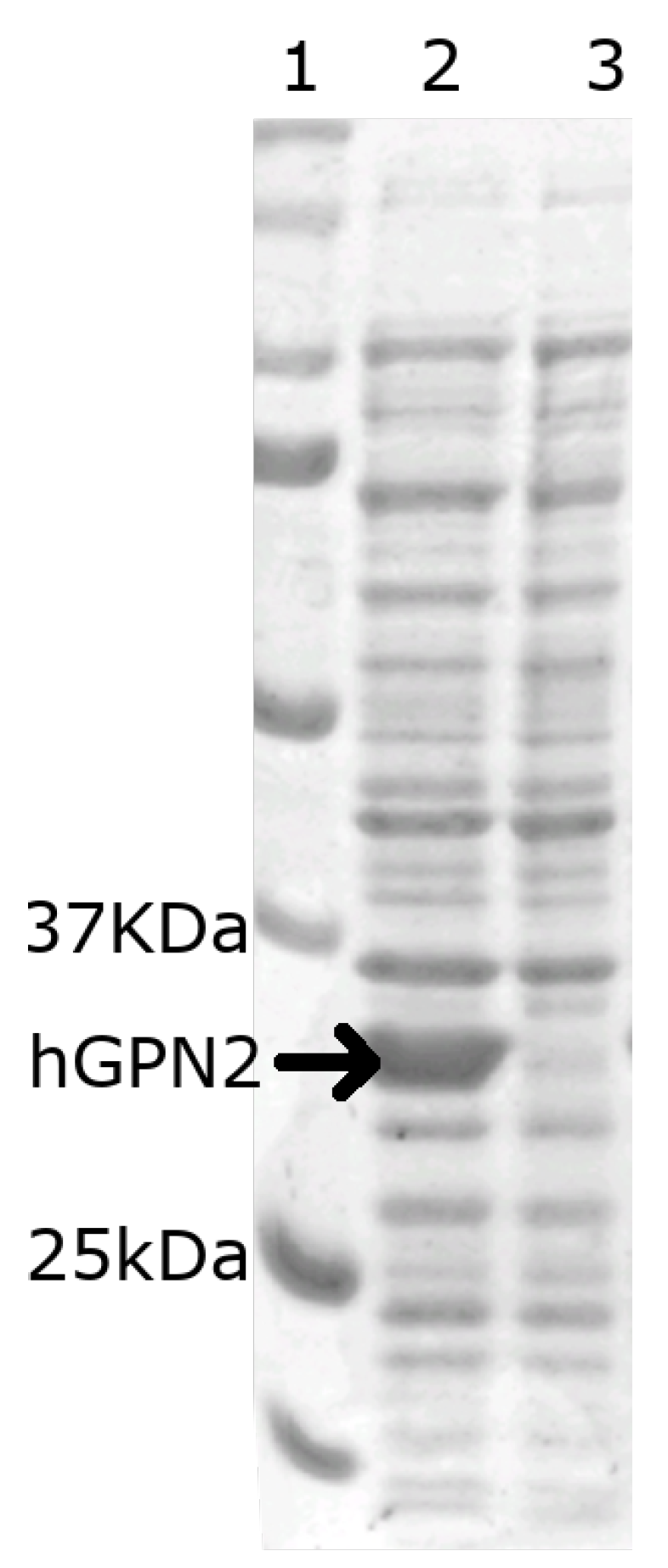
3.1. Construction of the Expression Vector for hGPN2 and Expression Assay
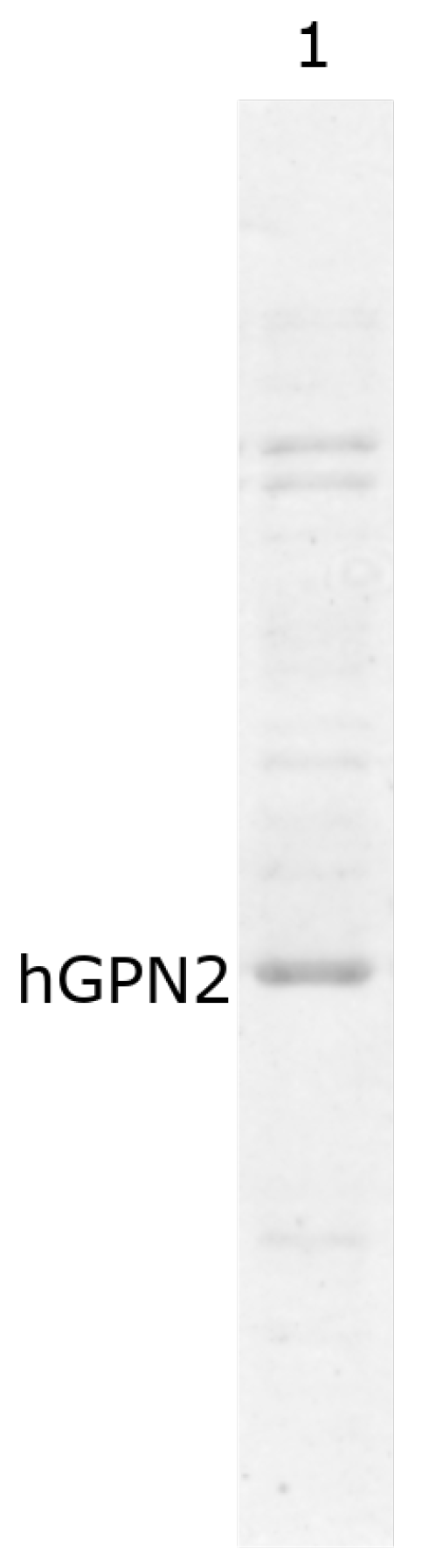
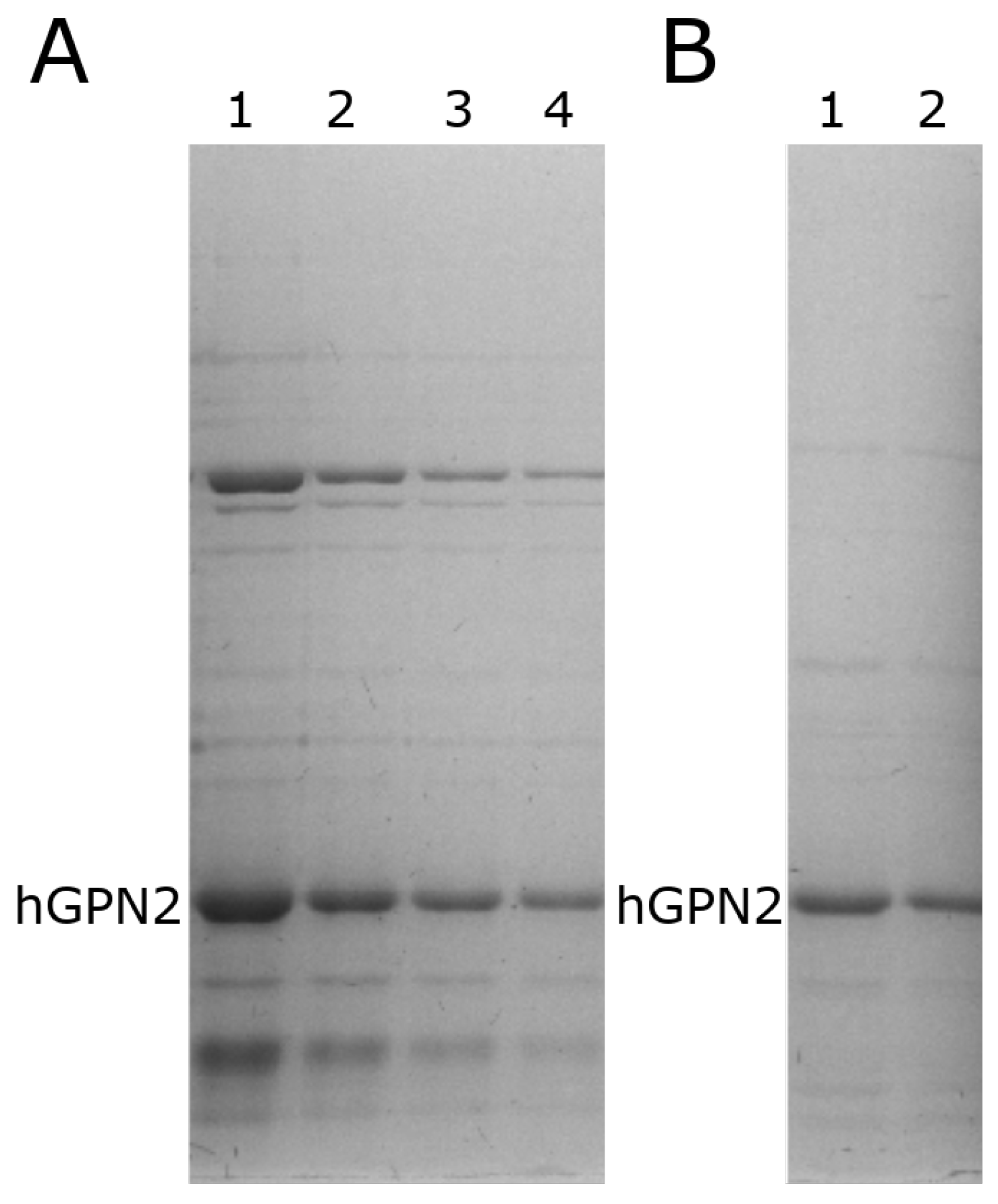
3.2. His-Bind Column Preparation
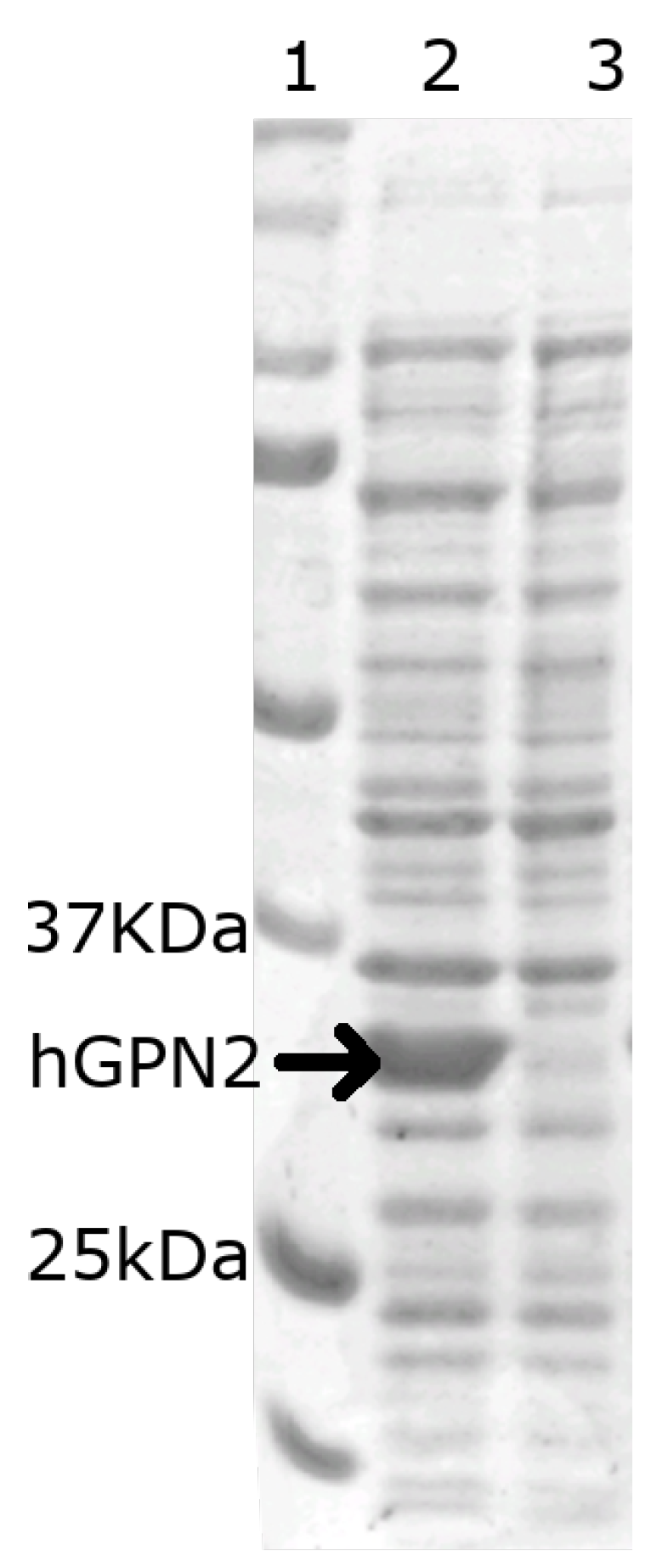
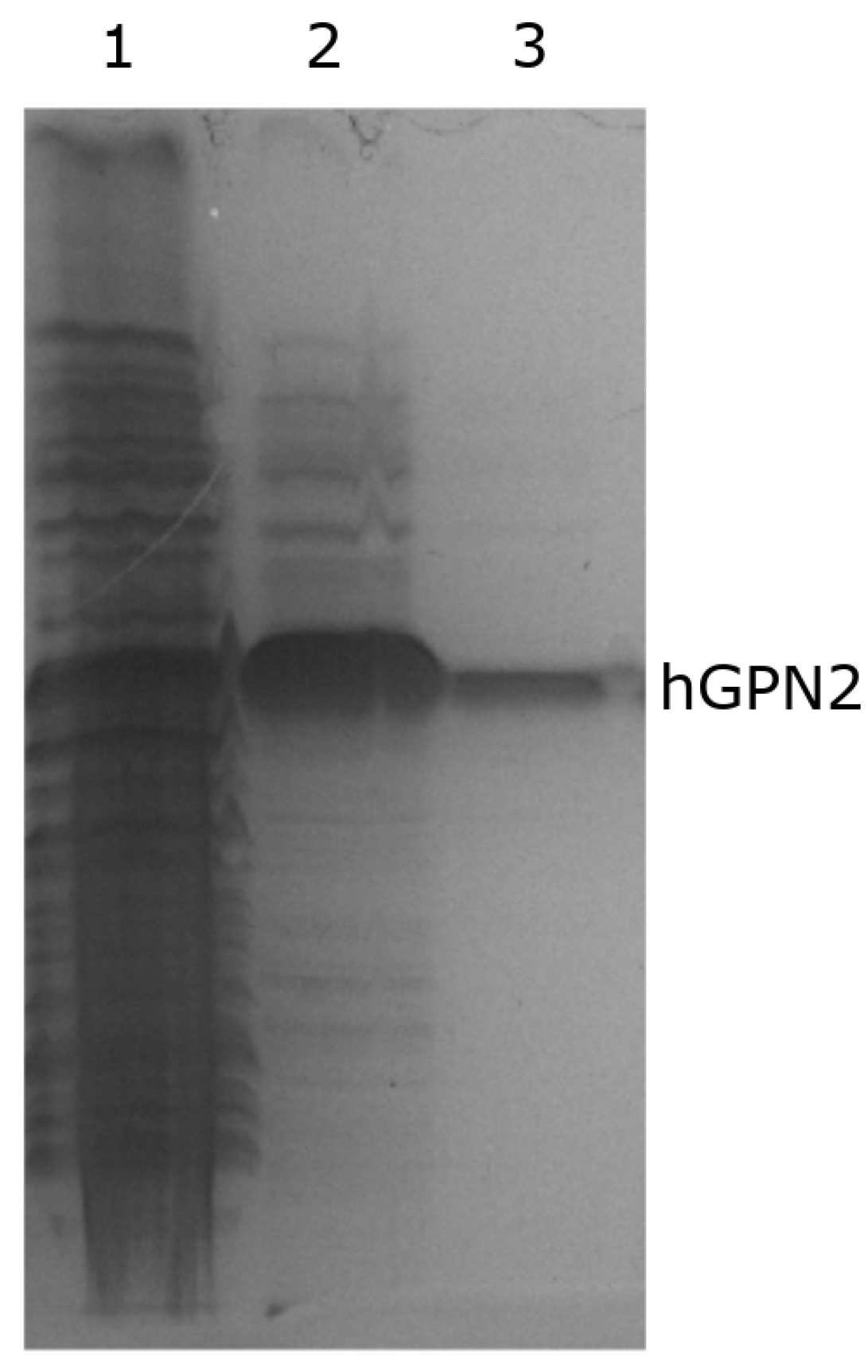
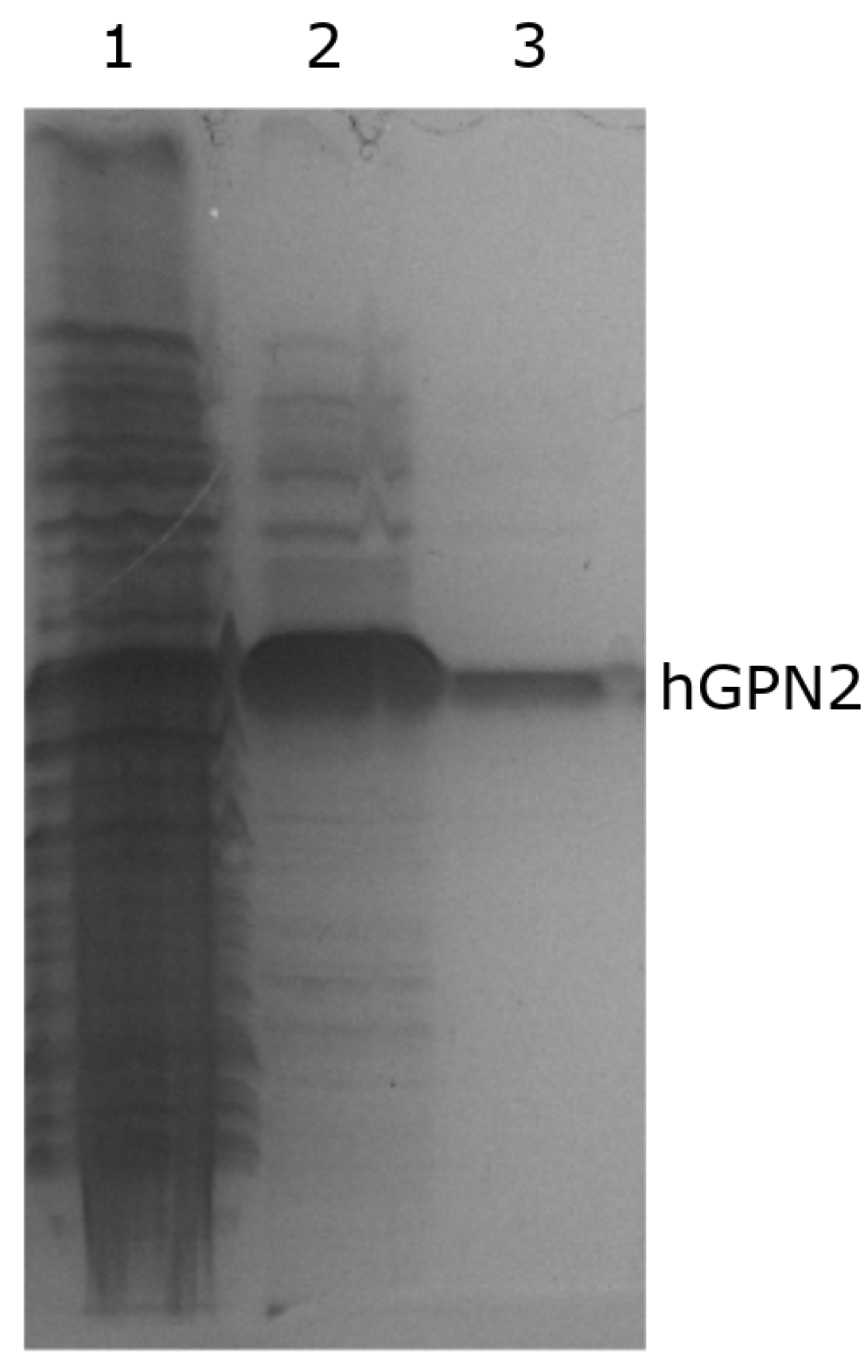
3.3. hGPN2 Protein Expression and Growth at Different Temperatures
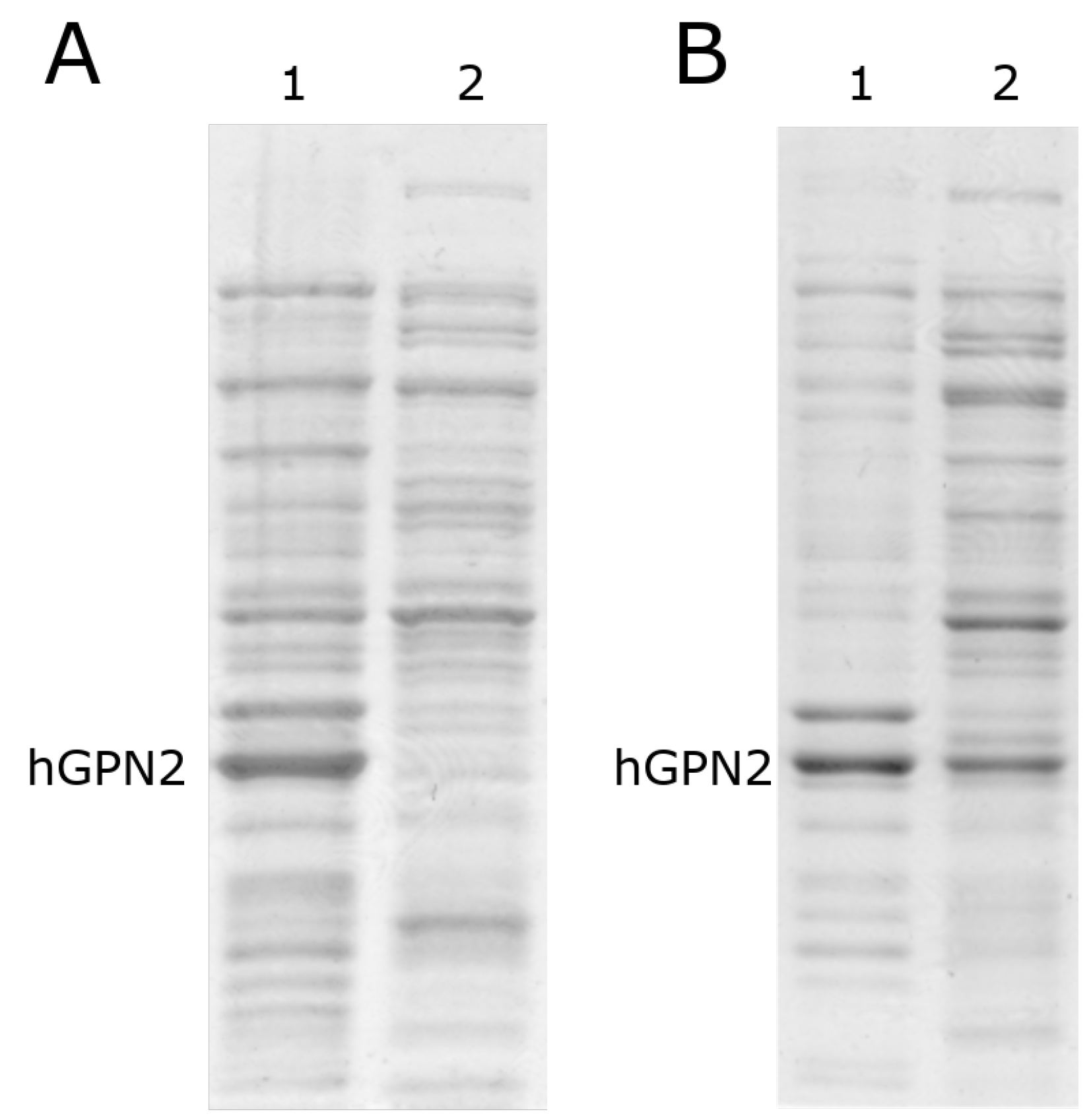
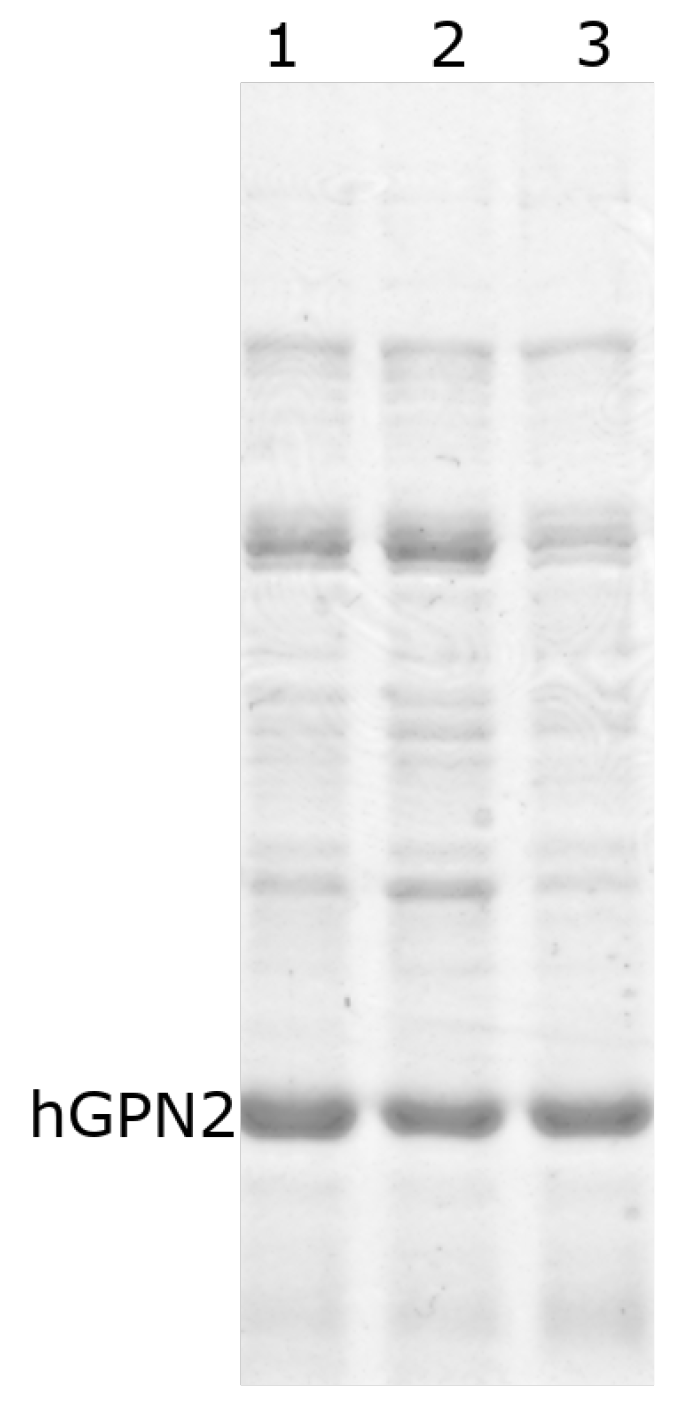
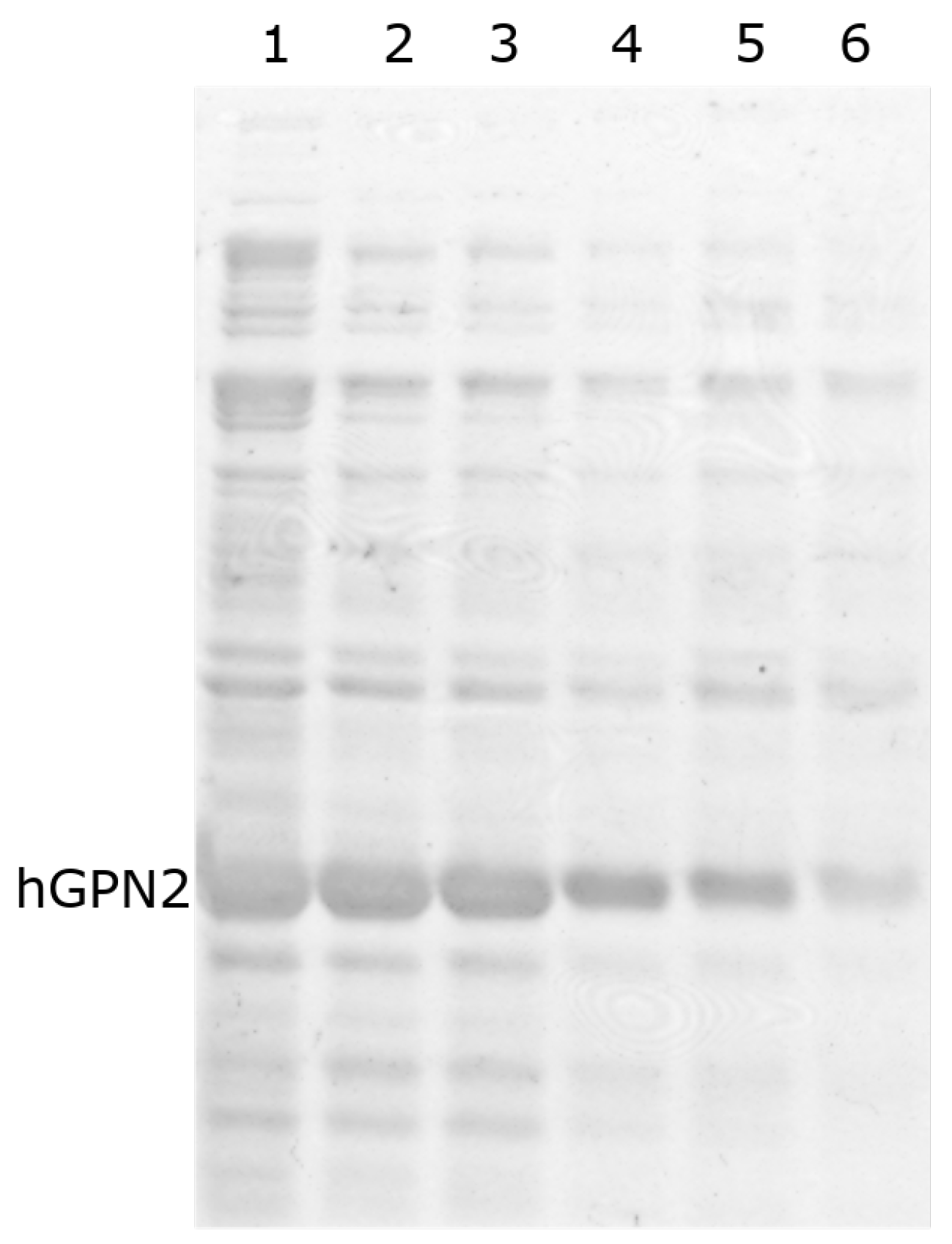
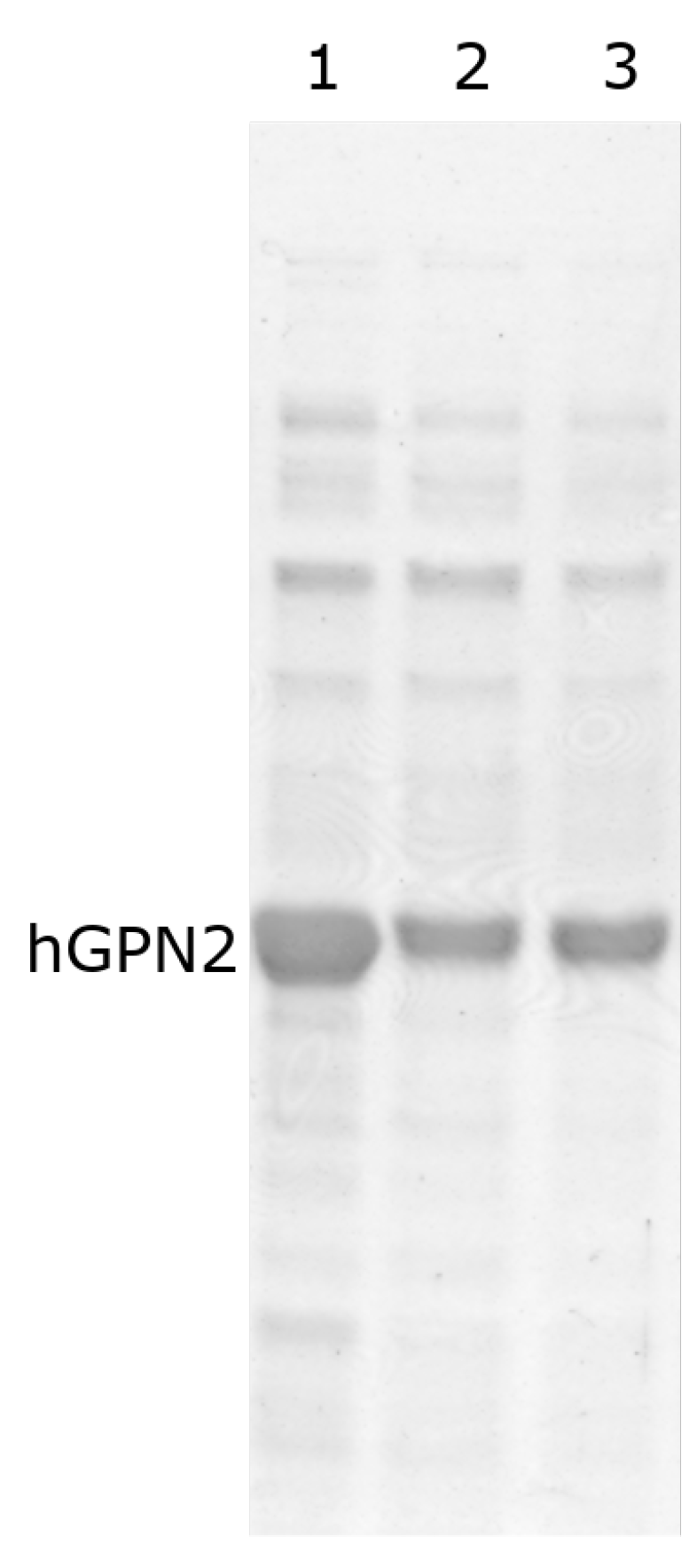
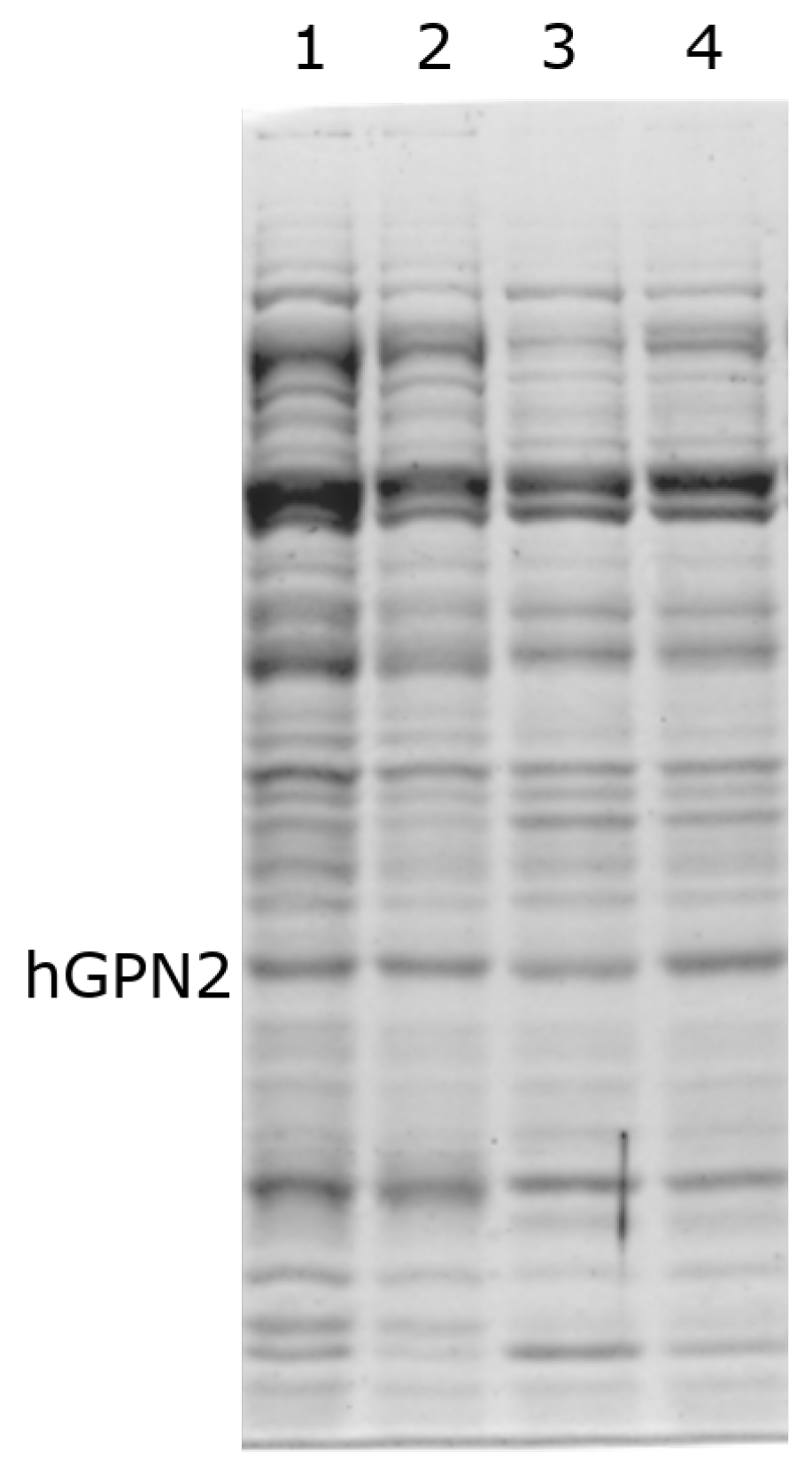
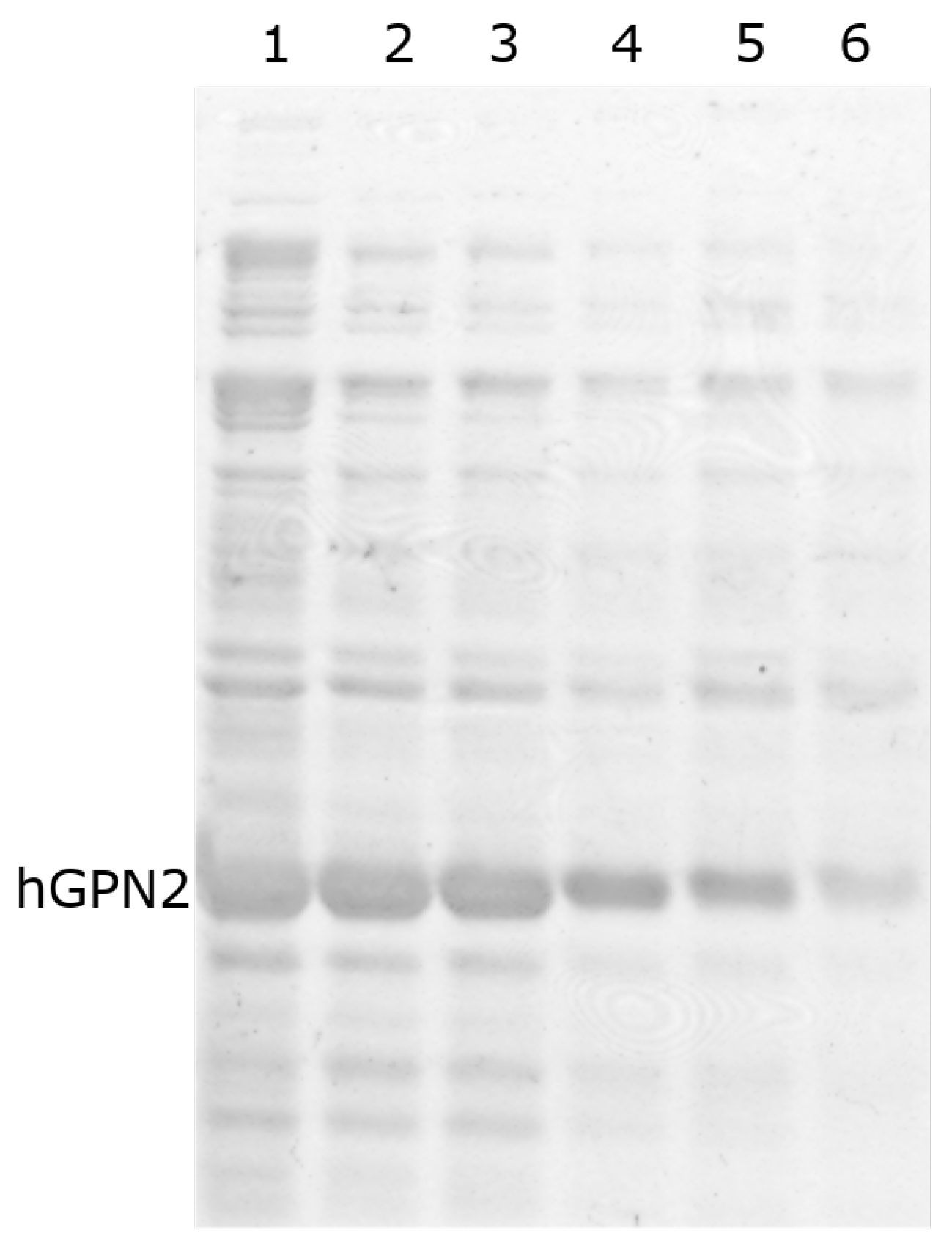
3.4. Protein Purification of Recombinant Protein with NaCl
3.5. Protein Purification of Recombinant Protein with TRIS
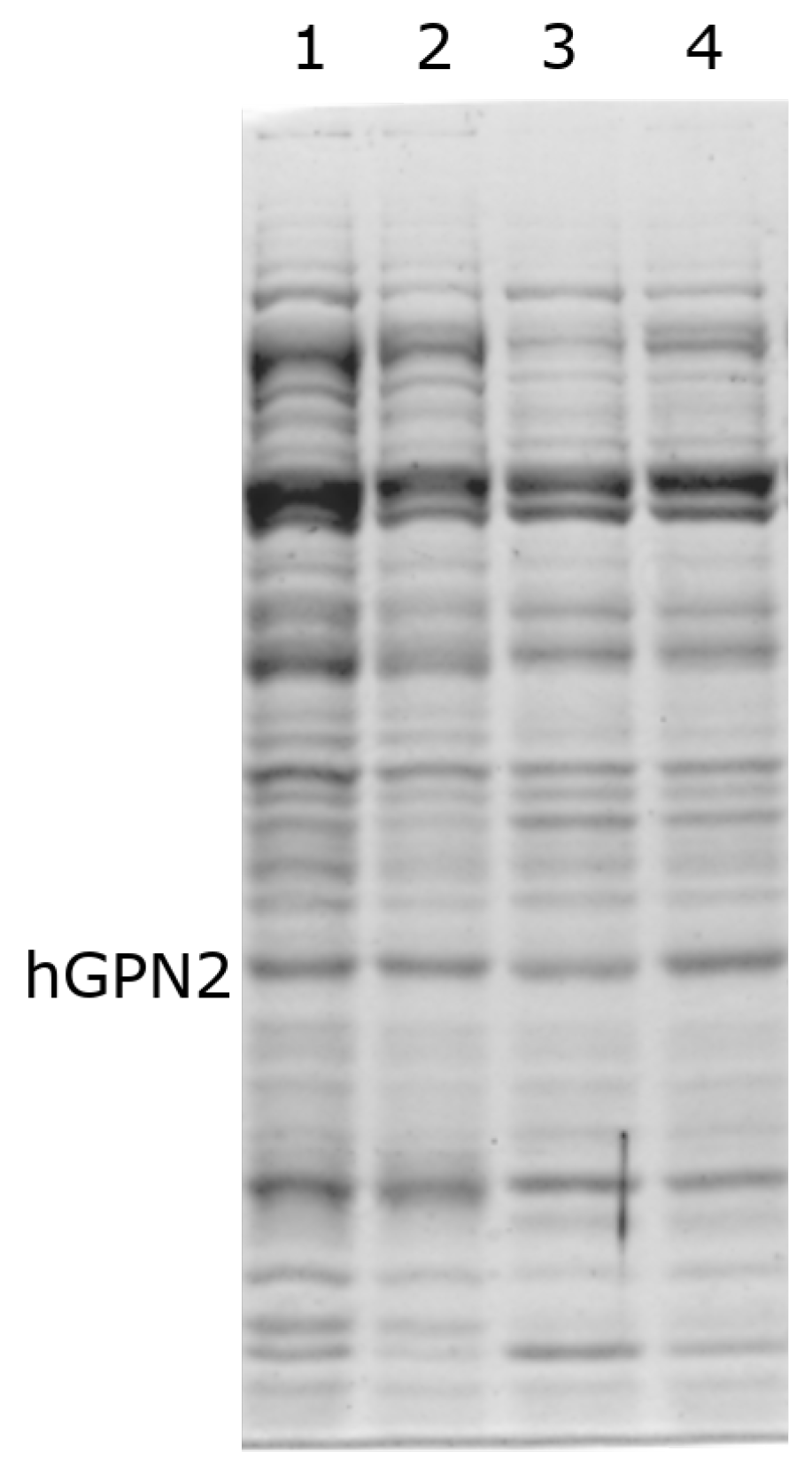
3.6. Protein Purification of Recombinant Protein with EDTA
3.7. Protein Purification of Recombinant Protein with DTT
3.8. Purification with Amino Acids
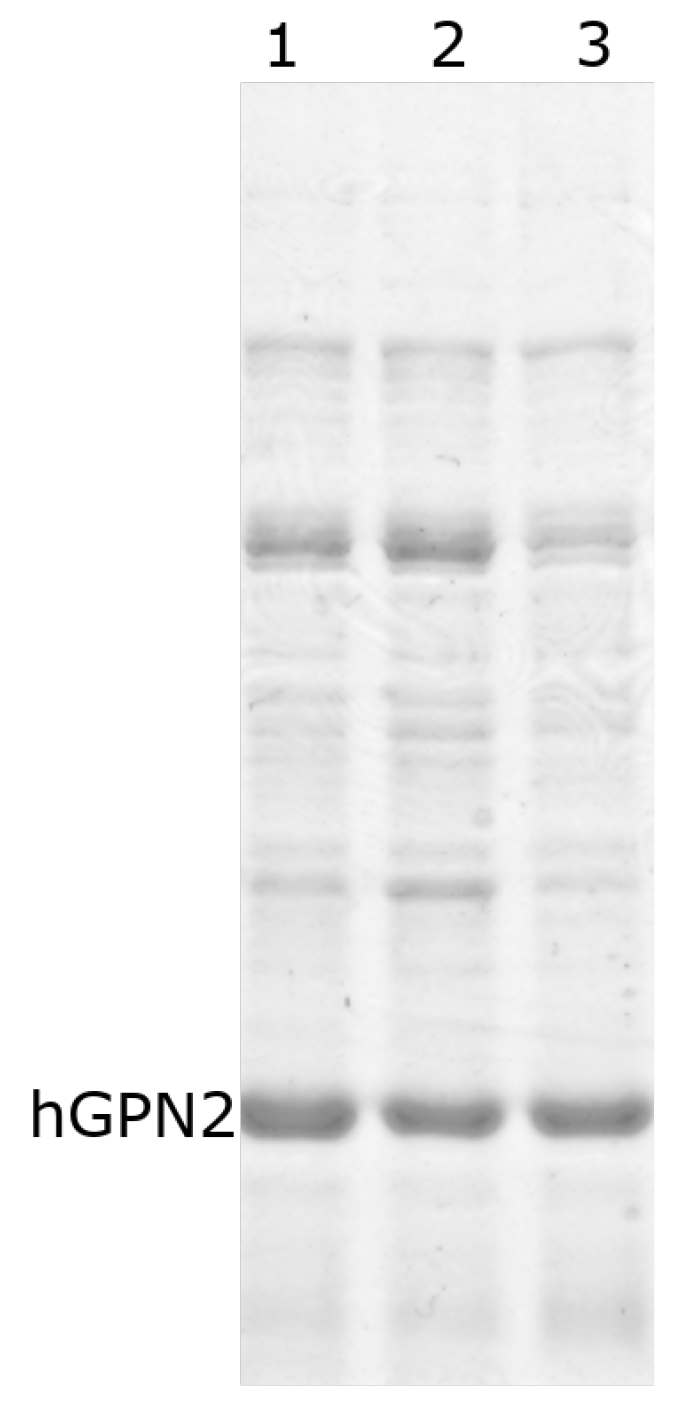
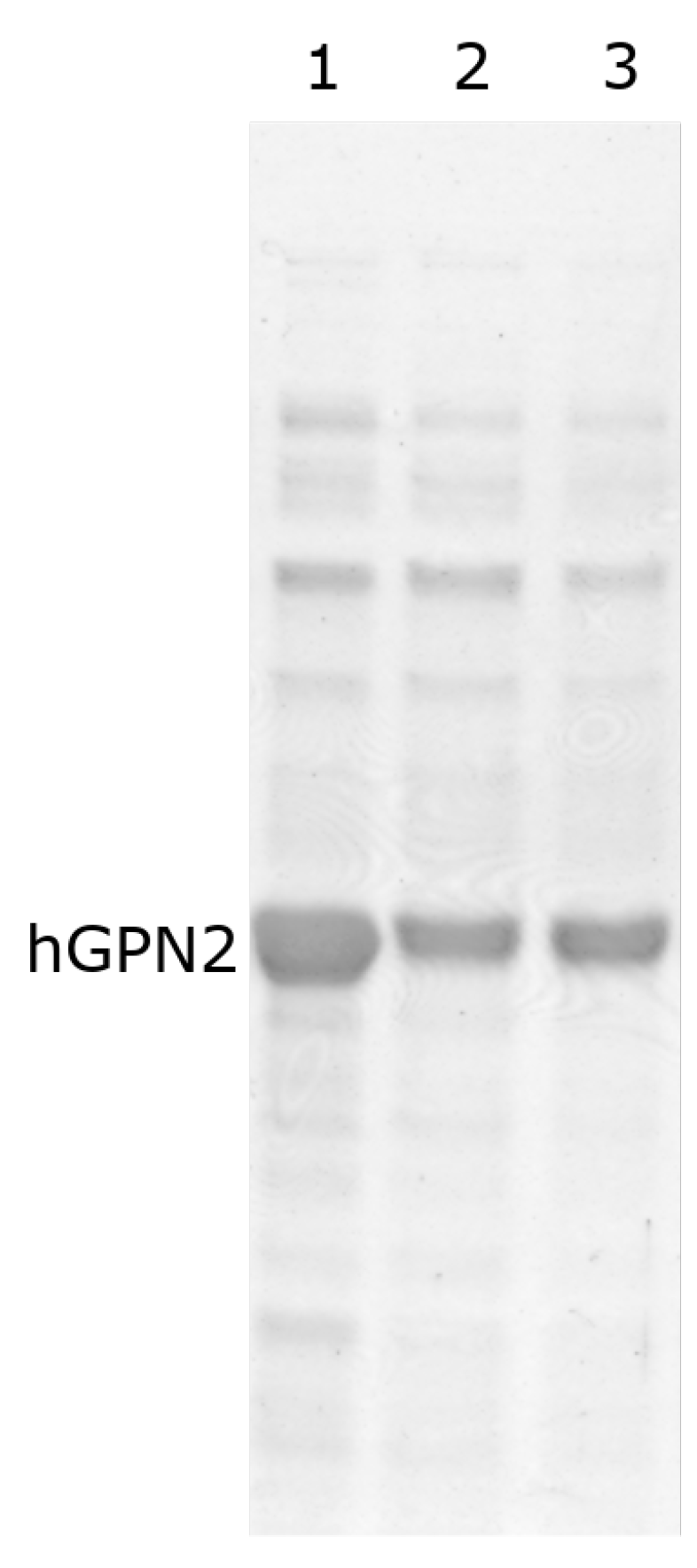
3.9. Purification with Final Buffer
3.10. Quantification of the hGPN2 Protein
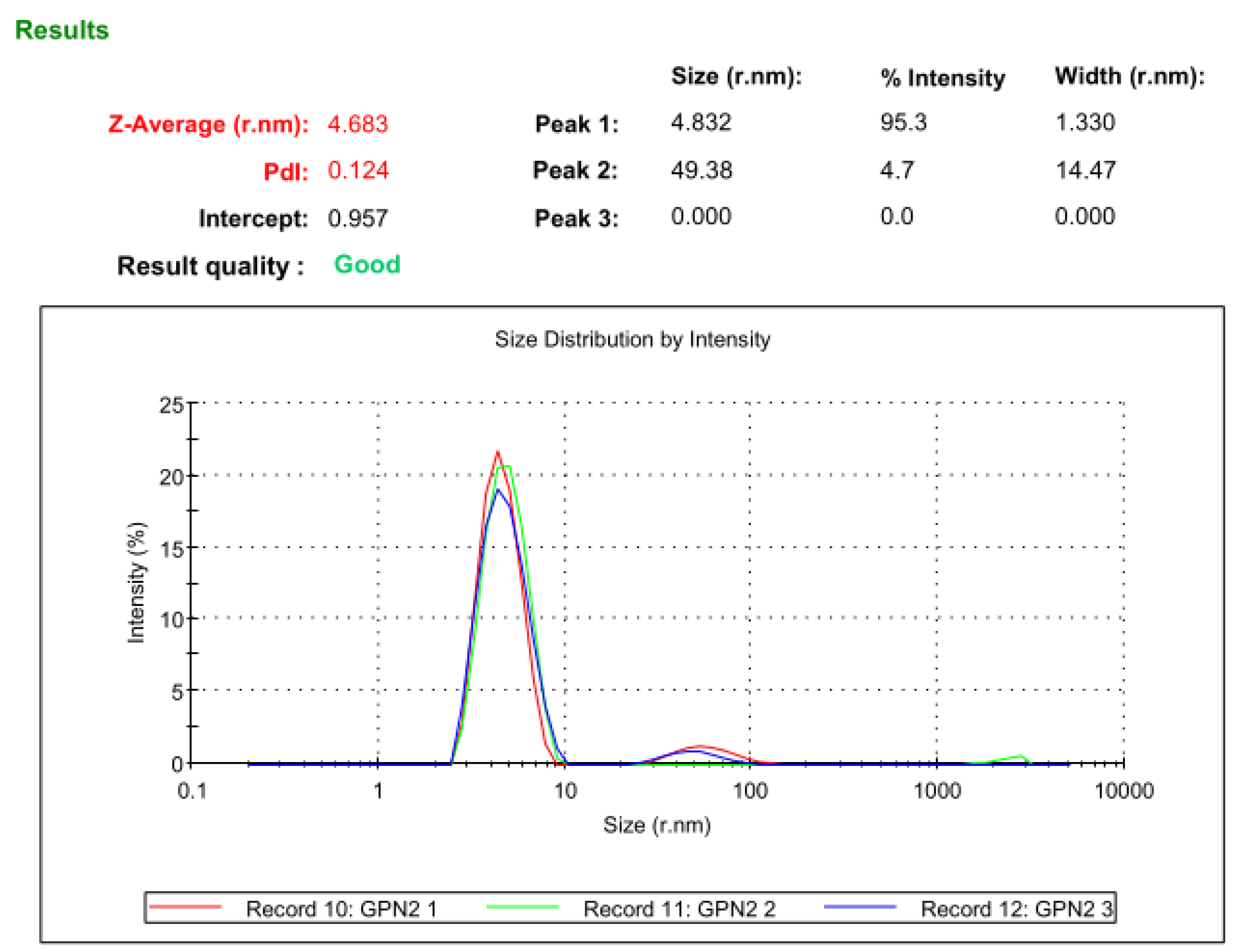
3.11. Dynamic Light Scattering Analysis
4. Discussion
| MAGAAPTTAFGQAVTGPPGSGKTTYCLGMSEFLRALGRRV |
| AVVNLDPANEGLPYECAVDVGELVGLGDVMDALRLGPNGG |
| LLYCMEYLEANLDWLRAKLDPLRGHYFLFDCPGQVELCTH |
| HGALRSIFSQMAQWDLRLTAVHLVDSHYCTDPAKFISVLC |
| TSLATMLHVELPHINLLSKMDLIEHYGKLAFNLDYYTEVL |
| DLSYLLDHLASDPFFRHYRQLNEKLVQLIEDYSLVSFIPL |
| NIQDKESIQRVLQAVDKANGYCFRAQEQRSLEAMMSAAMG |
| ADFHFSSTLGIQEKYLAPSNQSVEQEAMQL |
5. Conclusions
6. Patents
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berezovsky, I.N.; Guarnera, E.; Zheng, Z. Basic units of protein structure, folding, and function. Prog. Biophys. Mol. Biol. 2017, 128, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Feltrup, T.M.; Kukreja, R.V.; Patel, K.B.; Cai, S.; Singh, B.R. Evolutionary Features in the Structure and Function of Bacterial Toxins. Toxins 2019, 11, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GE Healthcare. ÄKTAdesign Purification. Retrieved from Method Handbook. Available online: https://kirschner.med.harvard.edu/files/protocols/GE_AKTApurificationdesign.pdf (accessed on 26 October 2021).
- Ghosh, R.; Gilda, J.E.; Gomes, A.V. The necessity of and strategies for improving confidence in the accuracy of western blots. Exp. Rev. Proteom. 2014, 11, 549–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, M.; Tiwari, S.; Gomes, A.V. Protein purification and analysis: Next generation Western blotting techniques. Exp. Rev. Proteom. 2017, 11, 1037–1053. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Hwang, S.M.; Gomes, A.V. Identification of the immunoproteasome as a novel regulator of skeletal muscle differentiation. Mol. Cell. Biol. 2014, 34, 96–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bass, J.J.; Wilkinson, D.J.; Rankin, D.; Phillips, B.E.; Szewczyk, N.J.; Smith, K.; Atherton, P.J. An overview of technical considerations for Western blotting applications to physiological research. Scand J. Med. Sci. Sport. 2017, 27, 4–25. [Google Scholar] [CrossRef] [Green Version]
- Janes, K.A. An analysis of critical factors for quantitative immunoblotting. Sci. Signal 2015, 8, 371. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; de Gannes, M.K.; Luchetti, G.; Pilsner, J.R. Rapid method for the isolation of mammalian sperm DNA. BioTechniques 2015, 58, 293–300. [Google Scholar] [CrossRef] [Green Version]
- Calbiochem. Detergents. Retrieved from A Guide to the Properties and Uses of Detergents in Biology and Biochemistry. Available online: http://wolfson.huji.ac.il/purification/PDF/detergents/CALBIOCHEM-DetergentsIV.pdf (accessed on 26 October 2021).
- Taylor, S.C.; Posch, A. The design of a quantitative western blot experiment. Biomed Res. Int. 2014, 2014, 361590. [Google Scholar] [CrossRef]
- Jancarik, J.; Pufan, R.; Hong, C.; Kim, S.H.; Kim, R. Optimum solubility (OS) screening: An efficient method to optimize buffer conditions for homogeneity and crystallization of proteins. Acta Crystallogr. D Bio. Crystallogr. 2004, 60, 1670–1673. [Google Scholar] [CrossRef]
- Gutiérrez, R.; Martin del Valle, E.; Galán, M. Immobilized Metal-Ion Affinity Chromatography: Status and Trends. Sep. Purif. Rev. 2007, 36, 71–111. [Google Scholar] [CrossRef]
- Ruprecht, B.; Koch, H.; Medard, G.; Mundt, M.; Kuster, B.; Lemeer, S. Comprehensive and reproducible phosphopeptide enrichment using iron immobilized metal ion affinity chromatography (Fe-IMAC) columns. Mol. Cell. Proteom. 2015, 14, 205–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tishchenko, K.; Hodrova, B.; Simunek, J.; Bleha, M. Nickel and copper complexes of a chelatind methacrylate sorbent in the purification of chitinases and specific immunoglobulin G1 by immobilized metal ion affinity chromatography. J. Chromatogr. A. 2003, 983, 125–132. [Google Scholar] [CrossRef]
- Rosano, G.L.; Ceccarelli, E.A. Recombinant protein expression in E. coli: Advances and challenges. Front. Microbiol. 2014, 5, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.M.; Panda, A.K. Solubilization and refolding of bacterial inclusion body proteins. J. Biosci. Bioeng. 2005, 99, 303–310. [Google Scholar] [CrossRef] [Green Version]
- Tsumoto, K.; Ejima, D.; Kumagai, I.; Arakawa, T. Practical considerations in refolding proteins from inclusion bodies. Protein Expres. Purif. 2003, 28, 1–8. [Google Scholar] [CrossRef]
- Gräslund, S.; Nordlund, P.; Weigelt, J.; Hallberg, B.M.; Bray, J.; Gileadi, O. Protein production and purification. Nat. Methods 2008, 5, 135–146. [Google Scholar]
- Garber-Porekar, V.; Menart, V. Perspectives of immobilized-metal affinity chromatography. J. Biochem. Biophys. 2001, 49, 335–360. [Google Scholar] [CrossRef]
- Cramer, P.; Armache, K.J.; Baumli, S.; Benkert, S.; Brueckner, F.; Buchen, C.; Vannini, A. Structure of eukaryotic RNA polymerases. Annu. Rev. Biophys. 2008, 37, 337–352. [Google Scholar] [CrossRef] [Green Version]
- Niesser, J.; Wagner, F.R.; Kostrewa, D.; Muhlbacher, W.; Cramer, P. Structure of GPN-Loop GTPase Npa3 and implications for RNA polymerase II assembly. Mol. Cell. Biol. 2015, 49, 820–831. [Google Scholar] [CrossRef] [Green Version]
- Baugh, E.H.; Lyskov, S.; Weitzner, B.D.; Gray, J.J. Real-Time PyMOL Visualization for Rosetta and PyRosetta. PLoS ONE 2011, 6, e21931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacket, O. Rapid purification of tubulin from tissue and tissue culture cells using solid-phase ion exchange. Anal. Biochem. 1995, 228, 343–348. [Google Scholar] [CrossRef]
- Arakawa, T.; Timasheff, S.N. The mechanism of action of Na glutamate, lysine HCl, and piperazine-N, N’-bis (2-ethanesulfonic acid) in the stabilization of tubulin and microtubule formation. J. Biol. Chem. 1984, 259, 4979–4986. [Google Scholar] [CrossRef]
- Golovanov, A.P.; Hautbergue, G.M.; Wilson, S.A.; Lian, L.Y. A simple method for improving protein solubility and Long-Term Stability. J. Am. Chem. Soc. 2004, 126, 8933–8939. [Google Scholar] [CrossRef] [PubMed]
- Blobel, J.; Brath, U.; Bernadó, P.; Diehl, C.; Ballester, L.; Sornosa, A.; Pons, M. Protein loop compaction and the origin of the effect of arginine and glutamic acid mixtures on solubility, stability and transient oligomerization of proteins. Eur. Biophys. J. 2011, 40, 1327–1338. [Google Scholar] [CrossRef] [Green Version]
- Shukla, D.; Trout, B. Understanding the synergistic effect of arginine and glutamic acid mixtures on protein solubility. J. Phys. Chem. B. 2011, 115, 11831–11839. [Google Scholar] [CrossRef]
- Abe, R.; Kudou, M.; Tanaka, Y.; Arakawa, T.; Tsumoto, K. Immobilized metal affinity chromatography in the presence of arginine. Biochem. Biophys. Res. Commun. 2009, 381, 306–310. [Google Scholar] [CrossRef]
- Braga, V. Signaling by small GTPases at Cell-Cell Junctions: Protein interactions building control and networks. Cold Spring Harb. Perspect. Biol. 2018, 10, a028746. [Google Scholar] [CrossRef] [Green Version]
- Feiguelman, G.; Fu, Y.; Yalovsky, S. ROP GTPases structure-function and signaling pathways. Plant Physiolpogy 2018, 176, 57–79. [Google Scholar] [CrossRef]
- Song, S.; Cong, W.; Zhou, S.; Shi, Y.; Dai, W.; Zhang, H.; Wang, X.; He, B.; Zhang, Q. Small GTPases: Structure, biologica function, and its interactions with nanoparticles. Asian J. Pharm. Sci. 2019, 14, 30–39. [Google Scholar] [CrossRef]
- Wang, X.; Wang, J.; Chen, F.; Zhong, Z.; Qi, L. Detection of K-ras gene mutations in feces by magnetic nanoprobe in patients with pancreatic cancer: A preliminary study. Exp. Ther. Med. 2018, 15, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Lara-Chacón, B.; Guerrero-Rodríguez, S.L.; Ramírez-Hernández, K.J.; Robledo-Rivera, A.Y.; Velasco, M.A.; Sánchez-Olea, R.; Calera, M.R. Gpn3 Is Essential for Cell Proliferation of Breast Cancer Cells Independent of Their Malignancy Degree. Technol. Cancer Res. Treat. 2019, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gras, S.; Chaumont, V.; Fernandez, B.; Carpentier, P.; Charrier-Savournin, F.; Schmitt, S.; Pineau, C.; Flament, D.; Hecker, A.; Forterre, P.; et al. Structural insights into a new homodimeric self-activated GTPase family. EMBO Rep. 2007, 8, 569–575. [Google Scholar] [CrossRef] [Green Version]
- Minaker, S.; Filiatrault, M.; Ben-Aroya, S.; Hieter, P.; Stirling, P. Biogenesis of RNA Polymerases II and III Requires the Conserved GPN Small GTPases in Saccharomyces cerevisiae. Genetics 2013, 193, 853–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forget, D.; Lacombe, A.; Cloutier, P.; Al-Khoury, R.; Bouchard, A.; Lavall´ee-Adam, M.; Coulombe, B. The protein interaction network of the human transcription machinery reveals a role for the conserved GTPase RPAP4/hGPN1 and microtubule assembly in nuclear import and biogenesis of RNA polymerase II. Mol. Cell Proteom. 2010, 9, 2827–2839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulon, S.; Pradet-Balade, B.; Verheggen, C.; Molle, D.; Boireau, S.; Georgieva, M.; Bertrand, E. HSP90 and its R2TP/Prefoldin-like cochaperone are involved in the cytoplasmic assembly of RNA polymerase II. Mol. Cell. 2010, 39, 912–924. [Google Scholar] [CrossRef] [Green Version]
- Zeng, F.; Hua, Y.; Liu, X.; Liu, S.; Lao, K.; Zhang, Z.; Kong, D. Gpn2 and Rba50 Directly participate in the assembly of the Rpb3 subcomplex in the biogenesis of RNA polymerase II. Mol. Cell. Biol. 2018, 38, e00091-18. [Google Scholar] [CrossRef] [Green Version]
- González-González, R.; Guerra-Moreno, J.A.; Cristóbal-Mondragón, G.R.; Romero, V.; Peña-Gómez, S.G.; Montero-Morán, G.M.; Lara-González, S.; Hernández-Arana, A.; Fernández-Velasco, D.A.; Calera, M.R.; et al. Human Gpn1 purified from bacteria binds guanine nucleotides and hydrolyzes GTP as a protein dimer stabilized by its C-terminal tail. Protein Expr. Purif. 2017, 132, 85–96. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Salt Concentration (Molar) | Solubility µg/mL |
|---|---|
| 0 | 0 |
| 50 | 447 |
| 100 | 464 |
| 500 | 358 |
| 1000 | 308 |
| 2000 | 18 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juárez-Lucero, J.; Guevara-Villa, M.d.R.; Sánchez-Sánchez, A.; Díaz-Hernández, R.; Altamirano-Robles, L. Development of a Methodology to Adapt an Equilibrium Buffer/Wash Applied to the Purification of hGPN2 Protein Expressed in Escherichia coli Using an IMAC Immobilized Metal Affinity Chromatography System. Separations 2022, 9, 164. https://doi.org/10.3390/separations9070164
Juárez-Lucero J, Guevara-Villa MdR, Sánchez-Sánchez A, Díaz-Hernández R, Altamirano-Robles L. Development of a Methodology to Adapt an Equilibrium Buffer/Wash Applied to the Purification of hGPN2 Protein Expressed in Escherichia coli Using an IMAC Immobilized Metal Affinity Chromatography System. Separations. 2022; 9(7):164. https://doi.org/10.3390/separations9070164
Chicago/Turabian StyleJuárez-Lucero, Jorge, María del Rayo Guevara-Villa, Anabel Sánchez-Sánchez, Raquel Díaz-Hernández, and Leopoldo Altamirano-Robles. 2022. "Development of a Methodology to Adapt an Equilibrium Buffer/Wash Applied to the Purification of hGPN2 Protein Expressed in Escherichia coli Using an IMAC Immobilized Metal Affinity Chromatography System" Separations 9, no. 7: 164. https://doi.org/10.3390/separations9070164
APA StyleJuárez-Lucero, J., Guevara-Villa, M. d. R., Sánchez-Sánchez, A., Díaz-Hernández, R., & Altamirano-Robles, L. (2022). Development of a Methodology to Adapt an Equilibrium Buffer/Wash Applied to the Purification of hGPN2 Protein Expressed in Escherichia coli Using an IMAC Immobilized Metal Affinity Chromatography System. Separations, 9(7), 164. https://doi.org/10.3390/separations9070164