Perfluoroalkyl Substance Assessment in Turin Metropolitan Area and Correlation with Potential Sources of Pollution According to the Water Safety Plan Risk Management Approach


Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Area and Sampling
2.2. Reagents and Chemicals
2.3. Sample Preparation
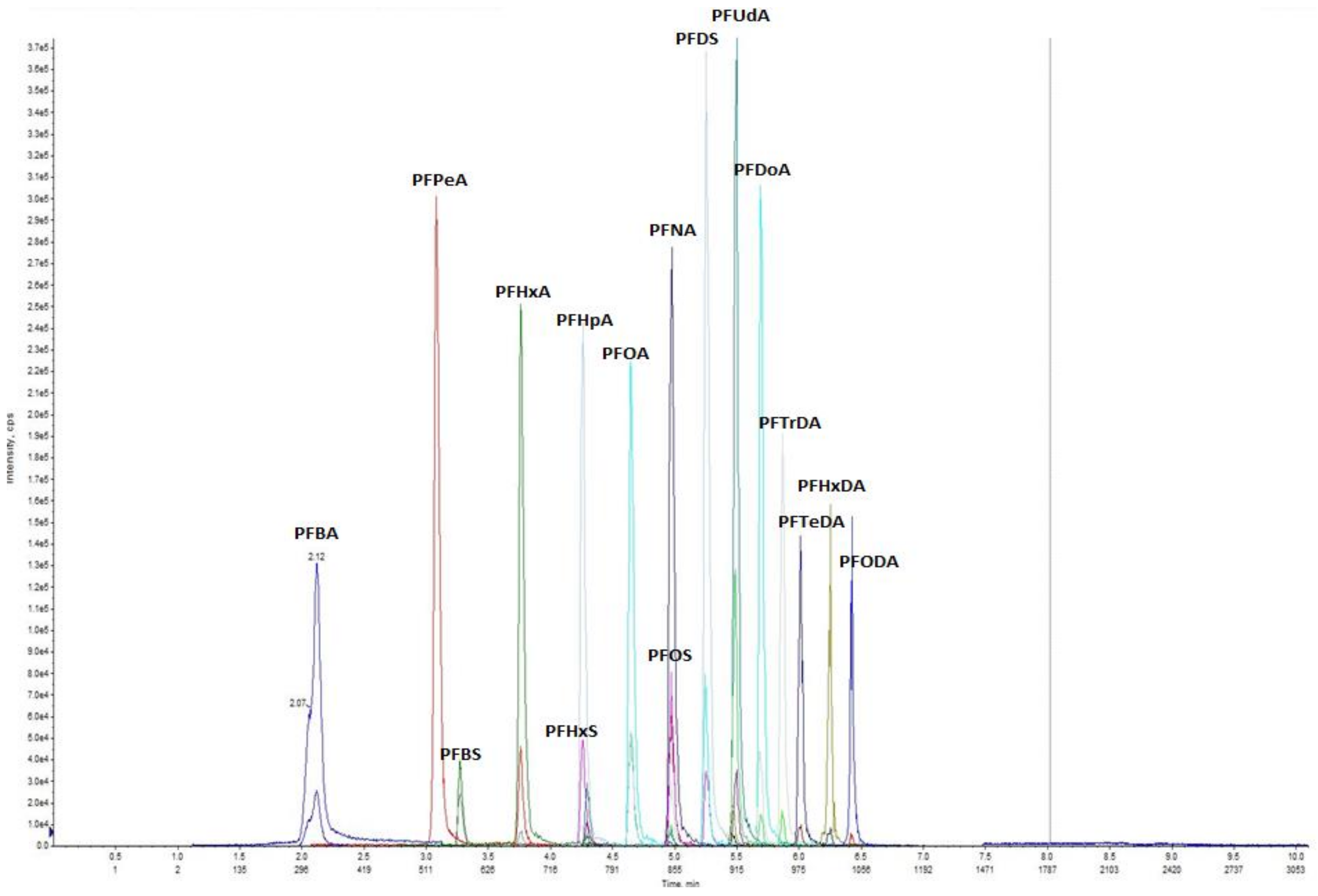
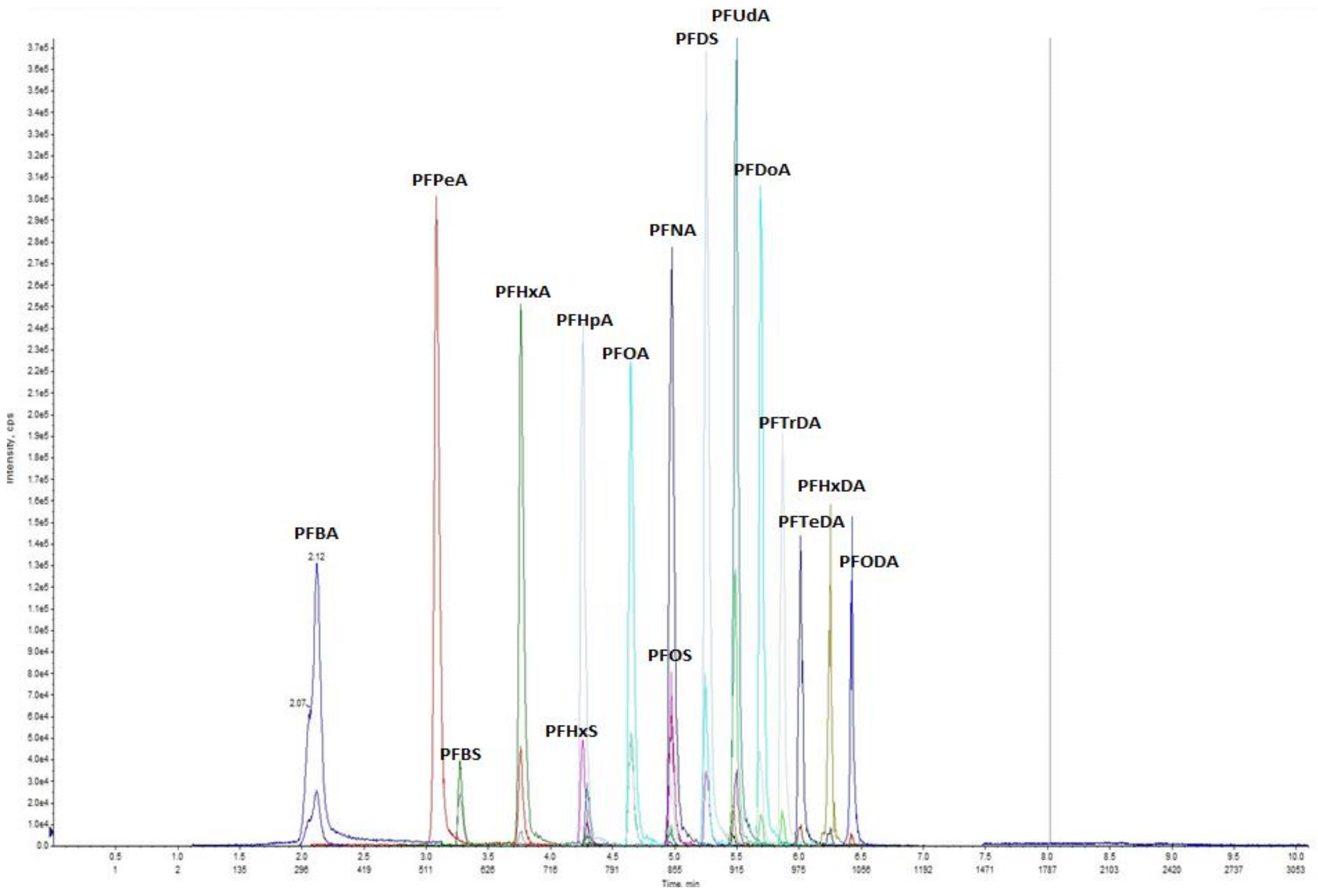
2.4. Instrumental Analysis
2.5. Method Validation and Quality Assurance
2.6. Spatial and Statistical Analysis
3. Results and Discussion
3.1. Cross Contamination
3.2. Method Optimization
3.3. Validation Study
3.3.1. Linearity
3.3.2. Accuracy, Recovery and Precision
3.3.3. Limits of Detection and Quantification
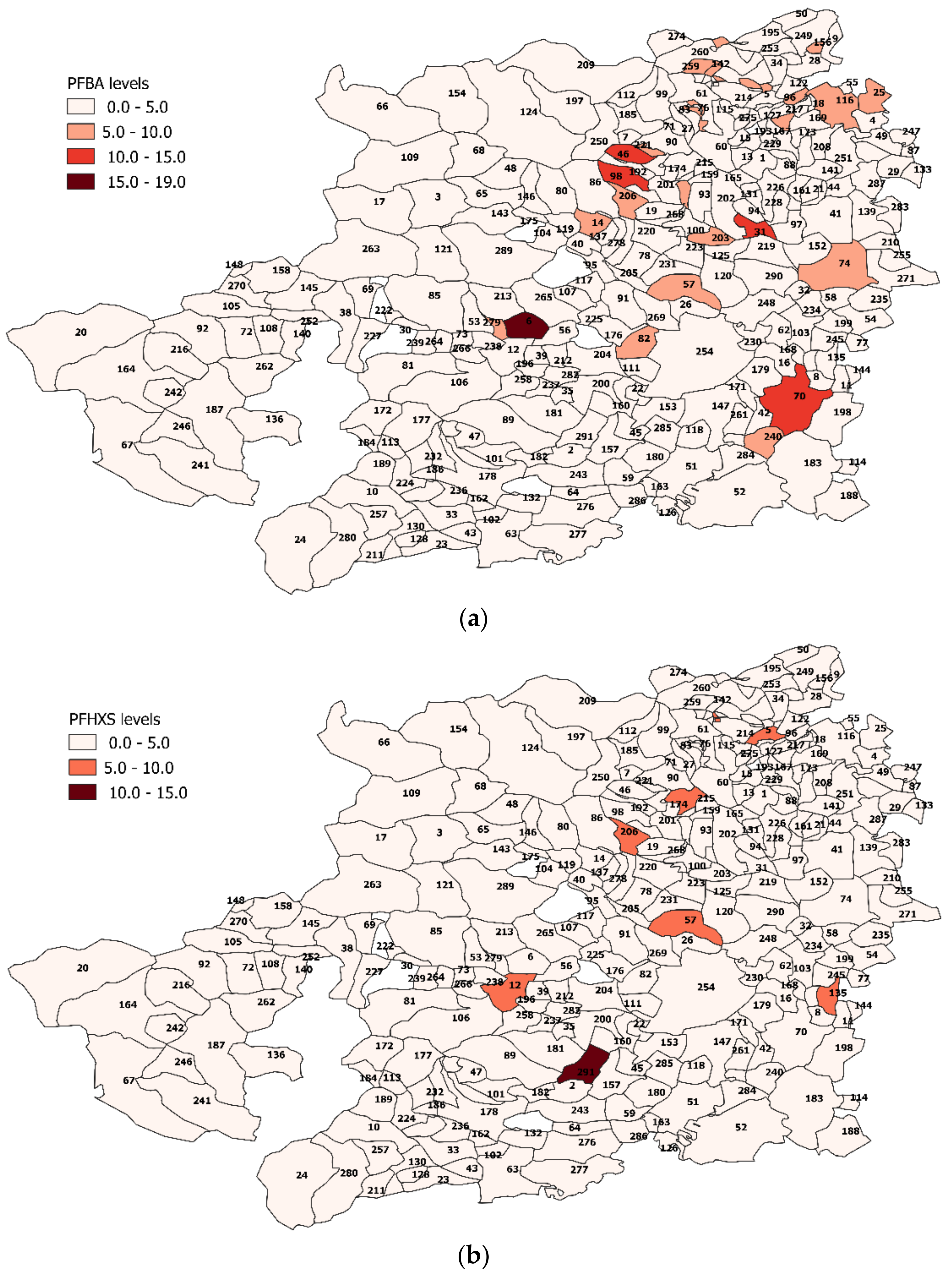
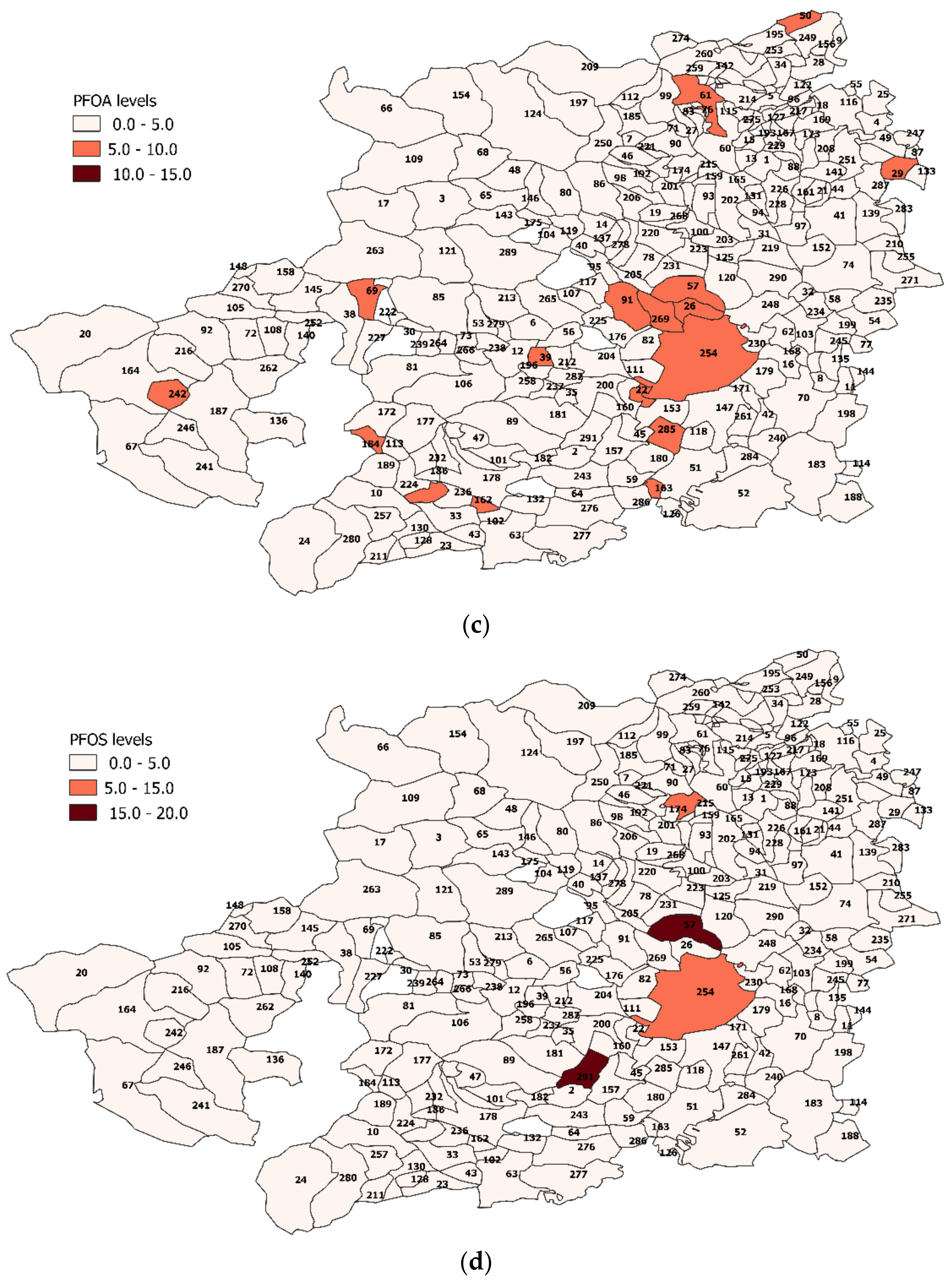
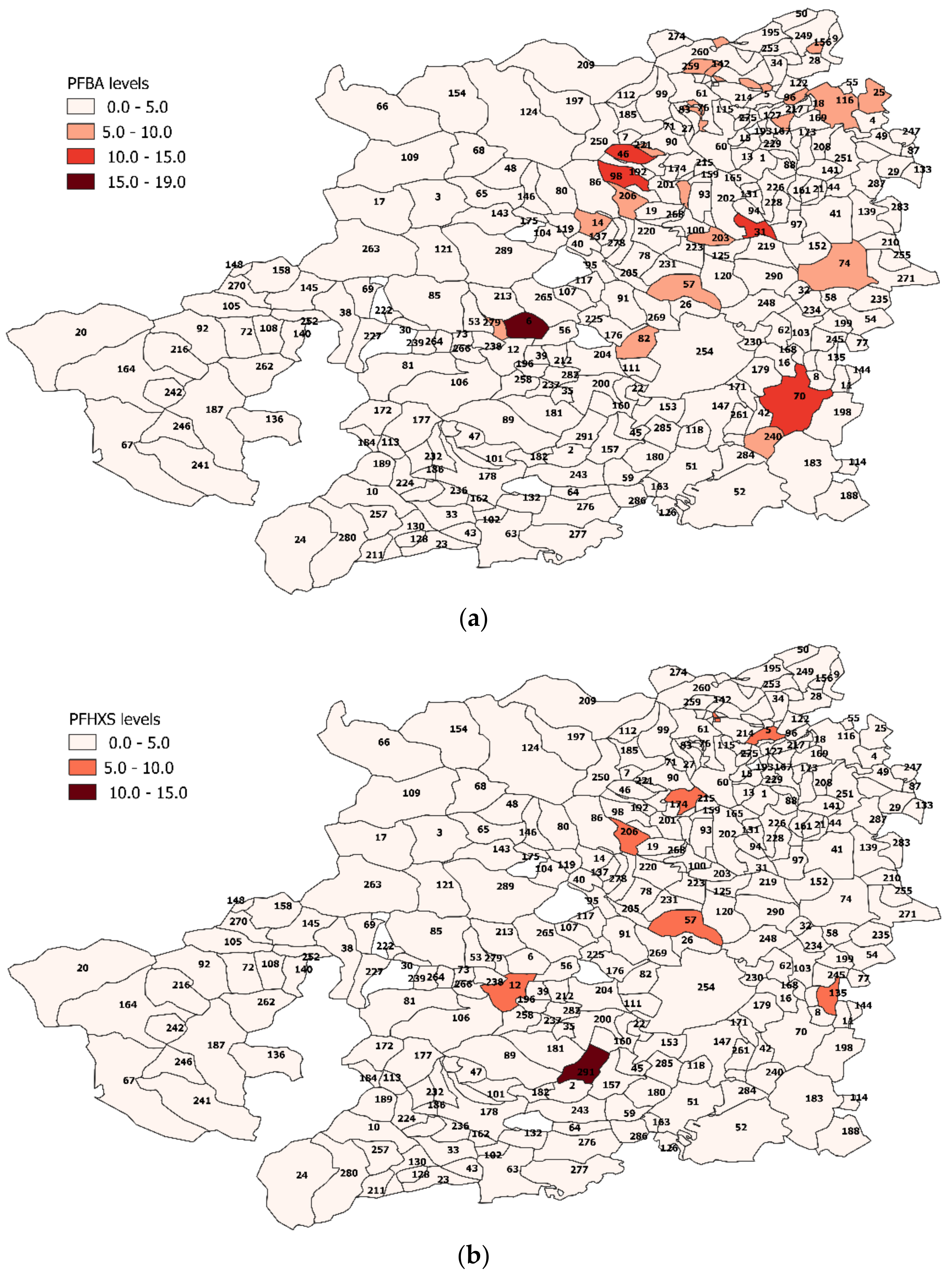
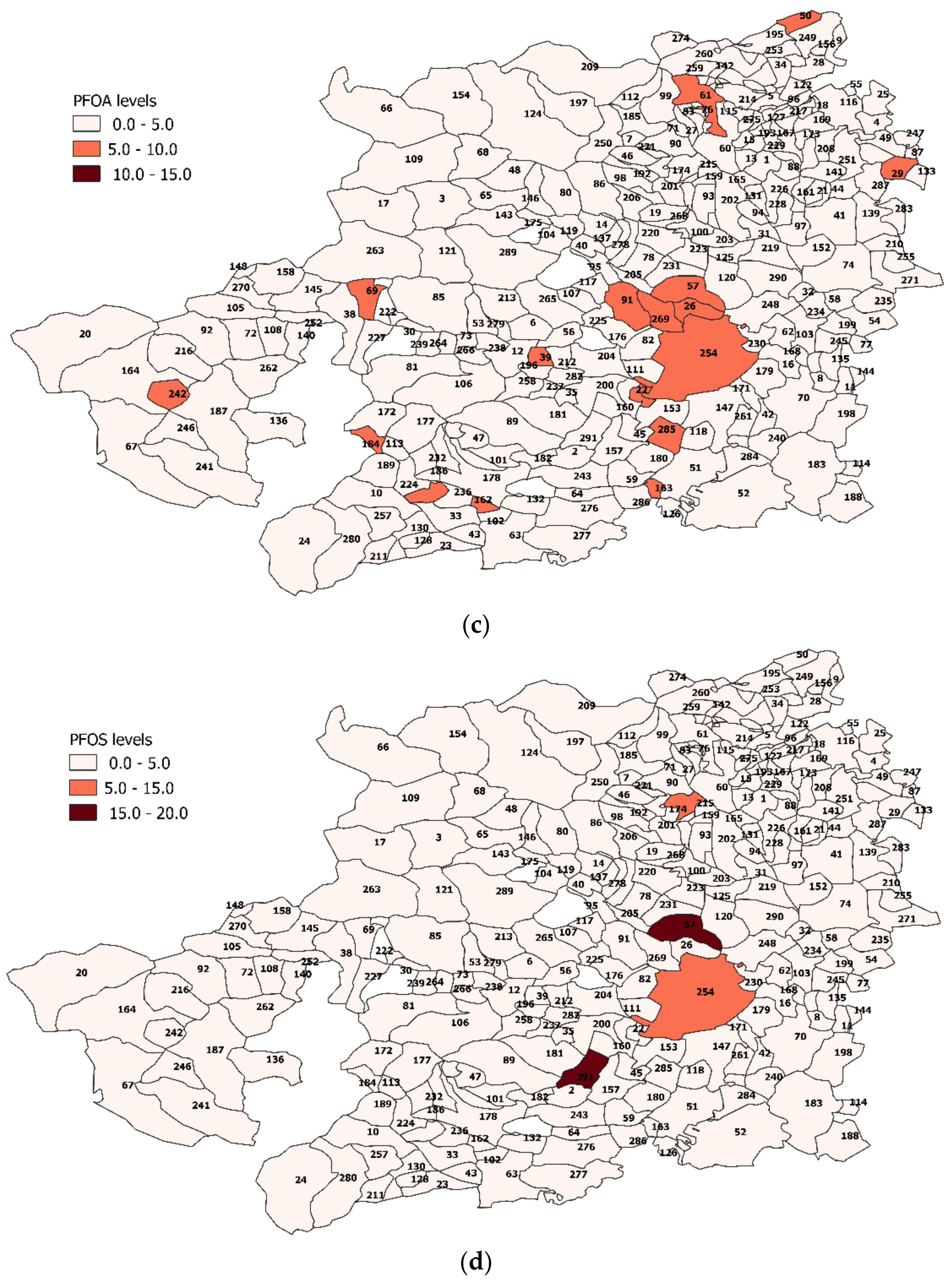
3.4. PFAS Assessment in Turin Metropolitan Area
Spatial Analysis Results
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Perfluoroalkane Sulfonates (PFSA) | Perfluoroalkyl Carboxylates (PFCA) | ||
|---|---|---|---|
| Short Chain n ≤ 5 e.g., PFBS | Long Chain n ≥ 6 e.g., PFHxS, PFOS and PFDS | Short Chain n ≤ 7 e.g., PFBA, PFPeA, PFHxA and PFHpA | Long Chain n ≥ 8 e.g., PFOA, PFNA, PFDA, PFUdA, PFDoA, PFTrDA, PFTeDA, PFHxDA and PFODA |
| Time (min) | Flow Rate (mL/min) | A % | B % |
|---|---|---|---|
| 0.000 | 0.550 | 98 | 2 |
| 0.000 | 0.550 | 98 | 2 |
| 0.500 | 0.550 | 98 | 2 |
| 1.000 | 0.550 | 70 | 30 |
| 6.000 | 0.550 | 0 | 100 |
| 7.500 | 0.550 | 0 | 100 |
| 7.600 | 0.550 | 98 | 2 |
| 10.000 | 0.550 | 98 | 2 |
| Parameter | Value |
|---|---|
| Polarity | Negative |
| Curtains Gas | 30 psi |
| Collision Gas | 30 psi |
| Ionspray Voltage | −4500 V |
| Temperature | 350 °C |
| GS1 | 50 psi |
| GS2 | 55 psi |
| Compound | Q1 m/z | Q3 m/z | RT |
|---|---|---|---|
| PFBA | 213 | 169 | 2.1 |
| PFPeA | 263 | 219 | 3.1 |
| PFHxA | 131 | 269 | 3.8 |
| PFHpA | 363 | 319 | 4.3 |
| PFOA | 413 | 369 | 4.6 |
| PFNA | 463 | 419 | 5.0 |
| PFDA | 513 | 469 | 6.9 |
| PFUdA | 563 | 519 | 5.5 |
| PFDoA | 613 | 569 | 5.7 |
| PFTrDA | 663 | 619 | 5.9 |
| PFTeDA | 713 | 669 | 6.0 |
| PFHxDA | 813 | 769 | 6.3 |
| PFODA | 913 | 869 | 6.5 |
| L-PFBS | 299 | 99 | 3.3 |
| L-PFHxS | 399 | 99 | 4.3 |
| L-PFOS | 499 | 99 | 5.0 |
| L-PFDS | 599 | 99 | 5.4 |
| MPFHxS | 403 | 10 | 4.3 |
| MPFOS | 503 | 99 | 5.0 |
| MPFBA | 217 | 172 | 2.1 |
| MPFHxA | 315 | 270 | 3.8 |
| MPFOA | 417 | 372 | 4.6 |
| MPFNA | 468 | 423 | 4.9 |
| MPFDA | 515 | 470 | 5.2 |
| MPFUdA | 565 | 520 | 5.5 |
| MPFDoA | 615 | 570 | 5.7 |
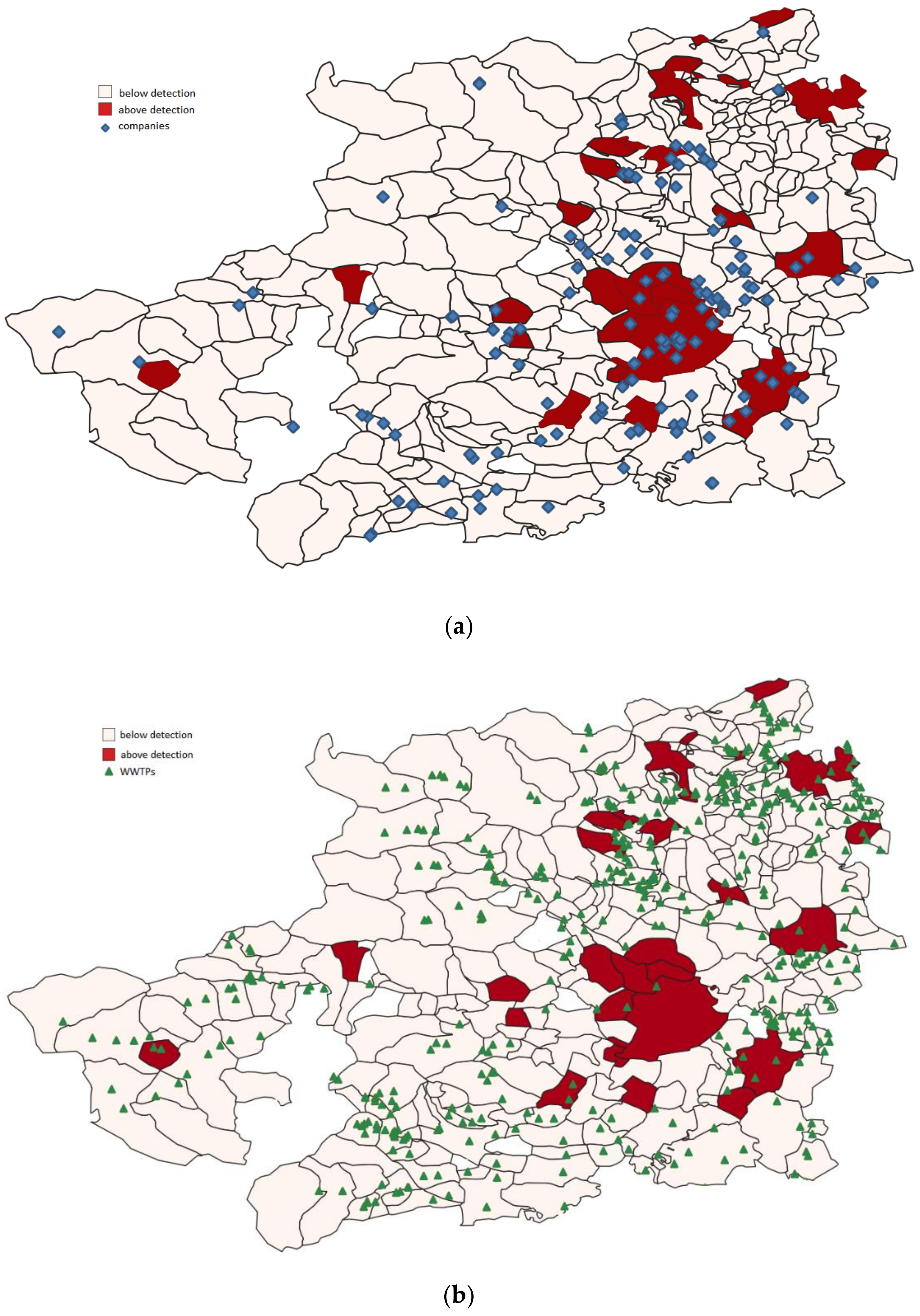
| Municipality Name ID | Number ID | Samples Analysed | Municipality Name ID | Number ID | Samples Analysed |
|---|---|---|---|---|---|
| AGLIE’ | 1 | 1 | MONCALIERI | 147 | 21 |
| AIRASCA | 2 | 1 | MONCENISIO | 148 | 1 |
| ALA DI STURA | 3 | 6 | MONTALDO TORINESE | 149 | 3 |
| ALBIANO D’IVREA | 4 | 3 | MONTALENGHE | 150 | 1 |
| ALICE SUPERIORE | 5 | 2 | MONTALTO DORA | 151 | 1 |
| ALMESE | 6 | 11 | MONTANARO | 152 | 2 |
| ALPETTE | 7 | 2 | NICHELINO | 153 | 6 |
| ANDEZENO | 8 | 2 | NOASCA | 154 | 4 |
| ANDRATE | 9 | 1 | NOLE | 155 | 3 |
| ANGROGNA | 10 | 2 | NOMAGLIO | 156 | 2 |
| ARIGNANO | 11 | 1 | NONE | 157 | 2 |
| AVIGLIANA | 12 | 7 | NOVALESA | 158 | 1 |
| BAIRO | 13 | 1 | OGLIANICO | 159 | 3 |
| BALANGERO | 14 | 3 | ORBASSANO | 160 | 2 |
| BALDISSERO CANAVESE | 15 | 1 | ORIO CANAVESE | 161 | 2 |
| BALDISSERO TORINESE | 16 | 3 | OSASCO | 162 | 2 |
| BALME | 17 | 2 | OSASIO | 163 | 2 |
| BANCHETTE | 18 | 1 | OULX | 164 | 2 |
| BARBANIA | 19 | 2 | OZEGNA | 165 | 1 |
| BARDONECCHIA | 20 | 1 | PANCALIERI | 166 | 1 |
| BARONE CANAVESE | 21 | 2 | PARELLA | 167 | 2 |
| BEINASCO | 22 | 9 | PAVAROLO | 168 | 2 |
| BIBIANA | 23 | 1 | PAVONE CANAVESE | 169 | 1 |
| BOBBIO PELLICE | 24 | 2 | PECCO | 170 | 1 |
| BOLLENGO | 25 | 3 | PECETTO TORINESE | 171 | 2 |
| BORGARO TORINESE | 26 | 12 | PEROSA ARGENTINA | 172 | 1 |
| BORGIALLO | 27 | 1 | PEROSA CANAVESE | 173 | 2 |
| BORGOFRANCO D’IVREA | 28 | 3 | PERTUSIO | 174 | 1 |
| BORGOMASINO | 29 | 1 | PESSINETTO | 175 | 2 |
| BORGONE SUSA | 30 | 4 | PIANEZZA | 176 | 8 |
| BOSCONERO | 31 | 7 | PINASCA | 177 | 1 |
| BRANDIZZO | 32 | 3 | PINEROLO | 178 | 1 |
| BRICHERASIO | 33 | 1 | PINO TORINESE | 179 | 1 |
| BROSSO | 34 | 1 | PIOBESI TORINESE | 180 | 1 |
| BRUINO | 35 | 7 | PIOSSASCO | 181 | 10 |
| BURIASCO | 36 | 1 | PISCINA | 182 | 1 |
| BUSANO | 37 | 2 | POIRINO | 183 | 5 |
| BUSSOLENO | 38 | 3 | POMARETTO | 184 | 2 |
| BUTTIGLIERA ALTA | 39 | 2 | PONT CANAVESE | 185 | 5 |
| CAFASSE | 40 | 2 | PORTE | 186 | 1 |
| CALUSO | 41 | 2 | PRAGELATO | 187 | 1 |
| CAMBIANO | 42 | 2 | PRALORMO | 188 | 2 |
| CAMPIGLIONE FENILE | 43 | 2 | PRAMOLLO | 189 | 1 |
| CANDIA CANAVESE | 44 | 9 | PRAROSTINO | 190 | 2 |
| CANDIOLO | 45 | 1 | PRASCORSANO | 191 | 2 |
| CANISCHIO | 46 | 2 | PRATIGLIONE | 192 | 1 |
| CANTALUPA | 47 | 1 | QUAGLIUZZO | 193 | 2 |
| CANTOIRA | 48 | 3 | QUASSOLO | 194 | 6 |
| CAPRIE | 49 | 4 | QUINCINETTO | 195 | 2 |
| CARAVINO | 50 | 3 | REANO | 196 | 2 |
| CAREMA | 51 | 5 | RIBORDONE | 197 | 6 |
| CARIGNANO | 52 | 6 | RIVA PRESSO CHIERI | 198 | 3 |
| CARMAGNOLA | 53 | 2 | RIVALBA | 199 | 1 |
| CASALBORGONE | 54 | 2 | RIVALTA DI TORINO | 200 | 2 |
| CASCINETTE D’IVREA | 55 | 3 | RIVARA | 201 | 1 |
| CASELETTE | 56 | 5 | RIVAROLO CANAVESE | 202 | 10 |
| CASELLE TORINESE | 57 | 5 | RIVAROSSA | 203 | 2 |
| CASTAGNETO PO | 58 | 2 | RIVOLI | 204 | 5 |
| CASTAGNOLE PIEMONTE | 59 | 2 | ROBASSOMERO | 205 | 1 |
| CASTELLAMONTE | 60 | 8 | ROCCA CANAVESE | 206 | 1 |
| CASTELNUOVO NIGRA | 61 | 5 | ROLETTO | 207 | 1 |
| CASTIGLIONE TORINESE | 62 | 3 | ROMANO CANAVESE | 208 | 2 |
| CAVOUR | 63 | 1 | RONCO CANAVESE | 209 | 9 |
| CERCENASCO | 64 | 1 | RONDISSONE | 210 | 1 |
| CERES | 65 | 5 | RORA’ | 211 | 2 |
| CERESOLE REALE | 66 | 4 | ROSTA | 212 | 1 |
| CESANA TORINESE | 67 | 1 | RUBIANA | 213 | 8 |
| CHIALAMBERTO | 68 | 5 | RUEGLIO | 214 | 2 |
| CHIANOCCO | 69 | 1 | SALASSA | 215 | 2 |
| CHIERI | 70 | 4 | SALBERTRAND | 216 | 2 |
| CHIESANUOVA | 71 | 1 | SALERANO CANAVESE | 217 | 1 |
| CHIOMONTE | 72 | 1 | SAMONE | 218 | 2 |
| CHIUSA DI SAN MICHELE | 73 | 2 | SAN BENIGNO CANAVESE | 219 | 3 |
| CHIVASSO | 74 | 8 | SAN CARLO CANAVESE | 220 | 2 |
| CICONIO | 75 | 1 | SAN COLOMBANO BELMONTE | 221 | 1 |
| CINTANO | 76 | 1 | SAN DIDERO | 222 | 2 |
| CINZANO | 77 | 1 | SAN FRANCESCO AL CAMPO | 223 | 1 |
| CIRIE’ | 78 | 8 | SAN GERMANO CHISONE | 224 | 1 |
| CLAVIERE | 79 | 1 | SAN GILLIO | 225 | 8 |
| COASSOLO TORINESE | 80 | 2 | SAN GIORGIO CANAVESE | 226 | 2 |
| COAZZE | 81 | 2 | SAN GIORIO DI SUSA | 227 | 3 |
| COLLEGNO | 82 | 13 | SAN GIUSTO CANAVESE | 228 | 1 |
| COLLERETTO CASTELNUOVO | 83 | 3 | SAN MARTINO CANAVESE | 229 | 2 |
| COLLERETTO GIACOSA | 84 | 1 | SAN MAURIZIO CANAVESE | 230 | 2 |
| CONDOVE | 85 | 1 | SAN MAURO TORINESE | 231 | 2 |
| CORIO | 86 | 2 | SAN PIETRO VAL LEMINA | 232 | 1 |
| COSSANO CANAVESE | 87 | 2 | SAN PONSO | 233 | 1 |
| CUCEGLIO | 88 | 2 | SAN RAFFAELE CIMENA | 234 | 2 |
| CUMIANA | 89 | 1 | SAN SEBASTIANO DA PO | 235 | 1 |
| CUORGNE’ | 90 | 3 | SAN SECONDO DI PINEROLO | 236 | 1 |
| DRUENTO | 91 | 8 | SANGANO | 237 | 5 |
| EXILLES | 92 | 1 | SANT’AMBROGIO DI TORINO | 238 | 4 |
| FAVRIA | 93 | 2 | SANT’ANTONINO DI SUSA | 239 | 9 |
| FELETTO | 94 | 2 | SANTENA | 240 | 2 |
| FIANO | 95 | 1 | SAUZE DI CESANA | 241 | 1 |
| FIORANO CANAVESE | 96 | 2 | SAUZE D’OULX | 242 | 2 |
| FOGLIZZO | 97 | 2 | SCALENGHE | 243 | 2 |
| FORNO CANAVESE | 98 | 3 | SCARMAGNO | 244 | 6 |
| FRASSINETTO | 99 | 8 | SCIOLZE | 245 | 1 |
| FRONT | 100 | 2 | SESTRIERE | 246 | 1 |
| FROSSASCO | 101 | 2 | SETTIMO ROTTARO | 247 | 2 |
| GARZIGLIANA | 102 | 1 | SETTIMO TORINESE | 248 | 11 |
| GASSINO TORINESE | 103 | 6 | SETTIMO VITTONE | 249 | 1 |
| GERMAGNANO | 104 | 1 | SPARONE | 250 | 1 |
| GIAGLIONE | 105 | 2 | STRAMBINO | 251 | 1 |
| GIAVENO | 106 | 10 | SUSA | 252 | 9 |
| GIVOLETTO | 107 | 3 | TAVAGNASCO | 253 | 3 |
| GRAVERE | 108 | 1 | TORINO | 254 | 54 |
| GROSCAVALLO | 109 | 1 | TORRAZZA PIEMONTE | 255 | 1 |
| GROSSO | 110 | 1 | TORRE CANAVESE | 256 | 2 |
| GRUGLIASCO | 111 | 7 | TORRE PELLICE | 257 | 3 |
| INGRIA | 112 | 1 | TRANA | 258 | 1 |
| INVERSO PINASCA | 113 | 1 | TRAUSELLA | 259 | 2 |
| ISOLABELLA | 114 | 1 | TRAVERSELLA | 260 | 6 |
| ISSIGLIO | 115 | 2 | TROFARELLO | 261 | 1 |
| IVREA | 116 | 16 | USSEAUX | 262 | 1 |
| LA CASSA | 117 | 2 | USSEGLIO | 263 | 7 |
| LA LOGGIA | 118 | 8 | VAIE | 264 | 3 |
| LANZO TORINESE | 119 | 3 | VAL DELLA TORRE | 265 | 5 |
| LEINI’ | 120 | 5 | VALGIOIE | 266 | 3 |
| LEMIE | 121 | 1 | VALPERGA | 267 | 3 |
| LESSOLO | 122 | 3 | VAUDA CANAVESE | 268 | 1 |
| LEVONE | 123 | 2 | VENARIA REALE | 269 | 30 |
| LOCANA | 124 | 15 | VENAUS | 270 | 1 |
| LOMBARDORE | 125 | 2 | VEROLENGO | 271 | 3 |
| LOMBRIASCO | 126 | 3 | VESTIGNE’ | 272 | 2 |
| LORANZE’ | 127 | 4 | VIALFRE’ | 273 | 1 |
| LUGNACCO | 128 | 2 | VICO CANAVESE | 274 | 1 |
| LUSERNA SAN GIOVANNI | 129 | 1 | VIDRACCO | 275 | 2 |
| LUSERNETTA | 130 | 1 | VIGONE | 276 | 1 |
| LUSIGLIE’ | 131 | 2 | VILLAFRANCA PIEMONTE | 277 | 1 |
| MACELLO | 132 | 1 | VILLANOVA CANAVESE | 278 | 1 |
| MAGLIONE | 133 | 1 | VILLAR DORA | 279 | 3 |
| MAPPANO | 134 | 1 | VILLAR PELLICE | 280 | 5 |
| MARENTINO | 135 | 4 | VILLAR PEROSA | 281 | 1 |
| MASSELLO | 136 | 1 | VILLARBASSE | 282 | 7 |
| MATHI | 137 | 2 | VILLAREGGIA | 283 | 2 |
| MATTIE | 138 | 4 | VILLASTELLONE | 284 | 3 |
| MAZZE’ | 139 | 1 | VINOVO | 285 | 6 |
| MEANA DI SUSA | 140 | 2 | VIRLE PIEMONTE | 286 | 1 |
| MERCENASCO | 141 | 1 | VISCHE | 287 | 2 |
| MEUGLIANO | 142 | 2 | VISTRORIO | 288 | 1 |
| MEZZENILE | 143 | 4 | VIU’ | 289 | 3 |
| MOMBELLO DI TORINO | 144 | 2 | VOLPIANO | 290 | 5 |
| MOMPANTERO | 145 | 1 | VOLVERA | 291 | 6 |
| MONASTERO DI LANZO | 146 | 2 |
| Standard Method | EPA 537 | ISO 25101:2029(E) | ASTM D7979-16 | ASTM D7868-14 |
|---|---|---|---|---|
| Sample volume | 250 mL | 500 mL | 5 mL | 2 g |
| Sample matrix | Drinking water | Drinking water, groundwater, surface water and seawater | Water; wastewater sludge, influent and effluent | Solid and biosolid |
| Analytes | PFAS and FOSAAs 14 PFAS | PFOS and PFOA | PFAS, FOSAAs, FTSs, n:2 FTUCAs and FTCAs | PFAS, FOSAAs, FTSs, n:2 FTUCAs and FTCAs |
| Preservation | Trizma for buffering and removal of free chlorine | Sodium thiosulfate pentahydrate for removal of free chlorine | None | None |
| Holding time | Before extraction: 14 days refrigerated at ≤6 °C Postextraction: 28 days at room temperature | 14 days at 4 ± 2 °C | 28 days at 0–6 °C | 28 days at 0–6 °C |
| Extraction Method | SPE-WAX (SPE Weak anion exchange) | SPE | Direct injection | Solvent extraction followed by filtration using polypropylene filters |
| Analytical instrument | LC-MS/MS (liquid chromatography tandem with mass spectrometry) | LC-MS/MS and LC/MS | LC-MS/MS | LC-MS/MS |
| Reporting limits | 2.9–14 ng/L | 2–10,000 ng/L | 10–400 ng/L | 25–1000 ng/L |
References
- Haukås, M.; Berger, U.; Hop, H.; Gulliksen, B.; Gabrielsen, G.W. Bioaccumulation of per- and polyfluorinated alkyl substances (PFAS) in selected species from the Barents Sea food web. Environ. Pollut. 2007, 148, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Hedlund, J. Per- and Polyfluoroalkyl Substances (PFASs) in Swedish Waters. 10 June 2016. Available online: https://stud.epsilon.slu.se/9187/ (accessed on 13 December 2018).
- Dasu, K.; Kempisty, D.M.; Millis, M.A. Per- and polufluoroalkyl substance analysis to support site characterization, exposure, and risk management. In Perfluoroalkyl Substances in the Environment | Theory, Practice, and Innovation, 1st ed.; Series: Environmental and occupational health series; Kempisty, D.M., Xing, Y., Racz, L., Eds.; CRC Press Taylor & Francis: Boca Raton, FL, USA, 2018; pp. 35–66. ISBN 9781498764186. [Google Scholar]
- Xiao, F. Emerging poly- and perfluoroalkyl substances in the aquatic environment: A review of current literature. Water Res. 2017, 124, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Buck, R.C.; Franklin, J.; Berger, U.; Conder, J.M.; Cousins, I.T.; De Voogt, P.; Jensen, A.A.; Kannan, K.; Mabury, S.A.; van Leeuwen, S.P. Perfluoroalkyl and polyfluoroalkyl substances in the environment: Terminology, classification, and origins. Integr. Environ. Assess. Manag. 2011, 7, 513–541. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, L. Polyfluoroalkyl compounds in the aquatic environment: A review of their occurrence and fate. J. Environ. Monit. 2011, 13, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Krafft, M.P.; Riess, J.G. Selected physicochemical aspects of poly- and perfluoroalkylated substances relevant to performance, environment and sustainability—Part one. Chemosphere 2015, 129, 4–19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, Y.; Taniyasu, S.; Yeung, L.W.; Lam, P.K.; Wang, J.; Li, X.; Yamashita, N.; Dai, J. Distribution and fate of perfluoroalkyl substances in municipal wastewater treatment plants in economically developed areas of China. Environ. Pollut. 2013, 176, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.G.; Jones, K.C.; Sweetman, A.J. A first global production, emission, and environmental inventory for perfluorooctane sulfonate. Environ. Sci. Technol. 2009, 43, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Kotthoff, M.; Bücking, M. Four chemical trends will shape the next decade’s directions in perfluoroalkyl and polyfluoroalkyl substances research. Front. Chem. 2018, 6, 103. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.T.; Reagen, W.K. Method and validation for the analysis of perfluorinated compounds in water by pre-sampling isotope dilution-direct injection-LC/MS/MS. Anal. Methods 2013, 5, 2444–2454. [Google Scholar] [CrossRef]
- Gosettia, F.; Chiuminatto, U.; Zampieri, D.; Mazzucco, E.; Robotti, E.; Calabrese, G.; Gennaro, M.C.; Marengo, E. Determination of perfluorochemicals in biological, environmental and food samples by an automated on-line solid phase extraction ultra high performance liquid chromatography tandem mass spectrometry method. J. Chromatogr. A 2010, 1217, 7864–7872. [Google Scholar] [CrossRef] [PubMed]
- Furdui, V.I.; Crozier, P.W.; Reiner, E.J.; Mabury, S.A. Trace level determination of perfluorinated compounds in water by direct injection. Chemosphere 2008, 73, S24–S30. [Google Scholar] [CrossRef] [PubMed]
- Ciofi, L.; Renai, L.; Rossini, D.; Ancillotti, C.; Falai, A.; Fibbi, D.; Bruzzoniti, M.C.; Santana-Rodriguez, J.J.; Orlandini, S.; Del Bubba, M. Applicability of the direct injection liquid chromatographic tandem mass spectrometric analytical approach to the sub-ng L−1 determination of perfluoro-alkyl acids in waste, surface, ground and drinking water samples. Talanta 2018, 176, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Liu, G.; Wang, J.; Aubry, A.-F.; Arnold, M.E. Selecting the correct weighting factors for linear and quadratic calibration curves with least-squares regression algorithm in bioanalytical LC-MS/MS assays and impacts of using incorrect weighting factors on curve stability, data quality, and assay performance. Anal. Chem. 2014, 86, 8959–8966. [Google Scholar] [PubMed]
- Taniyasu, S.; Kannan, K.; So, M.K.; Gulkowska, A.; Sinclair, E.; Okazawa, T.; Yamashita, N. Analysis of fluorotelomer alcohols, fluorotelomer acids, and short- and long-chain perfluorinated acids in water and biota. J. Chromatogr. A 2005, 1093, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, N.; Kannan, K.; Taniyasu, S.; Horii, Y.; Okazawa, T.; Petrick, G.; Gamo, T. Analysis of perfluorinated acids at parts-per-quadrillion levels in seawater using liquid chromatography-tandem mass spectrometry. Environ. Sci. Technol. 2004, 38, 5522–5528. [Google Scholar] [CrossRef] [PubMed]
- Wille, K.; Bussche, J.V.; Noppe, H.; De Wulf, E.; Van Caeter, P.; Janssen, C.R.; De Brabander, H.F.; Vanhaecke, L. A validated analytical method for the determination of perfluorinated compounds in surface-, sea- and sewagewater using liquid chromatography coupled to time-of-flight mass spectrometry. J. Chromatogr. A 2010, 1217, 6616–6622. [Google Scholar] [CrossRef] [PubMed]
- Pasáková, I.; Klimeš, J.; Sochor, J.; Hrabálek, A. Optimization of HPLC chromatographic conditions for determination of Transkarbam 12 and its degradation products. J. Pharm. Biomed. Anal. 2006, 42, 136–142. [Google Scholar] [CrossRef] [PubMed]
- GeoPortale ARPA Piemonte. Available online: http://webgis.arpa.piemonte.it/geoportale/ (accessed on 10 October 2018).
- Story Map Series. Available online: http://webgis.arpa.piemonte.it/monitoraggio_qualita_acque_mapseries/monitoraggio_qualita_acque_webapp/ (accessed on 13 October 2018).
- DIVA-GIS | Free, Simple & Effective. Available online: http://www.diva-gis.org/ (accessed on 11 November 2018).
- Gamerman, D.; Moreira, A.R.B. Multivariate spatial regression models. J. Multivar. Anal. 2004, 91, 262–281. [Google Scholar] [CrossRef]
- Hu, X.C.; Andrews, D.Q.; Lindstrom, A.B.; Bruton, T.A.; Schaider, L.A.; Grandjean, P.; Lohmann, R.; Carignan, C.C.; Blum, A.; Balan, S.A.; et al. Detection of poly- and perfluoroalkyl substances (PFASs) in U.S. drinking water linked to industrial sites, military fire training areas, and wastewater treatment plants. Environ. Sci. Technol. Lett. 2016, 3, 344–350. [Google Scholar] [CrossRef]
| Target Compounds (PFAC-MXB) | Internal Standard Compounds (MPFAC-MXA) | ||
|---|---|---|---|
| Full Name | Abbreviation | Full Name | Abbreviation |
| Perfluoro-n-butanoic acid | PFBA | Perfluoro-n-[13C4]butanoic acid | MPFBA |
| Perfluoro-n-pentanoic acid | PFPeA | Perfluoro-n-[1,2-13C2]hexanoic acid | MPFHxA |
| Perfluoro-n-hexanoic acid | PFHxA | Perfluoro-n-[1,2-13C2]hexanoic acid | MPFHxA |
| Perfluoro-n-heptanoic acid | PFHpA | Perfluoro-n-[1,2-13C2]hexanoic acid | MPFHxA |
| Perfluoro-n-octanoic acid | PFOA | Perfluoro-n-[1,2,3,4-13C4]octanoic acid | MPFOA |
| Perfluoro-n-nonanoic acid | PFNA | Perfluoro-n-[1,2,3,4,5-13C5]nonanoic acid | MPFNA |
| Perfluoro-n-decanoic acid | PFDA | Perfluoro-n-[1,2-13C2]decanoic acid | MPFDA |
| Perfluoro-n-undecanoic acid | PFUdA | Perfluoro-n-[1,2-13C2]undecanoic acid | MPFUdA |
| Perfluoro-n-dodecanoic acid | PFDoA | Perfluoro-n-[1,2-13C2]dodecanoic acid | MPFDoA |
| Perfluoro-n-tridecanoic acid | PFTrDA | Perfluoro-n-[1,2-13C2]dodecanoic acid | MPFDoA |
| Perfluoro-n-tetradecanoic acid | PFTeDA | Perfluoro-n-[1,2-13C2]dodecanoic acid | MPFDoA |
| Perfluoro-n-dexadecanoic acid | PFHxDA | Perfluoro-n-[1,2-13C2]decanoic acid | MPFDA |
| Perfluoro-n-octadecanoic acid | PFODA | Perfluoro-n-[1,2-13C2]decanoic acid | MPFDA |
| Potassium perfluoro-1-butanesulfonate | L-PFBS | Sodium perfluoro-1-hexane [18O2] sulfonate | MPFHxS |
| Sodium perfluoro-1-hexanesulfonate | L-PFHxS | Sodium perfluoro-1-hexane [18O2]sulfonate | MPFHxS |
| Sodium perfluoro-1-octanesulfonate | L-PFOS | Sodium perfluoro-1-[1,2,3,4-13C4] octanesulfonate | MPFOS |
| Sodium-1-decanesulfonate | L-PFDS | Sodium perfluoro-1-hexane [18O2]sulfonate | MPFHxS |
| Compounds | Spike ng L−1 | Accuracy % | Recovery % | Precision % | Linearity | LOQ |
|---|---|---|---|---|---|---|
| PFBA 1 | 50 | −8.6 | 91.38 | 3.132 | 0.997 | 5 |
| PFBS | 50 | −4.2 | 95.78 | 2.899 | 0.997 | 5 |
| PFPeA | 50 | 1.3 | 101.28 | 1.673 | 0.999 | 5 |
| PFHxA | 50 | −5.2 | 94.84 | 2.816 | 0.998 | 5 |
| PFHxS 1 | 50 | −3.9 | 96.12 | 6.065 | 0.997 | 5 |
| PFHpA | 50 | −4.7 | 95.28 | 3.632 | 0.999 | 5 |
| PFOA 1 | 50 | −3.7 | 96.28 | 4.357 | 0.998 | 5 |
| PFOS 1 | 50 | −8.0 | 92.04 | 1.956 | 0.992 | 5 |
| PFNA | 50 | −9.6 | 90.38 | 4.819 | 0.999 | 5 |
| PFDA | 50 | −14.4 | 85.58 | 7.591 | 0.995 | 5 |
| PFUdA | 50 | −5.3 | 94.72 | 9.637 | 0.998 | 5 |
| PFDoA | 50 | −7.2 | 92.78 | 8.715 | 0.999 | 5 |
| PFTrDA | 50 | −26.8 | 73.22 | 17.526 | 0.989 | 5 |
| PFTeDA | 50 | −30.0 | 69.98 | 13.758 | 0.987 | 5 |
| PFHxDA | 50 | −15.1 | 84.88 | 15.971 | 0.995 | 5 |
| PFODA | 50 | −13.7 | 86.34 | 5.477 | 0.980 | 5 |
| PFDS | 50 | −51.3 | 48.68 | 15.776 | 0.997 | 5 |
| Real Sample 1 | Real Sample 2 | Real Sample 3 | Real Sample 4 | ||
|---|---|---|---|---|---|
| Compounds | Spike ng L−1 | Recovery % | Recovery % | Recovery % | Recovery % |
| PFBA 1 | 50 | 107.52 | 106.06 | 90.84 | 107.05 |
| PFBS | 50 | 106.92 | 107.22 | 90.76 | 103.90 |
| PFPeA | 50 | 108.22 | 105.85 | 90.53 | 109.99 |
| PFHxA | 50 | 104.96 | 106.89 | 91.69 | 107.11 |
| PFHxS 1 | 50 | 104.96 | 106.73 | 90.88 | 105.98 |
| PFHpA | 50 | 102.19 | 102.93 | 92.57 | 105.56 |
| PFOA 1 | 50 | 103.63 | 114.74 | 92.09 | 107.14 |
| PFOS 1 | 50 | 95.92 | 97.31 | 86.33 | 96.11 |
| PFNA | 50 | 106.18 | 102.78 | 97.41 | 104.91 |
| PFDA | 50 | 95.29 | 90.22 | 81.93 | 90.39 |
| PFUdA | 50 | 93.49 | 83.01 | 81.57 | 80.78 |
| PFDoA | 50 | 93.19 | 100.73 | 96.98 | 97.45 |
| PFTrDA | 50 | 97.89 | 87.62 | 82.73 | 97.13 |
| PFTeDA | 50 | 101.03 | 84.89 | 94.62 | 88.16 |
| PFHxDA | 50 | 100.61 | 101.19 | 84.50 | 101.98 |
| PFODA | 50 | 86.63 | 90.16 | 87.91 | 93.71 |
| PFDS | 50 | 72.26 | 75.31 | 52.28 | 74.51 |
| Compounds | Industrial Sites | WWTPs | λ1 | R2 |
|---|---|---|---|---|
| PFBA | ||||
| coefficient | 1.1371 | 0.2795 | 0.092 | 0.66 |
| p-value | 0.001 | 0.001 | 0.05 | |
| PFHxS | ||||
| coefficient | 0.1688 | −0.0118 | 0.0329 | 0.30 |
| p-value | 0.002 | 0.523 | 0.07 | |
| PFOS | ||||
| coefficient | 0.2859 | −0.0123 | 0.0822 | 0.08 |
| p-value | 0.001 | 0.527 | 0.07 | |
| PFOA | ||||
| coefficient | 0.6166 | −0.0311 | 0.1586 | 0.24 |
| p-value | 0.001 | −0.151 | 0.07 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Binetti, R.; Calza, P.; Costantino, G.; Morgillo, S.; Papagiannaki, D. Perfluoroalkyl Substance Assessment in Turin Metropolitan Area and Correlation with Potential Sources of Pollution According to the Water Safety Plan Risk Management Approach. Separations 2019, 6, 17. https://doi.org/10.3390/separations6010017
Binetti R, Calza P, Costantino G, Morgillo S, Papagiannaki D. Perfluoroalkyl Substance Assessment in Turin Metropolitan Area and Correlation with Potential Sources of Pollution According to the Water Safety Plan Risk Management Approach. Separations. 2019; 6(1):17. https://doi.org/10.3390/separations6010017
Chicago/Turabian StyleBinetti, Rita, Paola Calza, Giovanni Costantino, Stefania Morgillo, and Dimitra Papagiannaki. 2019. "Perfluoroalkyl Substance Assessment in Turin Metropolitan Area and Correlation with Potential Sources of Pollution According to the Water Safety Plan Risk Management Approach" Separations 6, no. 1: 17. https://doi.org/10.3390/separations6010017
APA StyleBinetti, R., Calza, P., Costantino, G., Morgillo, S., & Papagiannaki, D. (2019). Perfluoroalkyl Substance Assessment in Turin Metropolitan Area and Correlation with Potential Sources of Pollution According to the Water Safety Plan Risk Management Approach. Separations, 6(1), 17. https://doi.org/10.3390/separations6010017