Abstract
Vitamin D and its analogues are fat-soluble vitamins that carry out important functions in human and animal organisms. Many studies have pointed out the relationship between the deficiency of these substances and the development of both skeletal- and extra-skeletal diseases. Although vitamin D is fundamentally derived from the bio-transformation of its precursor, 7-dehydrocholesterol, through the action of UV-B radiation in the skin, dietary intake also plays an important role in the regulation of its status in an organism. For this reason, the application of reliable methodologies that enable monitoring the content of vitamin D and its analogues in food and supplements constitutes an aspect of special relevance to establish adequate habits, which avoid the deficiency of these substances in organisms and, consequently, the appearance of related diseases. The use of chromatographic techniques in combination with conventional and novel sample pre-treatments has become a suitable strategy to achieve this aim. This review compiles the most relevant methodologies reported in the last ten years for vitamin D analogues analysis in food matrices. Particular attention has been paid to provide a general overview of the most suitable approaches in terms of reliability, sensitivity and simplicity, used in the field of food analysis.
1. Introduction
Vitamin D3 (cholecalciferol) (D3) and vitamin D2 (ergocalciferol) (D2) are fat-soluble vitamins with a steroidal structure. They play an important role in calcium and phosphate absorption, as well as bone metabolism, and have many other pleiotropic functions in humans and other species [1]. D3 is fundamentally derived from the transformation of its precursor, 7-dehydrocholesterol (7-DHC), present in the skin, through the action of ultraviolet radiation B (UV-B, 290–300 nm). It is converted into previtamin D3 (Pre-D3) which finally is thermally isomerized to D3. However, synthesis in the skin is not the only source of D3. In fact, around 10–20% of this substance is obtained through dietary intake by absorption in the intestine, although it also depends on the area and the season around the world [2]. In contrast, D2 is principally obtained from the consumption of edible plants, fungi or yeast, which also transform previtamin D2 (Pre-D2) into D2 by UV radiation action [1]. Both D3 and D2 are inactive substances that are transported through the blood stream to the liver, where the most stable metabolites, 25-hydroxyvitamin D (25-OHD) forms, are generated. Those are further hydroxylated into the most active metabolite, 1,25-dihydroxyvitamin D (1,25-OHD), in kidney and other organs such as breast, colon, brain, pancreas and prostate. Additionally, other less active forms, as, for example, 24,25-dihydroxyvitamin D (24,25-(OH)2D) or 1-hydroxyvitamin D (1-OHD) and their epimeric forms, are also generated [3]. Apart from hydroxylated metabolites, it is believed that ester forms are produced in different tissues of the organism, although the routes and tissues in which these metabolites are obtained and the roles that they play in the organism are not very well-known yet. Despite the fact that D2 is not produced by mammals, the active forms of D2 and D3 are considered equivalent, although this is an issue of controversy [4].
Currently, there is a global concern about the status of vitamin D and its metabolites in the population since it has been reported that insufficiency and deficiency in these vitamins in humans is linked not only to the development of bone diseases, such as rickets and osteoporosis, but also to many extra-skeletal diseases, including diabetes, hypertension, cardiovascular diseases, multiple sclerosis, psoriasis, Crohn’s disease, neuropsychiatric illnesses or even breast and colon cancer [3,5,6,7]. This is why increasing the consumption of foods which contain these vitamins, with a daily intake of 5–20 μg in adults, is essential to tackle this problem, especially for the population that lives at high latitudes, where the subcutaneous synthesis of vitamin D is absent during winter months. However, it should be emphasized that the recommendations for daily intake vary considerably between different population groups and advisory guidelines [3]. Besides, intake of excessive doses can cause other health problems, such as hypercalcemia, hypercalciuria or hyperphosphatemia [8].
Fatty fish, fish oil, meat or egg yolk are generally considered to be food items with a high content of vitamin D, although it can also be found in fruit, edible plants and seeds [8,9,10]. Nevertheless, the content of these substances in the diet is limited and their stability in food matrices is also questioned under certain conditions such as light, heat or oxidant conditions [1]. For this reason, there are international recommendations for food fortification including D3, D2 and 25-OHD analogues [11]. In this sense, diverse studies have been carried out in order to enrich widely consumed foodstuffs by the addition of these fat-soluble vitamins or by “bio-addition” enhancing the bio-production process in the products [2,11]. In the same direction, the US Food and Drug Administration (FDA) revised the food labelling guidelines in 2016 to make vitamin D content required information in conventional food and supplement packages with the aim of promoting vitamin D awareness among consumers [12]. Additionally, the Codex Alimentarius recommends that food for infants and young children is fortified with D2 and D3 [13], and many countries, including United Kingdom, Canada, Australia, United States, Finland, Denmark and Ireland, have mandatory or voluntary food fortification regulations [14]. This is why a strict evaluation of the presence of vitamin D and its metabolites in food items is an issue of special relevance for the scientific community and also for general society, in order to get an accurate knowledge of the population intake.
In this regard, the determination of vitamin D and its metabolites in food matrices has been traditionally carried out using bio-assay methods. However, these kinds of techniques are time-consuming and, additionally, cross-reactions occur as a result of the similar structures of the different analogues, hampering the analysis of the different individual bioactive compounds [10]. This is why the application of chromatographic techniques has become one of the most adequate alternatives since the 1980s because such applications allow the selective separation and analysis of each compound individually. Indeed, the official methods for determining D3 and D2 in dried skimmed milk and in general commodities, proposed by the International Dairy Federation (IDF) [15] and the European Commission (EC) [16], respectively, are based on the use of liquid chromatography (LC) combined with UV detection. In addition, the Association of Official Analytical Chemists (AOAC) has also proposed other methods for the determination of these two analytes in nutritional formulas using LC hyphenated with tandem mass spectrometry (MS/MS) [17,18], after the proposal of diverse methodologies based on high-performance liquid chromatography (HPLC)-UV analysis [19,20,21]. Apart from that, the Codex Alimentarius proposed in 2018 the endorsement of the AOAC Official Method 2016.05 as type II—the most suitable method—for the analysis and sampling of vitamin D in infant formulas displacing the EN 12821, based on HPLC-UV analysis, as type III, only necessary for verification in case of controversy. However, it is worth mentioning that most of the applications developed so far in this field have been focused on the determination of D2 and D3 forms [8,18,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39] and only a few applications have been performed for the analysis of hydroxylated and precursor forms [4,7,9,10,40,41,42], whereas the determination of ester forms has not been reported up to date in these kinds of matrices (see Table 1).

Table 1.
Structure of vitamin analogues commonly analyzed in food matrices.
This review article is aimed at providing an overview of the most recent advances in the analysis of vitamin D and its analogues in food matrices, paying special attention to the most promising extraction methods and their combination with chromatographic techniques for the suitable determination of this group of fat-soluble vitamins.
2. Recent Applications of Sample Preparation
The complexity of food matrices containing polysaccharides, proteins or lipids, among many other components, as well as the low concentration levels at which vitamin D analogues are present in such samples, make necessary the application of effective sample preparation procedures before the separation and determination of these substances using chromatographic techniques. However, their chemical instability, dealing with certain factors such as light, oxygen or pH, and their easy isomerization at high temperature make this step and the conditions at which it is developed, decisive aspects in the determination process [3].
In general terms, conventional solid-liquid extraction (SLE) and liquid-liquid extraction (LLE) have been the most widely applied techniques for the extraction of this vitamin and its metabolites from food matrices. However, solid-phase extraction (SPE) and preparative chromatography combined with SLE and LLE have also been applied as a common sample pre-treatment in this field. Additionally, a few applications of miniaturized techniques have been performed with the same aim, although to a lesser extent. In most cases, saponification in the presence of antioxidant agents was carried out at the beginning of the process in order to favor the separation of the analytes from the matrix and remove interferences [3], although other hydrolysis procedures or deproteinization have also been applied to accomplish this aim.
The application of saponification is focused on the chemical degradation of complex lipids present in this type of sample that can preclude the determination of vitamins. This procedure is carried out by a hydration reaction, in which ester bonds are broken by the action of an alkali agent, obtaining hydrosoluble free fatty acids and glycerol that can be easily removed in the aqueous phase [43,44]. With this aim, a certain concentration of KOH in ethanol (EtOH) was used in most cases as a strong alkali agent to carry out the reaction, and a high temperature (around 60–80 °C) was applied in order to reduce the process time [7,24,25,26,30,33,34,41] and avoid overnight reactions, which were necessary when room temperature was applied [9,10,42]. However, some authors have reported short reaction times (30–60 min) under room temperature with good results for the evaluation of milk-based formulas, fruit juice and vegetable beverages, after a thorough optimization of time and temperature factors [28,29]. In this sense, the work carried out by Kwak et al. [29] should be highlighted. In this case, saponification using 150 mg of KOH, 150 mg of NH3 and 2.4 g of NaCl was performed during 5 min at room temperature, after dilution and subsequently extraction using 10 mL of isopropanol (IPA) for the extraction of D3 from milk-based formulas. The extract was then evaporated and injected in a HPLC-MS/MS system after reconstitution. The methodology was compared with the AOAC official method [17], in which high-temperature (75 °C) saponification during 30 min was necessary for the determination of D2 and D3 in infant formula and adult nutritionals.
Additionally, due to the instability of these vitamins during the procedure [3], antioxidant agents are usually added to the reaction media to assure their correct determination. In this sense, ascorbic acid [9,24,34,35], pyrogallol [10,26], sodium ascorbate [7,36,42] and butylated hydroxytoluene (BHT) [23,28] have been the most commonly antioxidant agents applied with this aim.
2.1. Conventional Extraction Techniques
As previously indicated, despite the fact that conventional SLE and LLE are complex and time consuming, they have been the most widely applied extraction techniques for the evaluation of food matrices (see Table 2). Nevertheless, other alternative procedures have also been sporadically applied. In this sense, the use of the dilute-and-shoot strategy should be remarked. As an example, Byrdwell et al. [27] applied this pre-treatment in combination with HPLC-mass spectrometry (MS) and HPLC-UV determination for the evaluation of D3 in dietary supplements. The procedure avoids the application of complex steps, limiting the method to sample weighing, addition of an internal standard (IS) and dilution with the adequate solvent [43]. In this case, oil contained in the supplement gel caps was diluted to 100 mL using methanol (MeOH) and dichloromethane (DCM) in the composition MeOH/DCM (6/4, v/v), and was directly injected in the chromatographic system. This study demonstrated the suitability of the methodology for the evaluation of not only D3, but also triacylglycerols in supplement gel caps and oil samples in combination with both LC-MS and LC-UV systems, showing that UV detection provided lower sample-to-sample relative standard deviation than atmospheric pressure chemical ionization (APCI)-MS detection.
Table 2.
Recent applications of conventional extraction techniques for the analysis of vitamin D-related compounds in food matrices.
However, for other samples with higher complexity, the application of multiple step methodologies has been found to be unavoidable so far. SLE and LLE have been applied to a wide variety of food matrices including milk and milk-based formulas, rice, cereals, bread, different kinds of fish, mushrooms, fruit juice, vegetables beverages, oil or eggs, among others [4,7,22,23,24,25,26,28,29,40,41]. In those cases, a great range of solvents such as hexane, ethyl acetate (EtOAc), MeOH, isooctane, chloroform, petroleum ether, diethyl ether, EtOH, pentane, cyclohexane, isooctane or IPA, as well as mixtures of them have been used, applying volumes in the range 1–100 mL and carrying out several repetitions in most cases, in order to get a quantitative extraction of the target analytes [4,7,28]. In almost all cases reported so far, the application of not only saponification [7,24,25,26,28,29,41], but also other hydrolysis treatments for the evaluation of cereals and nutritional formulas [23] or deproteinization for different milk samples [4,40] has been carried out, prior to extraction, to favor the cleaning of the samples and to remove interferences. With respect to the efficiency of these three strategies, despite saponification being the one most extensively applied, some studies have demonstrated the higher effectiveness of deproteinization in releasing vitamins from milk matrices [4,40]. In this sense, it is worth mentioning the work carried out by Gomes et al. [40], who developed a careful comparative study of both strategies for the effective extraction of eight vitamin D analogues (i.e., D3, D2, 25-OHD2, 25-OHD2, 24,25-(OH)2D2, 24,25-(OH)2D3, 1,25-(OH)2D2 and 1,25-(OH)2D3) from human, cow, mare, goat and sheep milk samples prior to their determination by HPLC-MS/MS. With this aim, 4 mL of sample was deproteinized using 8 mL of acetonitrile (ACN), vortexed for 2 min, incubated for 15 min at room temperature and subsequently centrifuged. For saponification, 4 mL of 8 M ethanolic KOH solution and 2.8 mL of 1 M ascorbic acid were added and, after vortexing for 2 min, the sample was hatched at room temperature for 12 h, before centrifugation. In both cases, a subsequent extraction of the obtained supernatant was carried out using 12 mL of hexane/DCM (1/4, v/v) twice, prior to determination. As can be seen in Figure 1, larger efficiency in terms of concentration was obtained for all target analytes using the deproteinization treatment, which demonstrated the higher suitability of this procedure, at least for the analysis of milk-based matrices.
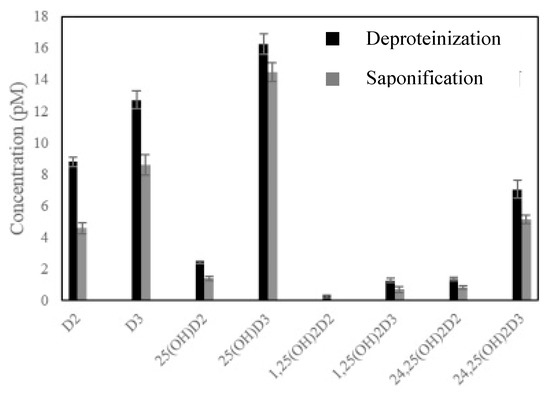
Figure 1.
Comparison of deproteinization and saponification effectiveness for the release of vitamin D analogues from milk samples. Reproduced from [40], with permission from Elsevier, 2020.
Although in general terms, saponification, hydrolysis or deproteinization in combination with only one extraction technique have been found sufficient for the correct release of the analytes from the complex food samples, in some cases, the performance of additional treatments or extraction procedures has been necessary [24,25]. As an example, Mohanty et al. [25] developed a methodology for the extraction of D3 from finfish and shellfish based on a modified Folch method using 450 mL of the mixture CHCl3/MeOH (2/1, v/v) followed by alkaline saponification under reflux for 30 min and LLE of fish oil using 50 mL of petroleum ether prior to determination by HPLC-UV. The thorough extraction developed and clean extract obtained from the application of the described method allowed the authors to determine D3 and other fat-soluble vitamins in a great variety of fishes with a high fat content and, therefore, the evaluation of the contribution of these food commodities to the vitamin D daily intake in the population with an occurrence in the range 7.72–23.28 μg/kg in seventeen especies evaluated, including marine and freshwater fishes and moluscs.
However, despite the necessity of carrying out strong digestive treatments to get an efficient release of vitamin D and their analogues from food samples, there exists a remarkable trend to avoid them in order to simplify the procedures and keep the whole information about the matrix vitamin forms distribution in this kind of analysis [23]. In this regard, the fact is that in only a few cases, LLE and SLE have been directly applied for the evaluation of these analytes in food matrices [22,24]. Especially remarkable is the work carried out by David et al. [22], in which the authors applied a previously developed methodology [45], for the determination of vitamin D, as well as other fat-soluble vitamins in rice samples from different parts of Nigeria and Thailand by direct extraction of the matrix using 4 mL of hexane under N2 steam for 5 min. After centrifugation, 1 mL of supernatant was evaporated, reconstituted in butanol and subsequently injected in the HPLC-UV system for the separation and determination of the target compounds. Although the Thailand species presented lower occurrence (13.8 mg/kg) than the Nigerian products (23.2–28.6 mg/kg), all species turned out to be poor sources of vitamin D, confirming the previous studies carried out in this area, in which the content of this vitamin has been associated with food commodities with a higher amount of fat such as fish, eggs and whole milk, among others [8,9,10].
Despite the fact that conventional extractions are the most commonly applied for the analysis of these analytes in food samples, the high amount of solvents used, as well as the complexity of the procedure, constitute clear disadvantages of their application. Apart from that, additional pre-treatments to carry out an effective extraction and clean-up of the sample, including hydrolysis or deproteinization, are still fundamental steps to achieve the successful analysis of compounds. For this reason, the search of new protocols that allow the miniaturization of such methods is an issue of special interest for the scientific community.
2.2. Solid-Phase Extraction
As shown in Table 3, SPE has been applied in this area for the extraction and/or sample clean-up of cereals and flour products [30], edible plants, seaweeds, fruit and fruit seeds [9,31], dietary supplements [31], porcine fat, meat and liver [42], cod liver supplements [8], beef, egg, chicken and fish [10]. The development of a previous digestion step, generally saponification, was the procedure most commonly carried out [9,10,30,42], although in some cases, the complexity of the evaluated samples also requires the performance of a solvent-based extraction between both stages [10,42], whereas in only one report, SPE has been applied directly after dilution of the sample using hexane [8]. In this last case, Bartoluccia et al. carried out a reliable methodology for the confirmation of the content of D3 in cod liver oil-based supplements after the detection of D3 intoxication in several consumers of this kind of pill. Authors pointed out the necessity of using a normal phase SPE with an apolar solvent eluent as the sample pre-treatment due to the lipidic nature of the sample since other alternatives, such as the application of C18 sorbent in combination with polar eluent, have not demonstrated to yield an adequate release of fat-soluble vitamins from this type of sample. With this aim, 0.2% of the content of each capsule was diluted up to 1 mL with hexane and the sample was loaded onto a NH2-propyl-SPE cartridge previously conditioned with the same solvent. After washing with hexane, the target compound was eluted with 1 mL of EtOAc. Finally, the extract was dried and the residue reconstituted with IPA prior to the separation and determination by HPLC-triple quadrupole (QqQ)-MS/MS. After validation of the methodology, the analysis of the samples showed a concentration of D3 three orders of magnitude higher than the one indicated in the product (1.5 μg/capsule) for some of the batches evaluated. These results emphasize the necessity of the development of selective and reliable methodologies that allow the correct evaluation of all kinds of commercialized dietary supplements which guarantees food safety and protects consumer health.
Table 3.
Recent applications of solid-phase extraction for the analysis of vitamin D-related compounds in food matrices.
Regarding the type of sorbent applied in the different methods developed so far, it should by highlighted that, as indicated in the previous example, the use of silica as normal phase sorbent [10,42] has been commonly applied for the evaluation of samples with a high fat content such as porcine fat and liver [42], as well as meat, fish and eggs [10]. Indeed, the AOAC proposed in 1996 the AOAC 995.05 method [20] for the determination of cholecalciferol in infant formulas and enteral products using a silica-SPE after saponification and extraction using hexane, which pointed out the improvement with respect to previously developed methods in which LLE or SLE had been applied [19]. On the contrary, non-polar cartridges based on C18 have been applied for the extraction of vitamin D analogues in cereal and flour products [30] or fruit-based commodities [31], and diatomeous earth for the evaluation of plants, seaweeds and fruit seeds [9]. The application of this sorbent in combination with 60 mL of petroleum ether as the elution solvent was used for the determination of D2, D3, 25-OHD2 and 25-OHD3 in several types of seaweeds and different parts of Australian plants like seeds, leaves or stems after saponification of the samples overnight at room temperature. The whole methodology was validated and applied for the determination of the target compounds with recovery values in the range 94–101% and a limit of detection (LOD) of 0.5 μg/kg. However, the authors remarked that the determination of 25-OHD3 in roasted and milled seeds of wattleseeds could not be successfully performed due to the high amount of matrix interferences obtained in the final extract, thus demonstrating the inefficiency of the selected sorbent to carry out the release of 25-OHD3 from this commodity.
As indicated at the beginning of this section, in some cases, the characteristics of the evaluated samples have made necessary the combination of SPE with other extraction and clean-up procedures to get an efficient extraction. Especially remarkable is the work carried out by Bilodeau et al. [10], in which the evaluation of D3 and its metabolite 25-OHD3 was carried out in pork, beef, egg, chicken, turkey, dolphinfish, salmon and tilapia, using a HPLC-diode array detector (DAD) for the quantification of D3 and HPLC-(QqQ)-MS/MS for 25-OHD3. Sample pre-treatment consisted of an initial alkaline saponification of the sample at room temperature using pyrogallol as the antioxidant agent, followed by LLE four times with 150 and 75 mL of petroleum ether/diethyl ether (2/8, v/v), washing with 5% KOH (one time) and water (twice), subsequent SPE, using 1 g of silica and 0.4–3% of IPA/DCM as the elution solvent, and semi-preparative HPLC using a normal phase column. Recovery values in the range 73–88% and an LOD of the method of 0.4 µg/kg demonstrated the suitability of the developed methodology. The reduction of matrix interferences and noise was considerable after de-application of the semi-preparative HPLC clean-up, especially for the determination of D3 by UV, as can be seen in Figure 2, in which the HPLC-UV chromatograms of the separation between D3 and the IS, used for its determination (D2), in a solvent standard and in a real sample submitted to the developed methodology are shown.
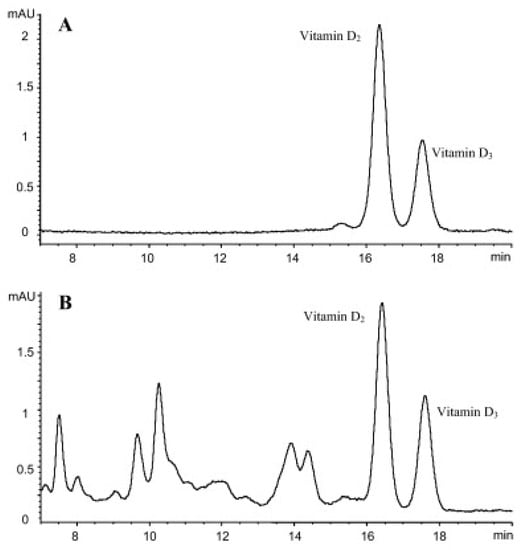
Figure 2.
HPLC-UV chromatograms of a solvent standard (A) and in a real sample extracted using the developed methodology (B). Reproduced from [10], with permission from Elsevier, 2020.
As it has been demonstrated throughout the discussed literature, the application of SPE involves an improvement in the extraction and clean-up efficiency for the analysis of vitamin D analogues. In fact, the AOAC included in 1996 this procedure as alternative to SLE and LLE, commonly used with this aim. However, despite the mentioned advantages, it is true that the application of hydrolysis treatments and even previous solvent-based extractions are still necessary in many of the developed approaches, as well as the use of a high amount of organic solvents as the eluents. It is remarkable the low variety of sorbents tested in this area, which is an aspect that could considerably enhance the relevance of this technique, extending their application to a wider variety of matrices and improving the extraction and clean-up efficiency achieved.
2.3. Preparative Chromatographic Techniques
Preparative liquid chromatography has also been proposed as a suitable technique to get an adequate cleaning of the sample and to avoid the presence of interferences that could preclude the determination of vitamin D and its analogues in food matrices (see Table 4). In fact, the AOAC Official Method 2002.05 [21] describes the use of this technique for the evaluation of D3 and D2 in fatty food, adding a UV detector at the outlet position that allows establishing the most accurate collection window and removing the highest amount of matrix interferences.
Table 4.
Recent applications of preparative chromatography for the analysis of vitamin D-related compounds in food matrices.
Following the guidelines of the official method, all applications of the technique carried out so far have been performed using silica as the stationary phase for the evaluation of cacao-based food [32], yogurt, dietary supplements and margarine [33], fish and seafood [34], and orange juice [35] and applying an initial step of saponification followed by LLE, before LC separation. As an exception, Byrdwell et al. [35] carried out the determination of D3 in fortified orange juice applying an LLE directly to the sample using ethyl ether and petroleum ether with ascorbic acid as the antioxidant agent and subsequent clean-up by preparative LC with a (25.0 cm × 4.6 mm, 5 mm) silica column using a mixture of IPA/methyl tert-butyl ether (MTBE)/cyclohexane/heptane (0.05/0.2/4.875/4.875, v/v/v/v) as the elution solvent. In this case, the particular characteristics of the sample, which presents a very low fat content in comparison with the rest of the analyzed matrices shown in Table 4, allowed the omission of a saponification step. The authors remarked that the application of LLE and preparative LC together with the use of D2 as IS and the analysis by HPLC-UV and HPLC-MS constitutes the best combination of elements taken from the AOAC Official Methods 992.26 and 200.05 for the determination of D3 in the selected matrix.
On the contrary, both Kühna et al. [32], who carried out the evaluation of cacao-based products, Nestola and Thellmann [33], who analyzed yogurt, dietary supplements and margarine, as well as Byrdwell et al. [34], who determined the content of vitamin D analogues in fish and seafood, described the necessity of applying a saponification step followed by LLE using hexane or a mixture of petroleum ether/diethyl ether to accomplish an adequate release of the analytes from the samples analyzed in each case. Especially remarkable is the evaluation of the influence of the saponification step carried out by Byrdwell et al., in which a comparison of of the amount of alkaline solution (30 and 60 mL of 50% KOH solution) effect on the reproducibility of the method was done, even though subsequently cleaning with the semi-preparative LC was applied. Results of the National Institute of Standards and Technology (NIST) standard reference material (SRM) of infant/adult nutritional formula and pulled salmon samples demonstrated that a stronger saponification increases the reproducibility between samples due to the increase in the matrix digestion [34].
This procedure constitutes an adequate complement step to both solvent and sorbent-based extractions in order to improve the clean-up step, particularly in fat content samples for which normal stationary phases have shown good results. In fact, it could be considered as an alternative to the application of SPE since the use of off-line set-ups usually simplifies the procedures and reduces the consumption of solvents.
2.4. Miniaturized Techniques
The current trends in analytical chemistry are focused on the development of environmentally sustainable methodologies that enable a decrease in the consumption of harmful chemicals and diminish the impact of chemical activity on the environment. In this sense, miniaturized techniques that allow a reduction of cost, time and toxic and hazardous organic solvents consumption, as well as which require lower amounts of samples, are being increasingly used [46]. However, despite the undeniable advantages obtained by the application of miniaturized techniques and their wide application in other areas, their use for the determination of vitamin D and vitamin D analogues is strikingly low. As it is shown in Table 5, there are only a few reports in the area for the determination of these substances in food samples using magnetic-micro-dispersive solid-phase extraction (m-μ-dSPE) [37,39] and dispersive liquid–liquid microextraction (DLLME) [36,38] for the evaluation of milk, infant formulas, green vegetables, milk/fruit-based beverages and cereals. It is worth mentioning that only in one case, saponification was necessary prior to the application of the extraction process [36], whereas deproteinization was applied in the case of cereals and fruit/milk-based beverages [36,39].
Table 5.
Recent applications of miniaturized extraction techniques for the analysis of vitamin D-related compounds in food matrices.
A fast and simple methodology was developed by Viñas et al. [38] based on the application of DLLME for the evaluation of infant formulas (50–250 mg) and green vegetables (i.e., spinach and cos, iceberg and lamb’s lettuce) (0.2–2.0 g). With this aim, an initial SLE using 3 mL of ACN as the extraction solvent was developed. Then, the extract obtained was used as the dispersant and mixed with 150 μL of carbon tetrachloride (extracting solvent). The mixture was rapidly injected in 6 mL of water and manually shaken for some seconds to favor the cloudy spread formation enabling the simultaneous preconcentration of the target analytes (D2, D3) and clean-up of the sample. After centrifugation, the small carbon tetrachloride drop (around 50 μL) containing the target compounds was taken from the bottom of the tube, dried and reconstituted in the same volume of ACN prior to injection in a HPLC-ion trap (IT)-MS. The good recovery values obtained in the range 88–103% and the LODs between 3.1 and 4.0 μg/L demonstrated the efficiency of the technique for the evaluation of the complex samples selected. In addition, the method demonstrated the simplification of the procedure in comparison with methods described until now and a reduction in the extraction time and organic solvents consumption.
The use of nanomaterials as sorbents in sample pre-treatments of food matrices is a strategy widely applied due to the particular characteristics of such materials that allows the miniaturization of the extraction and clean-up procedures, minimizing the sample and solvent amount requirements, simplifying the methodologies and providing very good efficiencies with a minimal environmental impact. Particular interest has reached the magnetic nanomaterials. Those not only provide a high surface area, increasing the active point, in which the sorbent can interact with the substances of interest and present high tuneable capacity, favoring the versatility and specificity of the extractions as occurs with the rest of nanomaterials, but their use also simplifies even more the procedures as a result of their magnetic properties, which allows the easy separation of the sorbent and the sample using an external magnet [47]. These advantages were precisely the ones provided by the sorbents developed by Jiao et al. [37] and Hu et al. [39] for the determination of vitamin D using m-μ-dSPE. Particularly simple was the methodology described by Jiao et al. [37] using polypyrrole (PPy)-coated Fe3O4 nanoparticles (PPy@Fe3O4) to carry out the extraction and preconcentration of D3 and D2 from milk samples prior to their determination by HPLC-UV. With this aim, 1 mL of sample was initially diluted with 9 mL of buffer phosphate solution. Then, the m-μ-dSPE was directly performed without any additional step by using 30 mg of PPy@Fe3O4. After stirring for 10 min, the sorbent was magnetically separated from the sample solution with the assistance of a magnet and then washed with 2 mL of water. Finally, the analytes were desorbed with 1 mL of ACN by ultrasonication for 5 min. The sorbent was again separated using an external magnet and the eluate dried, reconstituted and submitted to HPLC-UV analysis. The good recovery values obtained in the range 72–90% and the low LODs (0.02–0.05 µg/L) demonstrated the effectiveness of the π–π interactions between the sorbent commodities and the target analytes, as well as the good performance of the developed methodology with a pre-treatment time of only 15 min and avoiding the digestion steps.
The advantages of these techniques in terms of simplicity and chemicals consumption are undeniable and the great variety of sorbent and solvent materials that could be applied increases exponentially the range of application of these methods. However, their application to solid samples still involves the use of an initial pre-treatment using a high volume of solvents which could be considered as a disadvantage of their application. In this sense, only a few approaches have been applied so far. The application of other miniaturized procedures such as matrix solid phase dispersion could be an interesting alternative to solve this problem.
3. Recent Applications of Chromatographic Techniques
In the last thirty years, the use of chromatographic techniques for the separation and determination of vitamin D and its analogues has become a common trend in this area due to the advantages that these techniques offer with respect to bio-assay methods, particularly the possibility of carrying out the analysis of individual analytes, providing an accurate evaluation of the levels of each analogue in the organism [10].
Regarding the most recent applications, as shown in Table 1, Table 2, Table 3 and Table 4, the combination of LC with UV or MS detection have been the most common strategies [3], whereas gas chromatography (GC) applications have been hardly reported due to the easy isomerization of vitamin D analogues at high temperature [3], which would preclude the individual determination of this group of analytes. The application of other chromatographic techniques in the last ten years has been reduced to the use of supercritical fluid chromatography (SFC) hyphenated with MS. However, it has been applied in few cases owing to the current stage of development of this technique in terms of commercial instrumentation, etc. [48].
3.1. Liquid Chromatography Applications
Regarding the use of LC, different modalities have been applied, including HPLC [7,8,9,10,22,24,25,27,28,29,31,32,34,35,36,37,38,40,42], two dimensional-HPLC (2D-HPLC) [41], ultra-high performance liquid chromatography (UHPLC) [26,30] and capillary liquid chromatography (CLC) [38], although, among them, HPLC has been the most extended as a consequence of its higher availability in most laboratories. Aqueous–organic mixtures with ACN and MeOH, fundamentally, as well as IPA [7,8,9,10,22,24,26,29,30,31,32,34,38,40,41,42] or pure organic solvents and mixtures of them have also been used [25,27,28,35,36,37,39] as mobile phases without [8,9,10,22,25,27,28,30,34,35,36,37,38,39] or with additives including formic acid (FA) [7,26,40,41,42], ammonium formate [24,29] or both of them [32], acetic acid [10], methyl amine [42] and trifluoro acetic acid (TFA) [31], especially for LC-MS hyphenations. With respect to the columns usually selected for this type of application, despite the fact that the use of normal phases has been previously reported for the analysis of these compounds, the latest publications describe, fundamentally, the separation using reversed-phase columns with very good results. However, it is worth mentioning that in many cases, a clean-up step using polar sorbents in SPE or normal phases in preparative chromatography has been necessary in order to remove lipid interferences, especially for high-fat content sample analysis [1].
The combination of LC with UV detection or DAD was also very common in the previous decades. However, the high matrix effect that precludes the correct determination of these vitamins in food samples, as well as the limitations in terms of sensitivity and selectivity have brought about the necessity to move on to the use of more suitable systems such as LC-MS or LC-MS/MS which have become the gold tools for the simultaneous determination of vitamin D analogues [3]. In this sense, UV has been used for the determination of only one or two analytes simultaneously (D2 and D3) in most of cases [22,25,28,30,31,36,37,39,41] using, fundamentally, 265 nm as the most suitable wavelength, and their combination with MS has been necessary in some cases to reach the levels at which some analytes are present in the samples analyzed [10]. As a remarkable exception, Wittig et al. [7] carried out the determination of ten vitamin D analogues (i.e., D2, D4, previtamin-D2 (Pre-D2), previtamin-D4 (Pre-D4), tachysterol2, tachysterol4, lumisterol2, lumisterol4, ergosterol, 22,23-dihydroergostero), as well as D3 used as the surrogate, in oyster mushrooms after UV-B treatment by HPLC-UV. As can be seen in Figure 3, clean chromatograms of the evaluated samples treated with hot alkaline saponification followed by LLE and subsequent separation by HPLC were obtained with an acceptable separation of all analytes. The good recovery (98%) and the good sensitivity of the developed methodology (LODs: 0.02–0.06 mg/kg) enabled its successful application to the monitoring of these compounds in mushroom samples submitted to UV-B exposition in order to enhance their content in vitamin D-active metabolites with therapeutic functions. However, although analytes quantification was performed using UV detection, it should be highlighted that MS determination was also carried out to confirm their identification.
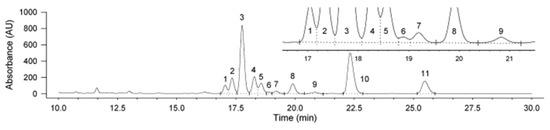
Figure 3.
HPLC-UV chromatogram of an oyster mushroom sample submitted to saponification and subsequent LLE after UV-B treatment. Analytes identification: (1) pre-D2; (2) tachysterol2; (3) D2; (4) D3 (IS); (5) lumisterol2; (6) pre-D4; (7) tachysterol4; (8) vitamin D4; (9) lumisterol4; (10) ergosterol (provitamin D2); (11) 22,23-dihydroergosterol (provitamin D4). Adapted from [7], with permission from Elsevier, 2020.
The development of methodologies based on the coupling of LC with MS systems has been carried out in combination with all kinds of sample pre-treatments previously described as a consequence of the advantages that this strategy provides in terms of sensitivity and selectivity. Both MS detection [27,34,35,38] and MS/MS [7,8,9,10,24,26,27,29,41,42] have been applied using QqQ in most cases [7,8,9,10,24,26,29,32,40,41,42]. Nevertheless, other analyzers such as tandem sector quadrupole (TSQ) [34,35], quadrupole linear-ion trap (QTrap) [32] and IT [35,38] have also been used recently.
Regarding the ionization sources, both electrospray ionization (ESI) [9,24,26,29,32,40,41,42] and APCI [7,8,10,27,34,35,38] in positive mode have been applied, using a great variety of additives as mentioned at the beginning of this section. However, FA has been preferred in the majority of cases for both modes.
Other usual practice in the evaluation of this type of compound by MS is the application of a derivatization step in order to increase the sensitivity, especially for those analogues which are present at a lower concentration and when the amount of sample available also is small. In recent years, 4-phenyl-1,2,4-triazoline-3,5-dione (PTAD) has been the chosen reagent in all cases in the area of food analysis since it presents a high effectiveness, increasing sensitivity between 7 and 100 times depending on the compound [40].
Finally, it should also be highlighted the use of deuterated [8,9,24,26,32,40,41] and non-deuterated [7,10,27,34] surrogates and ISs for the validation of a great number of the evaluated matrices using not only MS, but also UV or DAD detectors in order to guarantee the reliability of the obtained results.
3.2. Gas Chromatography
GC has been hardly applied as a separation technique for the analysis of vitamins in food matrices due to the low stability of these compounds at high termperature [33]. This was pointed out by Nestola and Thellmann in their work reported in 2015, in which the analysis of D2 and D3 was carried out using an online hyphenation between a prepartive HPLC and GC-time of flight (ToF)-MS, after saponification, in yogurt, dietary supplements and margarine [33]. Authors highlighted the simplicity of the online procedure and compared it to HPLC-MS/MS, indicating that similar results were obtained with less manual clean-up performance. However, the isomerization of the analytes as a consequence of the high temperature applied in the GC separations could not be avoided, generating double peaks which hindered the correct identification and quantification of vitamin D. This constraint made necessary to carry out the determination of the target analytes using their isomerized pyro-structures in combination with deuterated ISs by a “cross-contribution” procedure. Despite the advantages that the online procedure provided, the limitations of using GC for the analysis of thes kinds of substances are evident, which explains the higher use of other techniques such as LC.
3.3. Supercritical Fluid Chromatography
SFC is a separation technique which integrates the characteristics of LC and GC. Developed in the 1960s, it did not gain popularity until the 1980s due to infrastructure limitations. Since 2009, new instruments that allow the hyphenation of the technique with different detectors, as well as other improvements such as a reduction of dead volume, higher control of pressure or the use of stationary phases with smaller particle size have been developed by different companies, which has increased the number of applications and publications using this technique. SFC is based on the separation of the analytes using an eluent based on liquid and/or supercritical carbon dioxide mixed with a co-solvent or solvent mixture. Carbon dioxide is used as the eluent almost always owing to its particular advantages in terms of low toxicity, availability and accessible critical point, among others. The separation of analytes not only depends on the identity of the stationary phase ligand, the nature of the eluent constituents and how these are mixed in the gradient elution, but also on the density of the mobile phase which has an important influence on analytes solubility. The customized modification of such parameters allows the comprehensive separation of a wide variety of analytes with different polarities and also the selective separation of compounds with similar structures, as occur with enantiomers. The large range of polarities that cover this technique involving all compounds with Log P between −2.11 and −10.12 makes fat-soluble vitamins (Log P 6–9) ideal candidates for their separation using SFC [3,48,49,50,51].
Despite the recent development of equipment that guarantee the robustness and efficiency of this technique [3], in the area of food analysis and, particularly, for the evaluation of vitamin D analogues, the application of SFC is limited to a few studies developed in recent years for the evaluation of milk [4] and infant and adults nutritionals, as well as cereal samples [23]. In both cases, the hyphenation of ultra-high-performance supercritical fluid chromatography (UHPSFC) with MS has been applied using a particle size lower than 2 μm in order to improve the efficiency and rapidness of the technique, as well as decrease the consumption of solvents and reduce void volumes [51,52]. QqQ as analyzer and APCI ionization source in positive mode have been chosen on both publications. Besides, as a consequence of the particular characteristics of the mobile phase used in this technique, the coupling with MS involves the application of a make-up solvent added to favor the transfer of the compounds to the MS and guarantee the determination of the analytes. This solvent presents an important influence on the sensitivity of the methodology. In these cases, ammonium formate in MeOH was selected to achieve an adequate ionization of the analytes including D2 and D3 [4,23], as well as hydroxylated metabolites [4]. As described for LC separations, derivatization using PTAD was also selected for the determination of these analogues with low levels of concentration in the samples of interest [4,23].
Column selection is considered a big challenge in SFC since the specific interactions established between the analytes and stationary phases are important to achieve the successful separation of vitamin D isomers. In this sense, fluoro-phenyl columns with charged surface hybrid (CSH) particles [4] and 1-aminoanthracene (1-AA) columns [23] have been the ones selected so far. An interesting study about column selection was the one carried out by Oberson et al. [4]. In order to achieve the separation of PTAD-derivatized D2, D3, 25-OHD2, 3-epi-25-OHD2, 25-OHD3 and 3- epi-25-OHD3, the authors checked nine different columns with a great variety of stationary phases including fluoro-phenyl-CSH, ethylene bridged hybrid (BEH)-C18, BEH-2-ethylpyridine (2-EP), 1-AA-C18, high-strength silica (HSS)-C18, Diol-C18, 2,6-cholesteryl-C18, C30 and amylose tris(3,5-dimethylphenylcarbamate) (AD-3) columns. Most of them provided a poor separation of the analytes and low sensitivity, except in the cases of the fluoro-phenyl-CSH and AD-3 columns. However, the fluoro-phenyl-CSH column, using MeOH and water as the cosolvents and ammonium formate as the additive at 45 °C, was finally selected for the validation of the whole methodology. As can be seen in Figure 4, the baseline separation of isomers was obtained with better peak shape than the results obtained using the AD-3 phase, although double peaks were observed for the 3-epi-25-OHD forms.
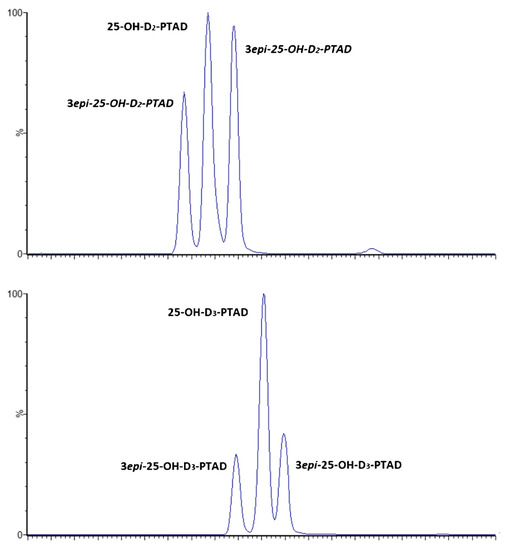
Figure 4.
UHPSFC-MS/MS chromatograms of the separation of PTAD-derivatized 25-OHD2, 3-epi-25OHD2, 25-OHD3 and 3-epi-25OHD3 using fluoro-phenyl column-CSH (1.7 μm, 3 × 100 mm). Gradient of MeOH/water (98/2, v/v) containing 10 mM ammonium formate as organic modifier on CO2. Column temperature: 45 °C. Atmospheric back pressure regulator: 128 bar. Make-up solvent was 10 Mm ammonium formate in MeOH, flow rate 0.4 mL/min. Reproduced from [4], with permission from Springer, 2020.
Finally, it should also be highlighted that mixtures of water and MeOH using ammonium formate as the additive were selected in the articles recently published in the area of vitamin D analysis in food matrices. The higher sensitivity provided by MeOH as compared with ACN has been demonstrated previously by some studies developed for the determination of these compounds by SFC-MS [4].
Although the application of this technique is still scarce, the great comprenhensivity of SFC and the excellent sensitivities obtained when it is hyphenated with MS systems makes this approach one of the most promising techniques for the analysis of vitamin D and its metabolites in food matrices.
4. Conclusions and Future Remarks
The important role that vitamin D and its metabolites play in several vital functions for human beings and the influence that their status in the organism has on the development of a great number of diseases have made vitamin D deficiency a global health concern. The low subcutaneous bio-production of vitamin D, especially for populations at high latitudes during winter months, makes the diet the main source of these compounds. For this reason, the determination of vitamin D and vitamin D analogues contents in commonly consumed food items, as well as the identification of food products that are especially rich in those substances, is of uttermost importance. Additionally, each time, the most common use of supplements or fortified foods that guarantee the adequate levels of these compounds in the population also urges a reliable assessment of these commercialized products for ensuring food safety.
All the abovementioned makes evident the necessity of developing reliable methodologies that enable an accurate determination of the content of vitamin D in foodstuffs. In this sense, chromatographic systems have become the most suitable techniques to carry out the individual evaluation of vitamin D and its analogues in food matrices displacing other methods initially applied such as bio-assays. Particularly, the hyphenation of different modalities of LC with conventional UV detectors and, most recently, with MS systems have been the most recurrent strategies due to the advantages that this technique presents with respect to GC in which analytes’ isomerization occurs at high temperature. In addition, the commercialization of robust SFC systems and the advantages that this technique provides in terms of comprehensiveness, selectivity and peak capacity has allowed the performance of suitable methodologies based on the use of supercritical fluids, which have become a promising alternative for the evaluation of vitamins.
Nevertheless, the great complexity of food matrices makes sample analysis difficult without pre-treatment. In this sense, the application of digestive treatments, such as saponification or deproteinization, is frequent in most applications. Those are usually combined with conventional SLE or LLE. However, other widely known techniques, such as SPE or preparative LC, have also been applied in recent years with considerable improvements in the efficiency of clean-up performance. However, the current trends in analytical chemistry based on the developed methodologies supported by the principles of green chemistry have come up with the application of miniaturized techniques also in this field, reducing the costs and time, as well as the sample and solvent amount requirements of conventional methodologies. So far, the techniques applied, DLLME and µ-dSPE, have provided promising results, although their use for the analysis of solid samples still requires previous pre-treatments that involve the use of high volumes of organic solvents. In this sense, it could be interesting the use of other approaches more suitable for this kind of sample such as matrix solid-phase extraction that would allow the direct application in the matrix and the accomplishment of green chemistry principles.
Ultimately, it is evident that there is much to be done in the field. There are limitations that hamper the individual and simultaneous determination of the different kinds of analogues including D3, D2, precursor forms, hydroxylated metabolites and ester analogues, for which there is scarce information up to date. Besides, the effective release of the analytes from the complex food samples without matrix interferences is still a big challenge for the scientific community. However, the current developments focus on the combination of simple, fast and effective sample pre-treatment with the state-of-the-art chromatographic equipment and the most sensitive and versatile detection systems constituting the most promising alternatives to achieve the goals posed in this area.
Author Contributions
All Authors contributed equally to the present work. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Carl Trygger Foundation (project: CTS 18:395).
Acknowledgments
B.S.-R. would like to thank Carl Trygger Foundation for funding her postdoctoral stipend. This work was supported by the Carl Trygger Foundation (project: CTS 18:395).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kasalová, E.; Aufartová, J.; Kujovská Krčmová, L.; Solichová, D.; Solich, P. Recent trends in the analysis of vitamin D and its metabolites in milk—A review. Food Chem. 2015, 171, 177–190. [Google Scholar] [CrossRef]
- Kühn, J.; Schutkowski, A.; Hirche, F.; Baur, A.C.; Mielenz, N.; Stangl, G.I. Non-linear increase of vitamin D content in eggs from chicks treated with increasing exposure times of ultraviolet light. J. Steroid Biochem. Mol. Biol. 2015, 148, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Yang, Y.; Wu, L.; Li, Y.; Sun, C. Recent advances in sample preparation and analysis methods for vitamin D and its analogues in different matrices. TrAC-Trend. Anal. Chem. 2019, 110, 204–220. [Google Scholar] [CrossRef]
- Oberson, J.M.; Bénet, S.; Redeuil, K.; Campos-Giménez, E. Quantitative analysis of vitamin D and its main metabolites in human milk by supercritical fluid chromatography coupled to tandem mass spectrometry. Anal. Bioanal. Chem. 2020, 412, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Patrick, R.P.; Ames, B.N. Vitamin D hormone regulates serotonin synthesis. Part 1: Relevance for autism. FASEB J. 2014, 24, 2398–2413. [Google Scholar] [CrossRef] [PubMed]
- Patrick, R.P.; Ames, B.N. Vitamin D and the omega-3 fatty acids control serotonin synthesis and action, part 2: Relevance for ADHD, bipolar disorder, schizophrenia, and impulsive behavior. FASEB J. 2015, 29, 2207–2222. [Google Scholar] [CrossRef] [PubMed]
- Wittig, M.; Krings, U.; Berger, R.G. Single-run analysis of vitamin D photoproducts in oyster mushroom (Pleurotus ostreatus) after UV-B treatment. J. Food Comp. Anal. 2013, 31, 266–274. [Google Scholar] [CrossRef]
- Bartoluccia, G.; Giocaliere, E.; Boscaro, F.; Vannacci, A.; Gallo, E.; Pieraccini, G.; Moneti, G. Vitamin D3 quantification in a cod liver oil-based supplement. J. Pharm. Biomed. Anal. 2011, 55, 64–70. [Google Scholar] [CrossRef]
- Hughes, L.J.; Black, L.J.; Sherriff, J.L.; Dunlop, E.; Strobel, N.; Lucas, R.M.; Bornman, J.F. Vitamin D content of Australian native food plants and australian-grown edible seaweed. Nutrients 2018, 10, 876. [Google Scholar] [CrossRef]
- Bilodeau, L.; Dufresne, G.; Deeks, J.; Clément, G.; Bertrand, J.; Turcotte, S.; Robichaud, A.; Beraldin, F.; Fouquet, A. Determination of vitamin D3 and 25-hydroxyvitamin D3 in foodstuffs by HPLC UV-DAD and LC–MS/MS. J. Food Comp. Anal. 2011, 24, 441–448. [Google Scholar] [CrossRef]
- Lips, P.; Cashman, K.D.; Lamberg-Allardt, C.; Bischoff-Ferrari, H.A.; Obermayer-Pietsch, B.; Bianchi, M.L.; Stepan, J.; El-Hajj Fuleihan, G.; Bouillon, R. Current vitamin D status in European and Middle East countries and strategies to prevent vitamin D deficiency: A position statement of the European calcified tissue society. Eur. J. Endocrinol. 2019, 180, P23–P54. [Google Scholar] [CrossRef] [PubMed]
- FDA. Food labelling: Revision of the nutritional and supplement facts labels. Fed. Regist. 2016, 81, 33741. [Google Scholar]
- Codex Alimentaroux. Advisory Lists of Nutrient Compounds for Use in Foods for Special Dietary Uses Intended for Infants and Young Children, CAC/GL 10/1979, last amended 2008.
- Kiely, M.; Cashman, K.D. The ODIN project: Development of food-based approaches for prevention of vitamin D deficiency throughout life. Nutr. Bull. 2015, 40, 235–246. [Google Scholar] [CrossRef]
- International Standard ISO 14892|IDF 177. Dried Skimmed Milk –Determination of Vitamin D Content Using High-Performance Liquid Chromatography; ISO: Geneva, Switzerland, 2002. [Google Scholar]
- European Standard EN. 12821 Foodstuffs—Determination of Vitamin D by High Performance Liquid Chromatography—Measurement of Cholecalciferol (D3) or Ergocalciferol (D2). 2009. [Google Scholar]
- AOAC Official Method 2011.13. Vitamin D2 and D3 in Infant formula and Adult Nutritionals. LC–MS/MS, First Action 2011; AOAC International: Gaithersburg, MD, USA, 2012. [Google Scholar]
- AOAC Official Method 2016.05. Analysis of Vitamin D2 and Vitamin D3 in Fortified Milk Powders and Infant and Nutritional Formulas by Liquid Chromatography–Tandem Mass Spectrometry: Single-Laboratory Validation, First Action 2016; AOAC International: Gaithersburg, MD, USA, 2016. [Google Scholar]
- AOAC Official Method 992.26. Vitamin D3 (Cholecalciferol) in Ready-To-Feed Milk-Based Infant Formula. First Action 1992; AOAC International: Gaithersburg, MD, USA, 2006. [Google Scholar]
- AOAC Official Method 995.05. Vitamin D in Infant Formula and Enteral Products. Liquid Chromatography. First Action 1995; AOAC International: Gaithersburg, MD, USA, 2000. [Google Scholar]
- AOAC Official Method 2002.05. Determination of Cholecalciferol (Vitamin D3) in Selected Food. Liquid Chromatography. First Action 2002; AOAC International: Gaithersburg, MD, USA, 2003. [Google Scholar]
- David, E.; Eleazu, C.; Igweibor, N.; Ugwu, C.; Enwefa, G.; Nwigboji, N. Comparative study on the nutrients, heavy metals and pesticide composition of some locally produced and marketed rice varieties in Nigeria. Food Chem. 2019, 278, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Oberson, J.-M.; Campos-Giménez, E.; Rivière, J.; Martin, F. Application of supercritical fluid chromatography coupled to mass spectrometry to the determination of fat-soluble vitamins in selected food products. J. Chromatogr. B 2018, 1086, 118–129. [Google Scholar] [CrossRef]
- Lipkie, T.E.; Ferruzzi, M.G.; Weaver, C.M. Low bioaccessibility of vitamin D2 from yeast fortified bread compared to crystalline D2 bread and D3 from fluid milks. Food Funct. 2016, 7, 4589–4596. [Google Scholar] [CrossRef]
- Mohanty, B.P.; Sankar, T.V.; Ganguly, S.; Mahanty, A.; Anandan, R.; Chakraborty, K.; Paul, B.N.; Sarma, D.; Dayal, J.S.; Mathew, S.; et al. Micronutrient composition of 35 food fishes from India and their significance in human nutrition. Biol. Trace Elem. Res. 2016, 174, 448–458. [Google Scholar] [CrossRef]
- Gill, B.D.; Abernethy, G.A.; Green, R.J.; Indyk, H.E. Analysis of vitamin D2 and vitamin D3 in fortified milk powders and infant and nutritional formulas by liquid chromatography–tandem mass spectrometry: Single- laboratory validation, First Action 2016.05. J. AOAC Int. 2016, 99, 1321–1330. [Google Scholar] [CrossRef][Green Version]
- Byrdwell, W.C. Quadruple parallel mass spectrometry for analysis of vitamin D and triacylglycerols in a dietary supplement. J. Chromatogr. A 2013, 1320, 48–65. [Google Scholar] [CrossRef]
- Barba, F.J.; Esteve, M.J.; Frígola, A. Determination of vitamins E (α-, γ- and δ-tocopherol) and D (cholecalciferol and ergocalciferol) by liquid chromatography in milk, fruit juice and vegetable beverage. Eur. Food Res. Technol. 2011, 232, 829–836. [Google Scholar] [CrossRef]
- Kwak, B.-M.; Jeong, I.-S.; Lee, M.-S.; Ahn, J.-H.; Park, J.-S. Rapid determination of vitamin D3 in milk-based infant formulas by liquid chromatography-tandem mass spectrometry. Food Chem. 2014, 165, 569–574. [Google Scholar] [CrossRef]
- Rathi, D.-N.; Noh, M.F.M.; Abd Rashed, A.; Dasuki, I. Simultaneous analysis of vitamin D and K in processed food products via ultra high- performance liquid chromatography (UHPLC). J. Food Meas. Charact. 2019, 13, 1947–1957. [Google Scholar] [CrossRef]
- Płonka, J.; Toczek, A.; Tomczyk, V. Multivitamin analysis of fruits, fruit–vegetable juices, and diet supplements. Food Anal. Methods 2012, 5, 1167–1176. [Google Scholar] [CrossRef]
- Kühna, J.; Schröter, A.; Hartmann, B.M.; Stangla, G.I. Cocoa and chocolate are sources of vitamin D2. Food Chem. 2018, 269, 318–320. [Google Scholar] [CrossRef] [PubMed]
- Nestola, M.; Thellmann, A. Determination of vitamins D2 and D3 in selected food matrices by online high-performance liquid chromatography–gas chromatography–mass spectrometry (HPLC-GC-MS). Anal. Bioanal. Chem. 2015, 407, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Byrdwell, W.C.; Horst, R.L.; Phillips, K.M.; Holden, J.M.; Patterson, K.Y.; Harnly, J.M.; Exler, J. Vitamin D levels in fish and shellfish determined by liquid chromatography with ultraviolet detection and mass spectrometry. J. Food Comp. Anal. 2013, 30, 109–119. [Google Scholar] [CrossRef]
- Byrdwell, W.C.; Exler, J.; Gebhardt, S.E.; Harnly, J.M.; Holden, J.M.; Horst, R.L.; Patterson, K.Y.; Phillips, K.M.; Wolf, W.R. Liquid chromatography with ultraviolet and dual parallel mass spectrometric detection for analysis of vitamin D in retail fortified orange juice. J. Food Comp. Anal. 2011, 24, 299–306. [Google Scholar] [CrossRef]
- Kamankesh, M.; Mohammadi, A.; Mollahosseini, A.; Jazaeri, S.; Shahdoostkhany, M. Vitamin D3: Preconcentration and determination in cereal samples using ultrasonic-assisted extraction and microextraction method. Cereal Chem. 2017, 94, 532–538. [Google Scholar] [CrossRef]
- Jiao, Z.; Zhang, Y.; Fan, H. Ultrasonic-microwave method in preparation of polypyrrole-coatedmagnetic particles for vitamin D extraction in milk. J. Chromatogr. A 2016, 1457, 7–13. [Google Scholar] [CrossRef]
- Viñas, P.; Bravo-Bravo, M.; López-García, I.; Hernández-Córdoba, M. Dispersive liquid–liquid microextraction for the determination of vitamins D and K in foods by liquid chromatography with diode-array and atmospheric pressure chemical ionization-mass spectrometry detection. Talanta 2013, 115, 806–813. [Google Scholar] [CrossRef]
- Hu, C.; Jia, L.; Liu, Q.; Zhang, S. Development of magnetic octadecylsilane particles as solid-phase extraction adsorbent for the determination of fat-soluble vitamins in fruit juice-milk beverage by capillary liquid chromatography A. J. Sep. Sci. 2010, 33, 2145–2152. [Google Scholar] [CrossRef] [PubMed]
- Gomes, F.P.; Shaw, P.N.; Whitfield, K.; Hewavitharana, A.K. Simultaneous quantitative analysis of eight vitamin D analogues in milk using liquid chromatography tandem mass spectrometry. Anal. Chim. Acta 2015, 891, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Schadt, H.S.; Gössl, R.; Seibel, N.; Aebischer, C.-P. Quantification of vitamin D3 in feed, food, and pharmaceuticals using high-performance liquid chromatography/tandem mass spectrometry. J. AOAC Int. 2012, 95, 1487–1494. [Google Scholar] [CrossRef] [PubMed]
- Burild, A.; Frandsen, H.L.; Poulsen, M.; Jakobsen, J. Quantification of physiological levels of vitamin D3 and 25-hydroxyvitamin D3 in porcine fat and liver in subgram sample sizes. J. Sep. Sci. 2014, 37, 2659–2663. [Google Scholar] [CrossRef]
- Byrdwell, W.C. “Dilute-and-shoot” triple parallel mass spectrometry method for analysis of vitamin D and triacylglycerols in dietary supplements. Anal. Bioanal. Chem. 2011, 401, 3317–3334. [Google Scholar] [CrossRef]
- Prabu, S.L.; Suriya Prakash, T.N.K.; Thirumurugan, R. Cleaning validation and its regulatory aspects in the pharmaceutical industry. In Developments in Surface Contamination and Cleaning, 1st ed.; Kohli, R., Mittal, K.L., Eds.; Elsevier Inc.: Oxford, UK, 2015; Volume 7, pp. 129–186. [Google Scholar]
- Qian, H.; Sheng, M. Simultaneous determination of fat-soluble vitamins A, D and E and pro-vitamin D in animal feeds by one-step extraction and high-performance liquid chromatography analysis. J. Chromatogr. A 1998, 825, 127–133. [Google Scholar] [CrossRef]
- Kabir, A.; Locatelli, M.; Ulusoy, H.I. Recent Trends in microextraction techniques employed in analytical and bioanalytical sample preparation. Separations 2017, 4, 36. [Google Scholar] [CrossRef]
- Socas-Rodríguez, B.; González-Sálamo, J.; Hernández-Borges, J.; Rodríguez-Delgado, M.Á. Recent applications of nanomaterials in food safety. TrAC-Trend. Anal. Chem. 2017, 96, 172–200. [Google Scholar] [CrossRef]
- Fanali, C.; D’Orazio, G.; Fanali, S.; Gentili, A. Advanced analytical techniques for fat-soluble vitamin analysis. TrAC-Trend. Anal. Chem. 2017, 87, 82–97. [Google Scholar] [CrossRef]
- Taguri, K.; Fukusaki, E.; Bamba, T. Simultaneous analysis for water- and fat-soluble vitamins by a novel single chromatography technique unifying supercritical fluid chromatography and liquid chromatography. J. Chromatogr. A 2014, 1362, 270–277. [Google Scholar]
- Tarafder, A. Metamorphosos of supercritical fluid chromatography to SFC: An overview. TrAC-Trend. Anal. Chem. 2016, 81, 3–10. [Google Scholar] [CrossRef]
- Losacco, G.L.; Veuthey, J.-L.; Guillarme, D. Supercritical fluid chromatography e Mass spectrometry: Recent evolution and current trends. TrAC-Trend. Anal. Chem. 2019, 118, 731–738. [Google Scholar] [CrossRef]
- Nováková, L.; Grand-Guillaume Perrenoud, A.; Francois, I.; West, C.; Leseiller, E.; Guillerme, D. Modern analytical supercritical fluid chromatography using columns packed with sub-2 μm particles: A tutorial. Anal. Chim. Acta 2014, 824, 18–25. [Google Scholar] [CrossRef] [PubMed]













© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).