Determination of Hydrophilic UV Filters in Real Matrices Using New-Generation Bar Adsorptive Microextraction Devices


Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents, Standards, and Samples
2.2. Experimental Set-Up
2.2.1. Optimization Assays
2.2.2. Validation Assays
2.2.3. Real Samples Assays
2.3. Instrumental Set-Up
3. Results and Discussion
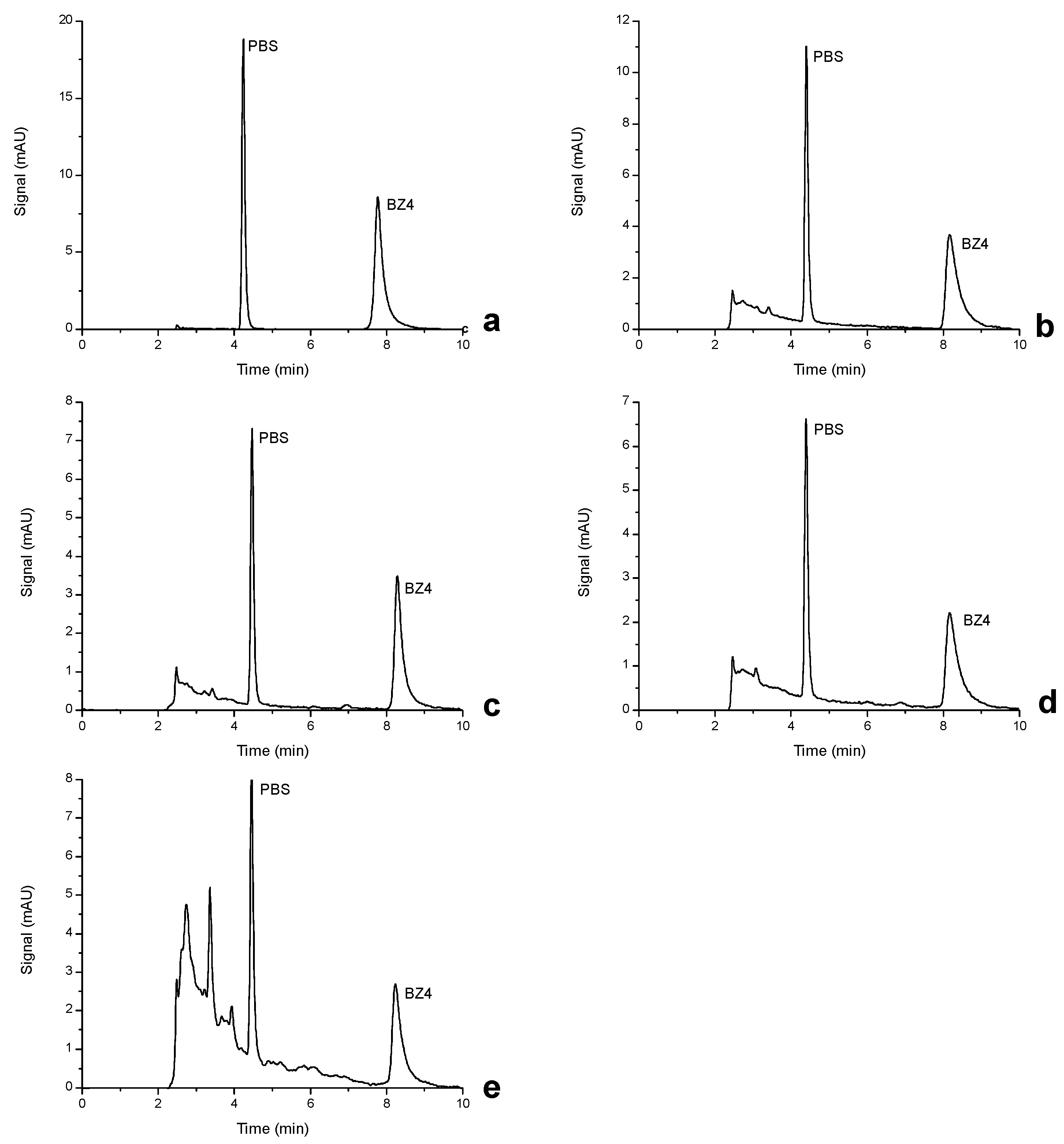
3.1. HPLC–DAD Optimization
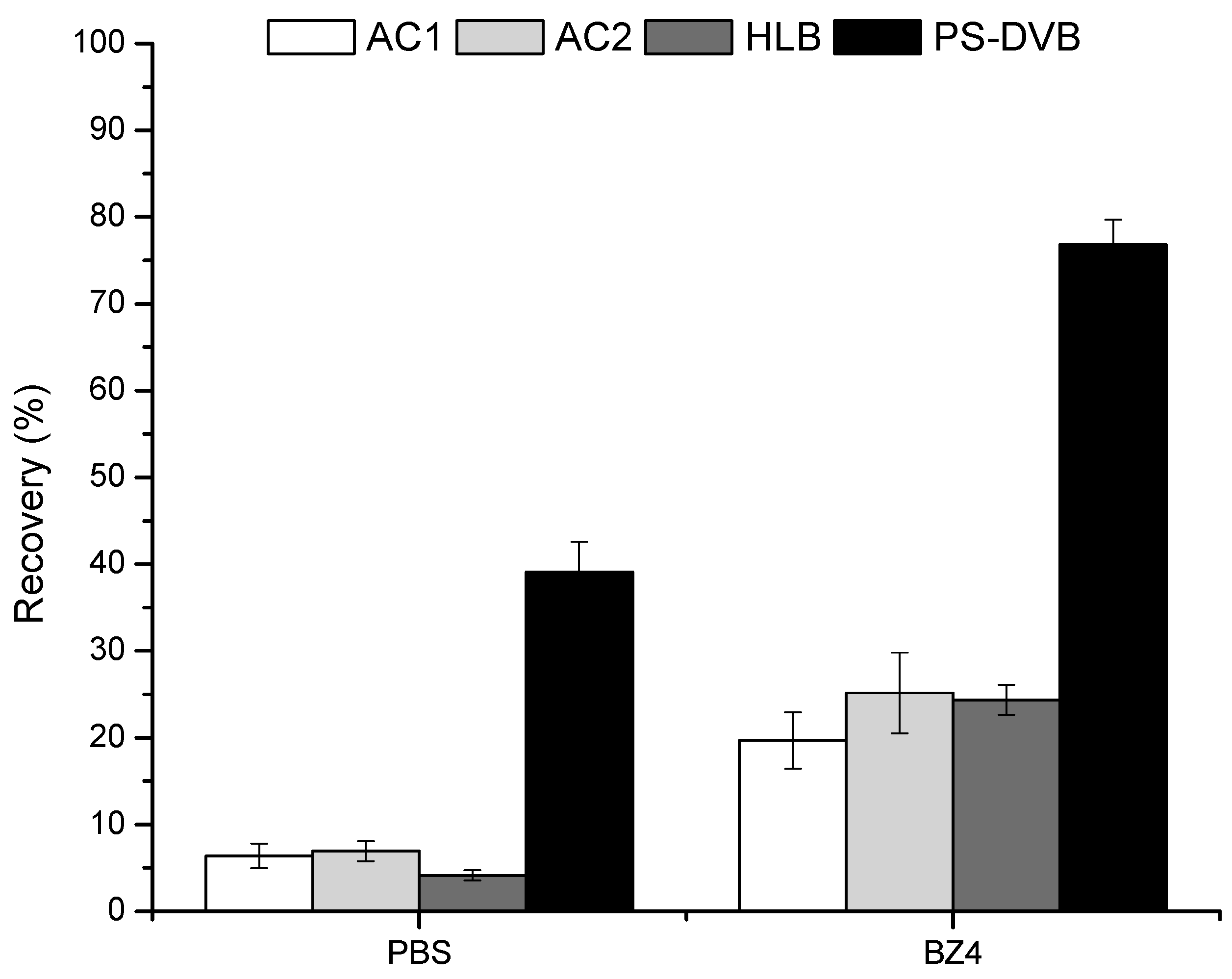
3.2. Selection of the BAµE Coating Phase
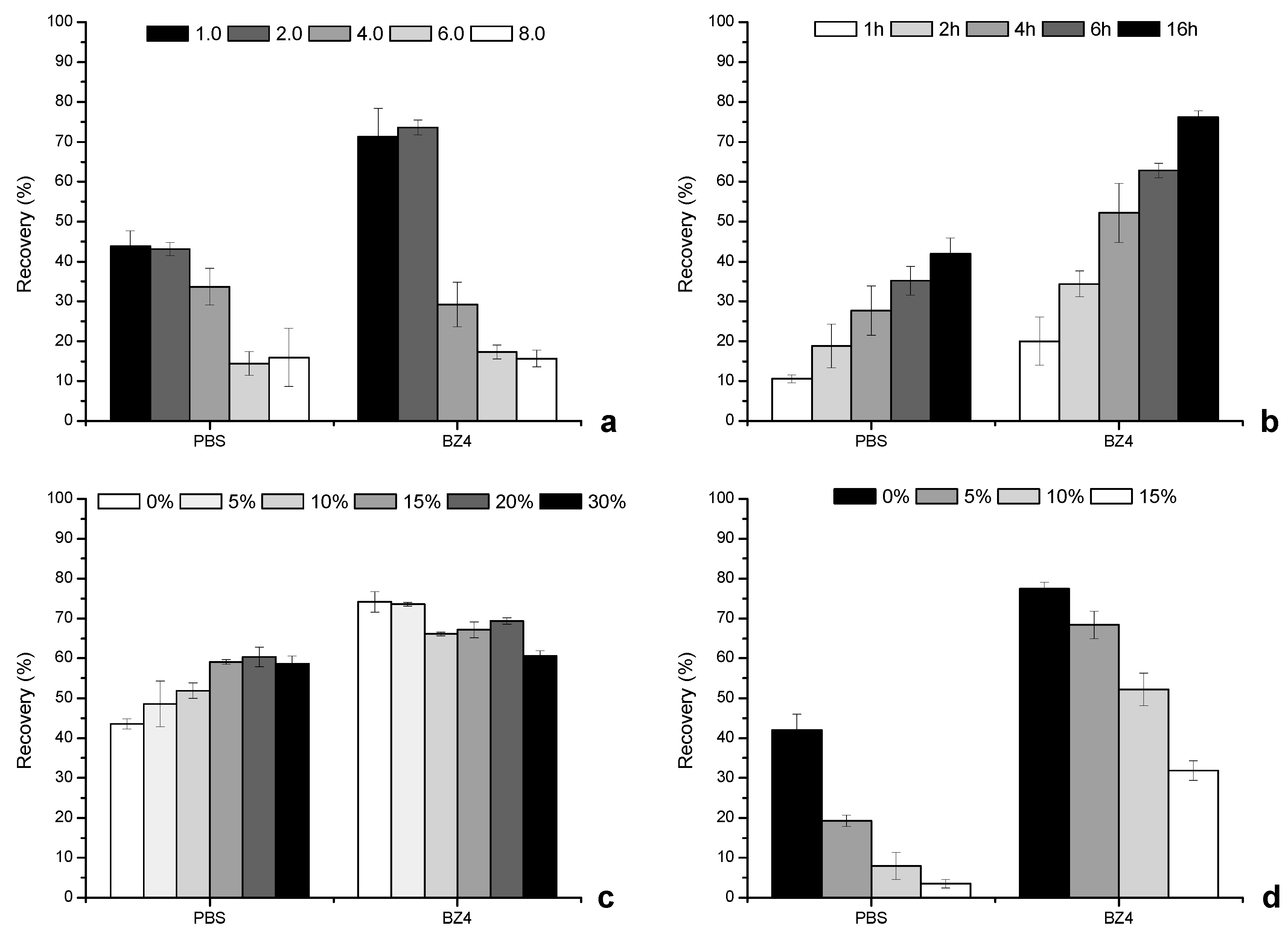
3.3. Back-Extraction Stage
3.4. Microextraction Stage
3.5. Method Validation
3.6. Application to Real Matrices
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BAµE | bar adsorptive microextraction |
| BZ4 | 5-benzoyl-4-hydroxy-2-methoxy-benzenesulfonic acid |
| DAD | diode array detector |
| GAC | green analytical chemistry |
| LD | liquid desorption |
| MeOH | methanol |
| PBS | 2-phenyl-5-benzemidazolesulfonic acid |
| PS-DVB | polystyrene-divinylbenzene |
| SAM | standard addition method |
| SBSE | stir bar sorptive extraction |
| SPME | solid phase microextraction |
References
- Arthur, C.L.; Pawliszyn, J. Solid phase microextraction with thermal desorption using fused silica optical fibers. Anal. Chem. 1990, 62, 2145–2148. [Google Scholar] [CrossRef]
- Baltussen, E.; Sandra, P. Stir bar sorptive extraction (SBSE), a novel extraction technique for aqueous samples: Theory and principles. J. Microcolumn. 1999, 11, 737–747. [Google Scholar] [CrossRef]
- Neng, N.R.; Silva, A.R.M.; Nogueira, J.M.F. Adsorptive micro-extraction techniques-novel analytical tools for trace levels of polar solutes in aqueous media. J. Chromatogr. A 2010, 1217, 7303–7310. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, J.M.F. Novel sorption-based methodologies for static microextraction analysis: A review on sbse and related techniques. Anal. Chim. Acta 2012, 757, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ide, A.H.; Nogueira, J.M.F. New-generation bar adsorptive microextraction (BAμE) devices for a better eco-user-friendly analytical approach–Application for the determination of antidepressant pharmaceuticals in biological fluids. J. Pharm. Biomed. Anal. 2018, 153, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Kameda, Y.; Kimura, K.; Miyazaki, M. Occurrence and profiles of organic sun-blocking agents in surface waters and sediments in japanese rivers and lakes. Environ. Pollut. 2011, 159, 1570–1576. [Google Scholar] [CrossRef] [PubMed]
- Tsui, M.M.; Leung, H.W.; Wai, T.C.; Yamashita, N.; Taniyasu, S.; Liu, W.; Lam, P.K.; Murphy, M.B. Occurrence, distribution and ecological risk assessment of multiple classes of UV filters in surface waters from different countries. Water Res. 2014, 67, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Román, I.P.; Alberto, A.C.; Canals, A. Dispersive solid-phase extraction based on oleic acid-coated magnetic nanoparticles followed by gas chromatography-mass spectrometry for UV-filter determination in water samples. J. Chromatogr. A 2011, 1218, 2467–2475. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.T.; Scapolla, C.; Di Carro, M.; Magi, E. Rapid and selective determination of UV filters in seawater by liquid chromatography-tandem mass spectrometry combined with stir bar sorptive extraction. Talanta 2011, 85, 2375–2384. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lee, H.K. Ionic liquid-based ultrasound-assisted dispersive liquid-liquid microextraction followed high-performance liquid chromatography for the determination of ultraviolet filters in environmental water samples. Anal. Chim. Acta 2012, 750, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Vila, M.; Celeiro, M.; Lamas, J.P.; Dagnac, T.; Llompart, M.; Garcia-jares, C. Determination of fourteen UV filters in bathing water by headspace solid-phase microextraction and gas chromatography-tandem mass spectrometry. Anal. Methods 2016, 8, 7069–7079. [Google Scholar] [CrossRef]
- Negreira, N.; Rodríguez, I.; Ramil, M.; Rubí, E.; Cela, R. Solid-phase extraction followed by liquid chromatography-tandem mass spectrometry for the determination of hydroxylated benzophenone UV absorbers in environmental water samples. Anal. Chim. Acta 2009, 654, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Bratkovics, S.; Sapozhnikova, Y. Determination of seven commonly used organic UV filters in fresh and saline waters by liquid chromatography-tandem mass spectrometry. Anal. Methods 2011, 3, 2943. [Google Scholar] [CrossRef]
- Benedé, J.L.; Chisvert, A.; Giokas, D.L.; Salvador, A. Stir bar sorptive-dispersive microextraction mediated by magnetic nanoparticles-nylon 6 composite for the extraction of hydrophilic organic compounds in aqueous media. Anal. Chim. Acta 2016, 926, 63–71. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| UV Filter | Chemical Structure | Log KOW | pKa |
|---|---|---|---|
| PBS | 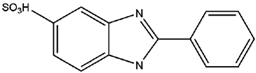 | −0.234 | −0.87 |
| BZ4 | 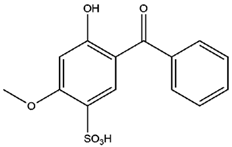 | 0.993 | −0.70 |
| UV Filter | Extraction Efficiency (%) | LOD (µg L−1) | LOQ (µg L−1) | Dynamic Range | r2 | Slope |
|---|---|---|---|---|---|---|
| PBS | 61.8 ± 9.1 | 0.04 | 0.16 | 0.16–16.0 | 0.9985 | 9.25 |
| BZ4 | 69.5 ± 4.8 | 0.20 | 0.80 | 0.80–80.0 | 0.9993 | 2.47 |
| UV Filter | Tap Water | Sea Water | Estuarine Water | Wastewater |
|---|---|---|---|---|
| r2 | ||||
| PBS | 0.9999 | 0.9998 | 0.9975 | 0.9942 |
| BZ4 | 0.9998 | 0.9998 | 0.9994 | 0.9940 |
| Slope | ||||
| PBS | 15.06 | 10.21 | 9.25 | 5.25 |
| BZ4 | 4.28 | 2.69 | 2.47 | 1.10 |
| Content (mg g−1) | ||||
| PBS | <LOD | <LOD | <LOD | <LOD |
| BZ4 | <LOD | <LOD | <LOD | <LOD |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ide, A.H.; Nogueira, J.M.F. Determination of Hydrophilic UV Filters in Real Matrices Using New-Generation Bar Adsorptive Microextraction Devices. Separations 2019, 6, 45. https://doi.org/10.3390/separations6040045
Ide AH, Nogueira JMF. Determination of Hydrophilic UV Filters in Real Matrices Using New-Generation Bar Adsorptive Microextraction Devices. Separations. 2019; 6(4):45. https://doi.org/10.3390/separations6040045
Chicago/Turabian StyleIde, Alessandra Honjo, and José Manuel Florêncio Nogueira. 2019. "Determination of Hydrophilic UV Filters in Real Matrices Using New-Generation Bar Adsorptive Microextraction Devices" Separations 6, no. 4: 45. https://doi.org/10.3390/separations6040045
APA StyleIde, A. H., & Nogueira, J. M. F. (2019). Determination of Hydrophilic UV Filters in Real Matrices Using New-Generation Bar Adsorptive Microextraction Devices. Separations, 6(4), 45. https://doi.org/10.3390/separations6040045