Food Sample Preparation for the Determination of Sulfonamides by High-Performance Liquid Chromatography: State-of-the-Art





Abstract
1. Introduction
1.1. Veterinary Drugs—Antibiotics
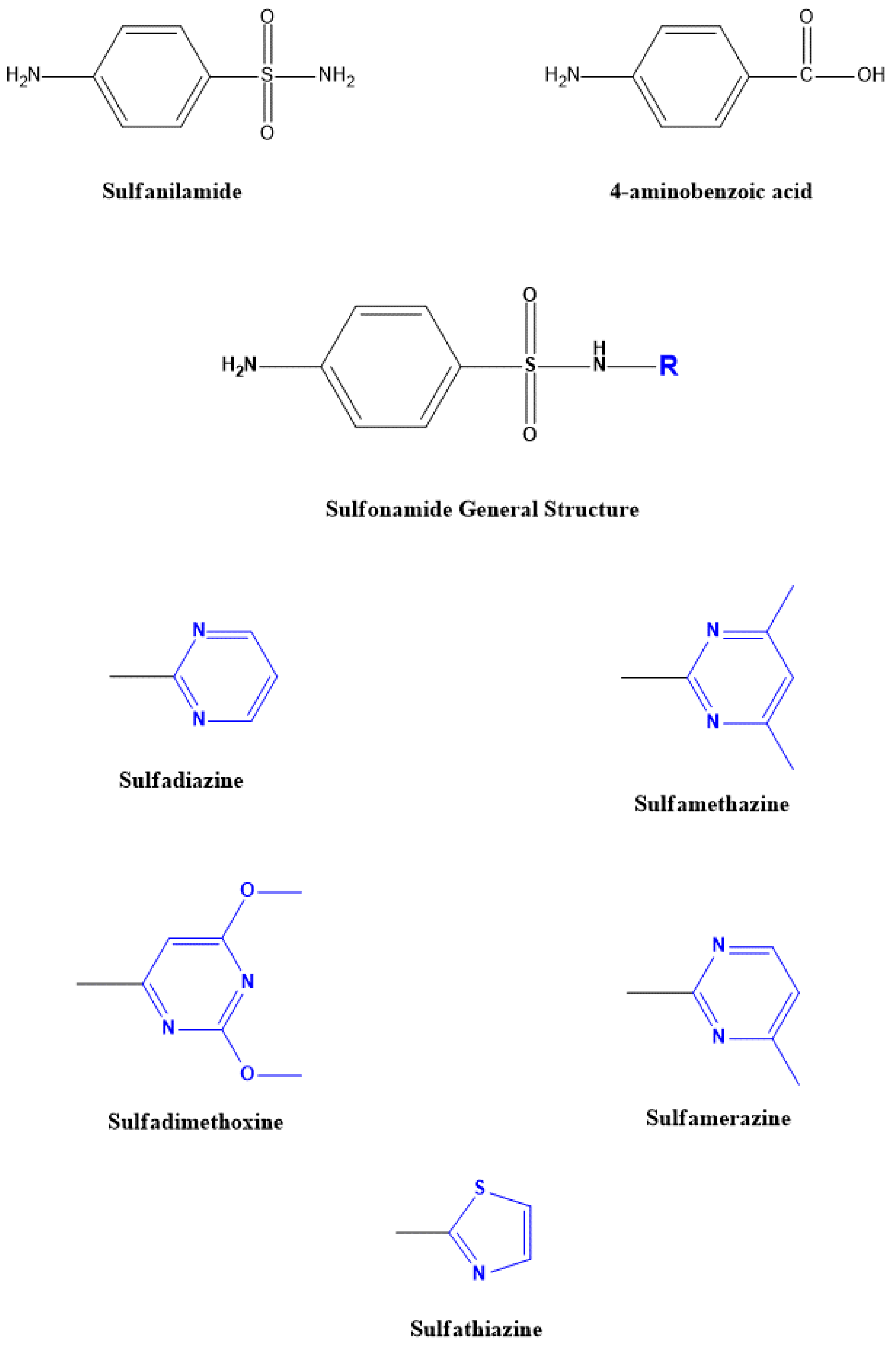
1.2. Sulfonamides
1.3. Sample Preparation
1.3.1. LLE
1.3.2. SLE
1.3.3. Salting-Out Extraction
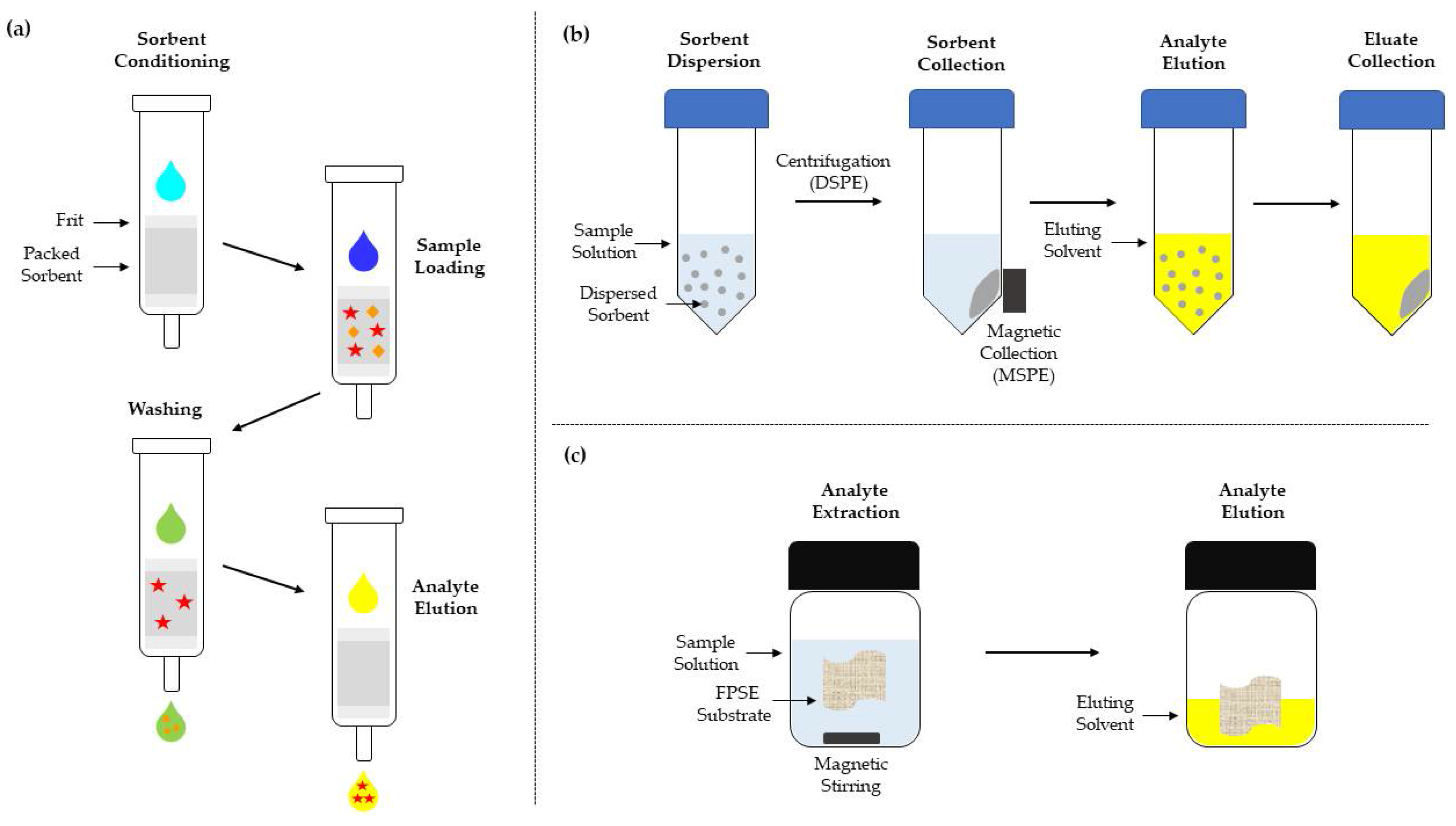
1.3.4. SPE
1.3.5. FPSE
1.4. Food Composition
1.5. Official Methods of Analysis
2. Extraction of Sulfonamides from Food Samples
2.1. Animal Tissue Samples
2.1.1. SLE
2.1.2. Salting-Out Extraction
2.1.3. SPE
2.2. Milk Samples
2.2.1. LLE
2.2.2. Salting-Out Extraction
2.2.3. SPE
2.2.4. Other Extraction Techniques
2.3. Milk Product Samples
2.4. Egg Samples
2.5. Honey Samples
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- The Council of The European Union. Council Directive 96/23/EC. Off. J. 1996, 125, 10–32. [Google Scholar]
- PAGE, S.W.; GAUTIER, P. Use of antimicrobial agents in livestock. Rev. Sci. Tech. OIE 2012, 31, 145–188. [Google Scholar] [CrossRef]
- Moreno, L.; Lanusse, C. Specific Veterinary Drug Residues of Concern in Meat Production. In New Aspects of Meat Quality; Elsevier: New York, NY, USA, 2017; pp. 605–627. ISBN 9780081005934. [Google Scholar]
- Croubels, S.; Daeseleire, E. Veterinary Drug Residues in Foods; Woodhead Publishing Limited: Sawston, UK, 2012; ISBN 9780857090584. [Google Scholar]
- Moreno, L.; Lanusse, C. Veterinary Drug Residues in Meat-Related Edible Tissues. In New Aspects of Meat Quality; Elsevier: New York, NY, USA, 2017; pp. 581–603. ISBN 9780081005934. [Google Scholar]
- Baran, W.; Adamek, E.; Ziemiańska, J.; Sobczak, A. Effects of the presence of sulfonamides in the environment and their influence on human health. J. Hazard. Mater. 2011, 196, 1–15. [Google Scholar] [CrossRef] [PubMed]
- EUR-Lex. Available online: http://eur-lex.europa.eu/legal-content/EN (accessed on 14 January 2017).
- Prescott, J.F. Sulfonamides, Diaminopyrimidines, and Their Combinations. In Antimicrobial Therapy in Veterinary Medicine; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2013; pp. 279–294. [Google Scholar]
- Reybroeck, W.; Daeseleire, E.; De Brabander, H.F.; Herman, L. Antimicrobials in beekeeping. Vet. Microbiol. 2012, 158, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Maximum Residue Limits, Codex Alimentarius. Available online: http://www.fao.org/fao-who-codexalimentarius/codex-texts/maximum-residue-limits/pt/ (accessed on 15 May 2018).
- U.S. Food & Drug Administration CFR, Code of Federal Regulations Title 21. Available online: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm?cfrpart=556 (accessed on 15 May 2018).
- Otles, S.; Ozyurt, V.H. Sampling and Sample Preparation. In Handbook of Food Chemistry; Springer: Berlin/Heidelberg, Germnay, 2015; pp. 151–164. [Google Scholar]
- Saha, S.; Singh, A.K.; Keshari, A.K.; Raj, V.; Rai, A.; Maity, S. Modern Extraction Techniques for Drugs and Medicinal Agents. In Ingredients Extraction by Physicochemical Methods in Food; Elsevier: New York, NY, USA, 2018; pp. 65–106. ISBN 9780128115213. [Google Scholar]
- Pérez-Rodríguez, M.; Pellerano, R.G.; Pezza, L.; Pezza, H.R. An overview of the main foodstuff sample preparation technologies for tetracycline residue determination. Talanta 2018, 182, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.Q.; Weng, N. Salting-out assisted liquid-liquid extraction for bioanalysis. Bioanalysis 2013, 5, 1583–1598. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.; Tao, Y.; Xie, S.; Zhu, Y.; Chen, D.; Wang, X.; Huang, L.; Peng, D.; Sattar, A.; Shabbir, M.A.B.; et al. Aqueous two-phase system (ATPS): An overview and advances in its applications. Biol. Proc. Online 2016, 18, 18. [Google Scholar] [CrossRef] [PubMed]
- Maciel, E.V.S.; de Toffoli, A.L.; Lanças, F.M. Recent trends in sorption-based sample preparation and liquid chromatography techniques for food analysis. Electrophoresis 2018, 1–48. [Google Scholar] [CrossRef] [PubMed]
- Bitas, D.; Samanidou, V. Molecularly imprinted polymers as extracting media for the chromatographic determination of antibiotics in milk. Molecules 2018, 23, 316. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, I.; Fernandes, C. Magnetic solid phase extraction for determination of drugs in biological matrices. TrAC Trends Anal. Chem. 2017, 89, 41–52. [Google Scholar] [CrossRef]
- Kazantzi, V.; Anthemidis, A. Fabric Sol–gel Phase Sorptive Extraction Technique: A Review. Separations 2017, 4, 20. [Google Scholar] [CrossRef]
- Cobos, Á.; Díaz, O. Chemical Composition of Meat and Meat Products. In Handbook of Food Chemistry; Springer: Berlin/Heidelberg, Germnay, 2015; pp. 471–510. [Google Scholar]
- Coppes Petricorena, Z. Chemical Composition of Fish and Fishery Products. In Handbook of Food Chemistry; Springer: Berlin/Heidelberg, Germnay, 2015; pp. 403–435. [Google Scholar]
- Mehta, B.M. Chemical Composition of Milk and Milk Products. In Handbook of Food Chemistry; Springer: Berlin/Heidelberg, Germnay, 2015; pp. 511–553. [Google Scholar]
- Sunwoo, H.H.; Gujral, N. Chemical Composition of Eggs and Egg Products. In Handbook of Food Chemistry; Springer: Berlin/Heidelberg, Germnay, 2015; pp. 331–363. [Google Scholar]
- U.S. Food and Drug Administration. Laboratory Methods, Drug & Chemical Residues Methods. Available online: https://www.fda.gov/Food/FoodScienceResearch/LaboratoryMethods/ucm2006950.htm (accessed on 17 May 2018).
- U.S. Food and Drug Administration. Laboratory Methods, Analytical Methods for Residues of Chloramphenicol & Related Compounds in Foods. Available online: https://www.fda.gov/Food/FoodScienceResearch/LaboratoryMethods/ucm113126.htm (accessed on 17 May 2018).
- FDA’s Center for Food Safety and Applied Nutrition. Laboratory Methods, Preparation and LC/MS/MS Analysis of Honey for Fluoroquinolone Residues. Available online: https://www.fda.gov/Food/FoodScienceResearch/LaboratoryMethods/ucm071495.htm (accessed on 17 May 2018).
- U.S. Food and Drug Administration. Field Science and Laboratories, Laboratory Information Bulletins. Available online: https://www.fda.gov/ScienceResearch/FieldScience/ucm231463.htm (accessed on 17 May 2018).
- FDA’s Center for Food Safety and Applied Nutrition. Laboratory Methods, Bacteriological Analytical Manual (BAM). Available online: https://www.fda.gov/Food/FoodScienceResearch/LaboratoryMethods/ucm2006949.htm (accessed on 17 May 2018).
- Horwitz, W. Official Methods of Analysis of AOAC International, 17th ed.; AOAC International: Gaithersburg, MD, USA, 2000. [Google Scholar]
- Freitas, A.; Barbosa, J.; Ramos, F. Multi-residue and multi-class method for the determination of antibiotics in bovine muscle by ultra-high-performance liquid chromatography tandem mass spectrometry. Meat Sci. 2014, 98, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Freitas, A.; Leston, S.; Rosa, J.J.J.; Castilho, M.; Barbosa, J.; Rema, P.; Pardal, M.Â.A.; Ramos, F. Multi-residue and multi-class determination of antibiotics in gilthead sea bream (Sparus aurata) by ultra high-performance liquid chromatography-tandem mass spectrometry. Food Addit. Contam. Part A 2014, 31, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Charitonos, S.; Samanidou, V.F.; Papadoyannis, I. Development of an HPLC-DAD Method for the Determination of Five Sulfonamides in Shrimps and Validation According to the European Decision 657/2002/EC. Food Anal. Methods 2017, 10, 2011–2017. [Google Scholar] [CrossRef]
- Konak, Ü.; Certel, M.; Şık, B.; Tongur, T. Development of an analysis method for determination of sulfonamides and their five acetylated metabolites in baby foods by ultra-high performance liquid chromatography coupled to high-resolution mass spectrometry (Orbitrap-MS). J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1057, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Du, J.; Chen, J.; Zhao, H. Determination of 19 antibiotic and 2 sulfonamide metabolite residues in wild fish muscle in mariculture areas of Laizhou Bay using accelerated solvent extraction and high performance liquid chromatography-tandem mass spectrometry. Se Pu Chin. J. Chromatogr. 2014, 32, 1320–1325. [Google Scholar] [CrossRef]
- Hoff, R.B.; Pizzolato, T.M.; Peralba, M.D.C.R.; Díaz-Cruz, M.S.; Barceló, D. Determination of sulfonamide antibiotics and metabolites in liver, muscle and kidney samples by pressurized liquid extraction or ultrasound-assisted extraction followed by liquid chromatography-quadrupole linear ion trap-tandem mass spectrometry (HPLC-QqL). Talanta 2015, 134, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, H.; Arnaudguilhem, C.; Jaber, F.; Lobinski, R. Multiresidue analysis of 22 sulfonamides and their metabolites in animal tissues using quick, easy, cheap, effective, rugged, and safe extraction and high resolution mass spectrometry (hybrid linear ion trap-Orbitrap). J. Chromatogr. A 2014, 1355, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Huertas-Pérez, J.F.; Arroyo-Manzanares, N.; Havlíková, L.; Gámiz-Gracia, L.; Solich, P.; García-Campaña, A.M. Method optimization and validation for the determination of eight sulfonamides in chicken muscle and eggs by modified QuEChERS and liquid chromatography with fluorescence detection. J. Pharm. Biomed. Anal. 2016, 124, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Shi, L.; Chu, X. Untargeted screening of sulfonamides and their metabolites in salmon using liquid chromatography coupled to quadrupole Orbitrap mass spectrometry. Food Chem. 2018, 239, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, H.; Zhang, J.; Liu, Y.; Wu, L. A novelty strategy for the fast analysis of sulfonamide antibiotics in fish tissue using magnetic separation with high-performance liquid chromatography–tandem mass spectrometry. Biomed. Chromatogr. 2016, 30, 1331–1337. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Huang, X.; Gu, R.; Hui, Y.; Tian, L.; Feng, B.; Zhang, X.; Yu, H. Determination of 14 sulfonamide residues in shrimps by high performance liquid chromatography coupled with post-column derivatization. Se Pu Chin. J. Chromatogr. 2014, 32, 874–879. [Google Scholar] [CrossRef]
- Freitas, A.; Barbosa, J.; Ramos, F. Multidetection of antibiotics in liver tissue by ultra-high-pressure-liquid-chromatography-tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2015, 976, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Liu, H.; Yan, Z. Analysis of sulfonamide residues in pork and chicken by high performance liquid chromatography coupled with solid-phase extraction using multiwalled carbon nanotubes as adsorbent. Se Pu Chin. J. Chromatogr. 2014, 32, 294–298. [Google Scholar] [CrossRef]
- Xia, L.; Liu, L.; Lv, X.; Qu, F.; Li, G.; You, J. Towards the determination of sulfonamides in meat samples: A magnetic and mesoporous metal-organic framework as an efficient sorbent for magnetic solid phase extraction combined with high-performance liquid chromatography. J. Chromatogr. A 2017, 1500, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Xie, M.; Huang, X.; Wu, H.; Zhu, Z.; Huang, F.; Lin, X.; Ouyang, G. Multiresidue analysis of 63 veterinary drugs in meat by dispersive solid-phase extraction and high performance liquid chromatography-tandem mass spectrometry. Chin. J. Chromatogr. 2015, 33, 354–362. [Google Scholar] [CrossRef]
- Wang, Z.; He, M.; Jiang, C.; Zhang, F.; Du, S.; Feng, W.; Zhang, H. Matrix solid-phase dispersion coupled with homogeneous ionic liquid microextraction for the determination of sulfonamides in animal tissues using high-performance liquid chromatography. J. Sep. Sci. 2015, 38, 4127–4135. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Xiao, X.; Li, G. Porous molecularly imprinted monolithic capillary column for on-line extraction coupled to high-performance liquid chromatography for trace analysis of antimicrobials in food samples. Talanta 2014, 123, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Chen, L.; Yuan, D. Development of monolith-based stir bar sorptive extraction and liquid chromatography tandem mass spectrometry method for sensitive determination of ten sulfonamides in pork and chicken samples. Anal. Bioanal. Chem. 2013, 405, 6885–6889. [Google Scholar] [CrossRef] [PubMed]
- Nebot, C.; Regal, P.; Miranda, J.M.; Fente, C.; Cepeda, A. Rapid method for quantification of nine sulfonamides in bovine milk using HPLC/MS/MS and without using SPE. Food Chem. 2013, 141, 2294–2299. [Google Scholar] [CrossRef] [PubMed]
- Arroyo-Manzanares, N.; Gámiz-Gracia, L.; García-Campaña, A.M. Alternative sample treatments for the determination of sulfonamides in milk by HPLC with fluorescence detection. Food Chem. 2014, 143, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Leung, D.; Chow, W.; Chang, J.; Wong, J.W. Development and Validation of a Multiclass Method for Analysis of Veterinary Drug Residues in Milk Using Ultrahigh Performance Liquid Chromatography Electrospray Ionization Quadrupole Orbitrap Mass Spectrometry. J. Agric. Food Chem. 2015, 63, 9175–9187. [Google Scholar] [CrossRef] [PubMed]
- Sereshti, H.; Khosraviani, M.; Sadegh Amini-Fazl, M. Miniaturized salting-out liquid-liquid extraction in a coupled-syringe system combined with HPLC-UV for extraction and determination of sulfanilamide. Talanta 2014, 121, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Cong, B.; Tan, Z.; Yan, Y. Synchronized separation, concentration and determination of trace sulfadiazine and sulfamethazine in food and environment by using polyoxyethylene lauryl ether-salt aqueous two-phase system coupled to high-performance liquid chromatography. Ecotoxicol. Environ. Saf. 2016, 133, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; Zhang, X.; Li, N.; Shi, J.; Zhang, H.; Wang, Z.; Zhang, H.; Yu, A.; Yu, Y. Ionic liquid-based aqueous two-phase system extraction of sulfonamides in milk. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 961, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.L.; Chen, G.; Zhu, L.; Yang, T.; Zhao, J.; Wang, L.; Wu, Y.L. Development and validation of an ultra high performance liquid chromatography tandem mass spectrometry method for simultaneous determination of sulfonamides, quinolones and benzimidazoles in bovine milk. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 962, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Kechagia, M.; Samanidou, V.; Kabir, A.; Furton, K.G. One-pot synthesis of a multi-template molecularly imprinted polymer for the extraction of six sulfonamide residues from milk before high-performance liquid chromatography with diode array detection. J. Sep. Sci. 2017, 41, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Ibarra, I.S.; Miranda, J.M.; Rodriguez, J.A.; Nebot, C.; Cepeda, A. Magnetic solid phase extraction followed by high-performance liquid chromatography for the determination of sulphonamides in milk samples. Food Chem. 2014, 157, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Tolmacheva, V.V.; Apyari, V.V.; Furletov, A.A.; Dmitrienko, S.G.; Zolotov, Y.A. Facile synthesis of magnetic hypercrosslinked polystyrene and its application in the magnetic solid-phase extraction of sulfonamides from water and milk samples before their HPLC determination. Talanta 2016, 152, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, X.; Li, Z.; Zhong, S.; Wang, W.; Wang, A.; Chen, J. Fabrication of CoFe2O4-graphene nanocomposite and its application in the magnetic solid phase extraction of sulfonamides from milk samples. Talanta 2015, 144, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Karageorgou, E.; Manousi, N.; Samanidou, V.; Kabir, A.; Furton, K.G. Fabric phase sorptive extraction for the fast isolation of sulfonamides residues from raw milk followed by high performance liquid chromatography with ultraviolet detection. Food Chem. 2016, 196, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Chatzimitakos, T.; Samanidou, V.; Stalikas, C.D. Graphene-functionalized melamine sponges for microextraction of sulfonamides from food and environmental samples. J. Chromatogr. A 2017, 1522, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Yan, H.; Sun, N. Water-compatible poly (hydroxyethyl methacrylate) polymer sorbent for miniaturized syringe assisted extraction of sulfonamides in milk. Anal. Chim. Acta 2013, 800, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Zulkoski, J.; Mastovska, K. Development and Validation of a Multiclass, Multiresidue Method for Veterinary Drug Analysis in Infant Formula and Related Ingredients Using UHPLC-MS/MS. J. Agric. Food Chem. 2017, 65, 7268–7287. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.L.G.; Romero-González, R.; Vidal, J.L.M.; Frenich, A.G. Analysis of veterinary drug residues in cheese by ultra-high-performance LC coupled to triple quadrupole MS/MS. J. Sep. Sci. 2013, 36, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Song, Y.; Hu, M.; Xu, X.; Zhang, H.; Yu, A.; Ma, Q.; Wang, Z. Determination of sulfonamides in butter samples by ionic liquid magnetic bar liquid-phase microextraction high-performance liquid chromatography. Anal. Bioanal. Chem. 2015, 407, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Summa, S.; Lo Magro, S.; Armentano, A.; Muscarella, M. Development and validation of an HPLC/DAD method for the determination of 13 sulphonamides in eggs. Food Chem. 2015, 187, 477–484. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Tan, L.; Wu, W.; Wang, J. Determination of sulfadiazine in eggs using molecularly imprinted solid-phase extraction coupled with high-performance liquid chromatography. J. Sep. Sci. 2016, 39, 2204–2212. [Google Scholar] [CrossRef] [PubMed]
- Sajid, M.; Na, N.; Safdar, M.; Lu, X.; Ma, L.; He, L.; Ouyang, J. Rapid trace level determination of sulfonamide residues in honey with online extraction using short C-18 column by high-performance liquid chromatography with fluorescence detection. J. Chromatogr. A 2013, 1314, 173–179. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Sulfonamide | Animal Species | MRL | Target Tissue |
|---|---|---|---|
| Commission Regulation (EU) No 37/2010 [7] | |||
| Sulfonamides | All food producing species | 100 μg/kg | Muscle Fat Liver Kidney |
| Bovine Ovine Caprine | 100 μg/kg | Milk | |
| Codex Alimentarius [10] | |||
| Sulfadimidine | Cattle | 25 μg/L | Milk |
| Not specified | 100 μg/kg | Muscle Liver Kidney Fat | |
| CFR—Code of Federal Regulations—U.S. Food & Drug Administration (FDA) [11] | |||
| Sulfabromomethazine sodium | Cattle | 100 μg/kg | Uncooked edible tissue |
| Not specified | 10 μg/L | Milk | |
| Sodium sulfachloropyrazine monohydrate | Chicken | 0 | Uncooked edible tissue |
| Sulfachlorpyridazine | Calves Swine | 100 μg/kg | Uncooked edible tissue |
| Sulfadimethoxine | Chickens Turkeys Cattle Ducks Salmonids Catfish Chukar partridges | 100 μg/kg | Uncooked edible tissue |
| Not specified | 10 μg/L | Milk | |
| Sulfaethoxypyridazine | Cattle | 100 μg/kg | Uncooked edible tissue |
| Swine | 0 | Uncooked edible tissue | |
| Not specified | 0 | Milk | |
| Sulfamerazine | Trout | 0 | Uncooked edible tissue |
| Sulfamethazine | Chickens Turkeys Cattle Swine | 100 μg/kg | Uncooked edible tissue |
| Sulfaquinoxaline | Chickens Turkeys Calves Cattle | 100 μg/kg | Uncooked edible tissue |
| Food Sample | Analytes | Sample Preparation | Analytical Technique/Run-Time | LOD-LOQ | Recovery (%) | Ref. |
|---|---|---|---|---|---|---|
| bovine tissue | 41 antibiotics (15 SAs) | SLE | UHPLC-MS/MS 12 min | CCα (μg/kg): 104–132 (SAs) CCβ (μg/kg): 108–164 (SAs) | 91–109 (SAs) | [31] |
| fish tissue | 41 antibiotics (15 SAs) | SLE | UHPLC-MS/MS 12 min | CCα (μg/kg): 110.6–126.9 (SAs) CCβ (μg/kg): 121.2–153.7 (SAs) | 92–111 (SAs) | [32] |
| shrimp tissue | SDZ, SMZ, SIX, SDMX and SQX | SLE | HPLC-DAD 40 min | LOD (μg/kg): 15 LOQ (μg/kg): 50 | 88.6–108.4 | [33] |
| baby foods (combinations of powdered milk, cereal, vegetable, honey and meat) | 15 SAs and metabolites | ASE | UHPLC-Orbitrap-MS 10 min | LOD (μg/kg): 0.03–0.17 LOQ (μg/kg): 0.10–0.55 | 75.5–96.6 | [34] |
| fish tissue | 19 antibiotics (9 SAs) | ASE | HPLC-MS/MS N/A | LOD (ng/g): 0.003–0.6 | 55.2–113 (all analytes) | [35] |
| chicken, sheep, fish and horse tissue | 16 SAs and metabolites | PLE, USE | HPLC-QqLIT-MS/MS 11 min | CCα (μg/kg): 111.2–161.4 (PLE), 119.3-142.7 (USE) CCβ (μg/kg): 122.4–222.8 (PLE), 138.6-185.5 (USE) | N/A | [36] |
| bovine, chicken, pork and sheep tissue | 22 SAs and metabolites | QuEChERS extraction | HPLC-HRMS 17 min | LOD (μg/kg): 3–26 LOQ (μg/kg): 11–88 CCα (μg/kg): 101–111 CCβ (μg/kg): 102–122 | 88–107 (beef muscle) | [37] |
| chicken tissue and egg | 8 SAs | QuEChERS extraction | HPLC-FLD 23 min | LOD (μg/kg): 5.8–19.9 (chicken muscle), 4.1–25.6 (egg) LOQ (μg/kg): 19.2–66.2 (chicken muscle), 13.6–85.4 (egg) | 66.9–86.8 (chicken muscle), 65.9–88.1 (egg) | [38] |
| salmon tissue | 27 SAs and metabolites | on-line QuEChERS extraction | UHPLC-ESI-Q-Orbitrap-MS 7 min | CCα (μg/kg): 0.04–1.34 CCβ (μg/kg): 0.07–2.33 | 83–109 | [39] |
| fish tissue | 8 SAs | SALLE and magnetic separation | HPLC-MS/MS 8 min | LOD (μg/kg): 2.5–10 LOQ (μg/kg): 5–25 | 74.87–104.74 | [40] |
| shrimp tissue | 14 SAs | SALLE | HPLC-FLD N/A | LOD (μg/kg): 1.0–5.0 | 77.8–103.6 | [41] |
| bovine tissue | 39 antibiotics (16 SAs) | SPE | UHPLC-MS/MS 12 min | CCα (μg/kg): 65–125 (SAs) CCβ (μg/kg): 81–150 (SAs) | 85–110 (SAs) | [42] |
| chicken and pork tissue | SAs | SPE | HPLC-UV N/A | LOD (mg/L): 0.003 LOQ (mg/L): 0.01 | > 70 | [43] |
| chicken, pork and shrimp tissue | SDZ, sulfathiazole, sulfamerazine, SMZ and SMP | MSPE | HPLC-DAD 16 min | LOD (ng/g): 1.73–5.23 LOQ (ng/g): 3.97–15.89 | 81.4–101.3 (chicken), 76.1–102.6 (pork), 79.2–102.5 (shrimp) | [44] |
| animal tissue | 63 veterinary drugs (21 SAs) | DSPE | HPLC-MS/MS N/A | LOD (μg/kg): 0.1–3.0 (all analytes) LOQ (μg/kg): 0.5–10.0 (all analytes) | 62.2–112.0 (all analytes) | [45] |
| bovine, chicken and pork tissue | 7 SAs | MSPD-HILME | HPLC-DAD 30 min | LOD (μg/kg): 4.3–13.4 LOQ (μg/kg): 14.2–44.8 | 85.4–95.1 (kidney), 104.5–118.0 (liver), 85.5–112.3 (muscle) | [46] |
| chicken and pork tissue and egg | 5 antimicrobials (sulfametoxydiazine, sulfamethoxazole and SQX) | on-line SPME | HPLC-UV 20 min | LOD (μg/L): 0.10–0.14 (SAs) LOQ (μg/L): 0.39–0.47 (SAs) | 84.1–99.6 (SAs in chicken), 87.0–108.2 (SAs in pork), 80.1–101.4 (SAs in egg) | [47] |
| chicken and pork tissue | 10 SAs | SBSE | HPLC-MS/MS N/A | LOD (μg/kg): 0.0012–0.0061 (pork), 0.0021–0.0146 (chicken) LOQ (μg/kg): 0.0040–0.0203 (pork), 0.0066–0.0487 (chicken) | 62.4–109.9 (pork), 55.2–109.1 (chicken) | [48] |
| Food Sample | Analytes | Sample Preparation | Analytical Technique/Run-Time | LOD-LOQ | Recovery (%) | Ref. |
|---|---|---|---|---|---|---|
| milk | 9 SAs | LLE | HPLC-MS/MS 30 min | LOQ (μg/kg): 12.5–45 CCα (μg/kg): 106–122 CCβ (μg/kg): 112–145 | 89–105 | [49] |
| milk | 9 SAs | DLLME, QuEChERS extraction | HPLC-FLD 15 min | LOD (μg/L): 0.60–1.21 (DLLME), 1.15–2.73 (QuEChERS) LOQ (μg/L): 2.01–4.02 (DLLME), 3.85–9.09 (QuEChERS) | 90.8–104.7 (DLLME), 83.6–104.8 (QuEChERS) | [50] |
| milk | 105 veterinary drugs (26 SAs) | SALLE and SPE | UHPLC-ESI-Q-Orbitrap-MS 14 min | LOQ (μg/kg): 1.0 (all analytes) | 71–120 (all analytes) | [51] |
| tea beverage, water, milk, honey, plasma, blood and urine | sulfonamide | miniaturized SALLE | HPLC-UV 7 min | LOD (ng/mL): 0.3 LOQ (ng/mL): 1.0 | 96.66 (tea beverage), 76.67 (milk), 43.33 (honey) | [52] |
| milk, egg and water | SDZ and SMZ | ATPS extraction | HPLC-UV N/A | LOD (pg/mL): 2.92–3.64 (milk), 2.90–3.49 (egg) LOQ (pg/mL): 9.73–12.15 (milk), 9.66–11.62 (egg) | 97.14–99.52 (milk), 96.90–99.30 (egg) | [53] |
| milk | 6 SAs | ATPS extraction | HPLC-UV 30 min | LOD (ng/mL): 2.04–2.84 LOQ (ng/mL): 6.73–9.37 | 72.32–108.96 | [54] |
| milk | 38 veterinary drugs (18 SAs) | SPE | UHPLC-ESI-MS/MS 13 min | CCα (μg/kg): 109–114 (SAs) CCβ (μg/kg): 116–123 (SAs) | 87–119 (all analytes) | [55] |
| milk | 6 SAs | SPE | HPLC-DAD 15.3 min | LOD (μg/kg): 1.9–13.3 LOQ (μg/kg): 5.6–42.2 CCα (μg/kg): 101.9–113.5 CCβ (μg/kg): 114.4–135.4 | N/A | [56] |
| milk | 9 SAs | MSPE | HPLC-DAD 35 min | LOD (μg/L): 7–14 CCα (μg/kg): 108.86–117.16 CCβ (μg/kg): 117.73–134.32 | 81.88–114.98 | [57] |
| milk and water | SMP, SMZ, sulfamethoxazole and sulfachloropyridazine | MSPE | HPLC-AD N/A | LOD (ng/mL): 2.0–2.5 (milk) LOQ (ng/mL): 6.0–7.5 (milk) | 92–105 (milk) | [58] |
| milk | 5 SAs | MSPE | HPLC-UV 8 min | LOD (μg/L): 1.16–1.59 LOQ (μg/L): 3.52–4.81 | 62.0–104.3 | [59] |
| milk | SMZ, SIX and SDMX | FPSE | HPLC-UV 6.5 min | CCα (μg/kg): 114.4–116.5 CCβ (μg/kg): 104.1–118.5 | 93–107 | [60] |
| milk, egg and water | 8 SAs | GMeS microextraction | HPLC-DAD 30 min | LOQ (μg/kg): 0.31–0.91 (milk), 0.96–1.32 (egg) | 90–105 (milk), 90–108 (egg) | [61] |
| milk | SDZ and sulfamonomethoxine | mini-SAE | HPLC-FLD N/A | LOD (ng/g): 0.19–0.26 LOQ (ng/g): 0.67–0.87 | 85.6–100.3 | [62] |
| baby formula | 150 veterinary drugs (24 SAs) | SLE | UHPLC-MS/MS 17.5 min | LOQ (ng/g): 1–10 (all analytes) | 50–120 (all analytes) | [63] |
| cheese | 17 veterinary drugs (sulfachloropyridazine, sulfadimidine, SDMX and SQX) | QuEChERS extraction | UHPLC-MS/MS 8.5 min | LOD (μg/kg): 0.2–1.7 (SAs) LOQ (μg/kg): 0.7–5.5 (SAs) CCα (μg/kg): 3.4–5.8 (SAs) CCβ (μg/kg): 5.7–10.2 (SAs) | 72.5–106.3 (SAs) | [64] |
| butter | 8 SAs | IL-MB-LPME | HPLC-UV 30 min | LOD (μg/kg): 1.20–2.17 LOQ (μg/kg): 4.00–7.25 | 73.25–103.85 | [65] |
| egg | 13 SAs | SPE | HPLC-DAD 45 min | LOD (μg/kg): 0.30–1.29 LOQ (μg/kg): 0.92–3.92 CCα (μg/kg): 11.3–18.5 CCβ (μg/kg): 13.2–27.3 | 45.2–87.5 | [66] |
| egg | SDZ | SPE | HPLC-DAD N/A | LOD (μg/L): 0.06 (egg yolk), 0.05 (egg white) LOQ (μg/L): 0.20 (egg yolk), 0.17 (egg white) | 78.22–86.10 | [67] |
| honey | 15 SAs | on-line SPE | HPLC-FLD 30 min | LOD (ng/g): 0.1–1.0 | 76–108 | [68] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bitas, D.; Kabir, A.; Locatelli, M.; Samanidou, V. Food Sample Preparation for the Determination of Sulfonamides by High-Performance Liquid Chromatography: State-of-the-Art. Separations 2018, 5, 31. https://doi.org/10.3390/separations5020031
Bitas D, Kabir A, Locatelli M, Samanidou V. Food Sample Preparation for the Determination of Sulfonamides by High-Performance Liquid Chromatography: State-of-the-Art. Separations. 2018; 5(2):31. https://doi.org/10.3390/separations5020031
Chicago/Turabian StyleBitas, Dimitrios, Abuzar Kabir, Marcello Locatelli, and Victoria Samanidou. 2018. "Food Sample Preparation for the Determination of Sulfonamides by High-Performance Liquid Chromatography: State-of-the-Art" Separations 5, no. 2: 31. https://doi.org/10.3390/separations5020031
APA StyleBitas, D., Kabir, A., Locatelli, M., & Samanidou, V. (2018). Food Sample Preparation for the Determination of Sulfonamides by High-Performance Liquid Chromatography: State-of-the-Art. Separations, 5(2), 31. https://doi.org/10.3390/separations5020031