
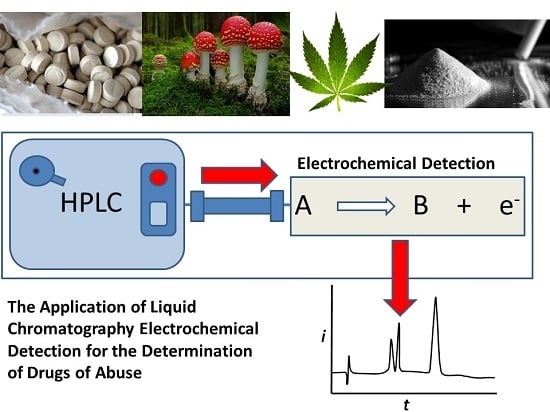
Review: The Application of Liquid Chromatography Electrochemical Detection for the Determination of Drugs of Abuse

Abstract
:
1. Introduction
2. Principles of Operation and Practical Considerations
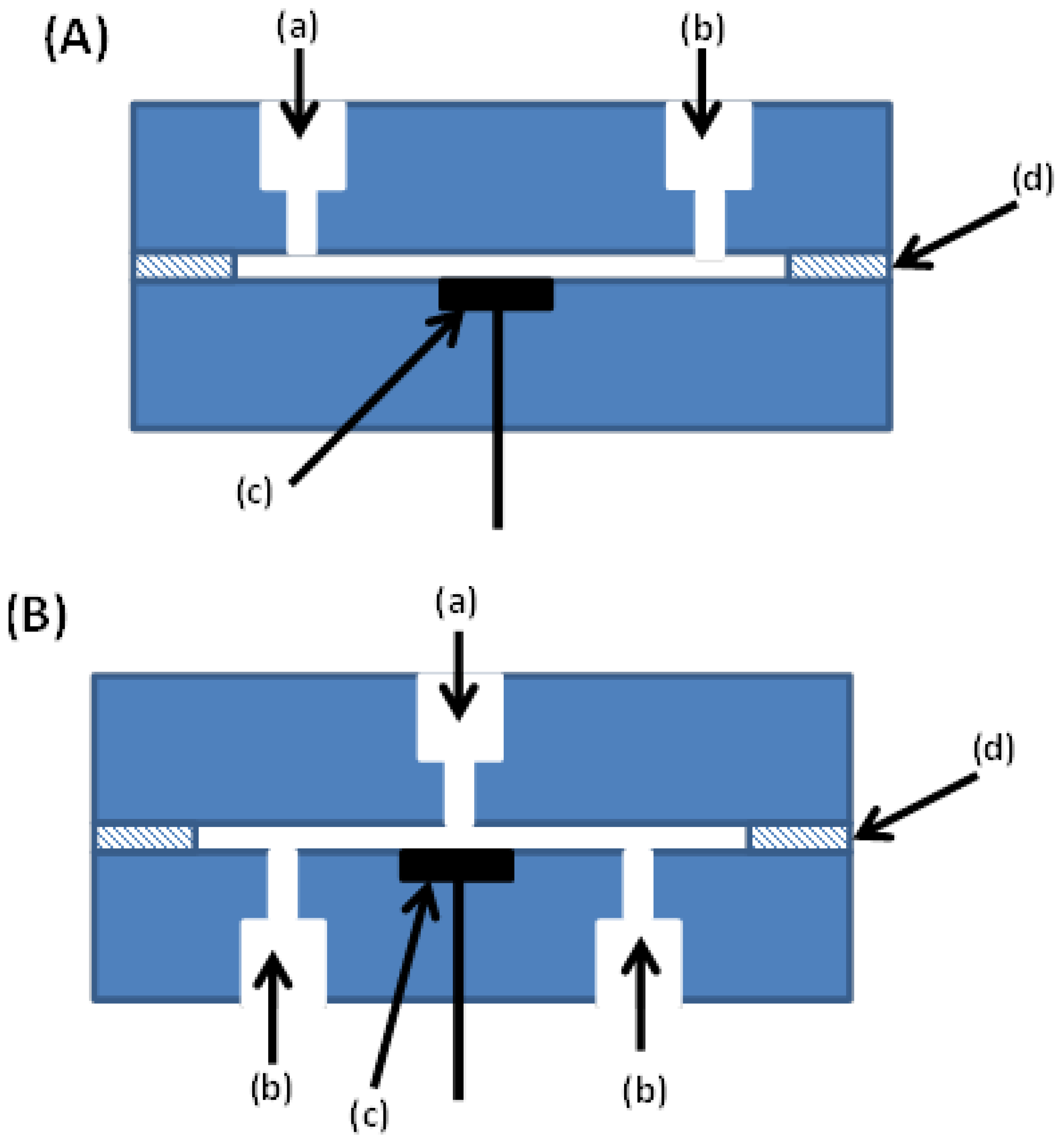
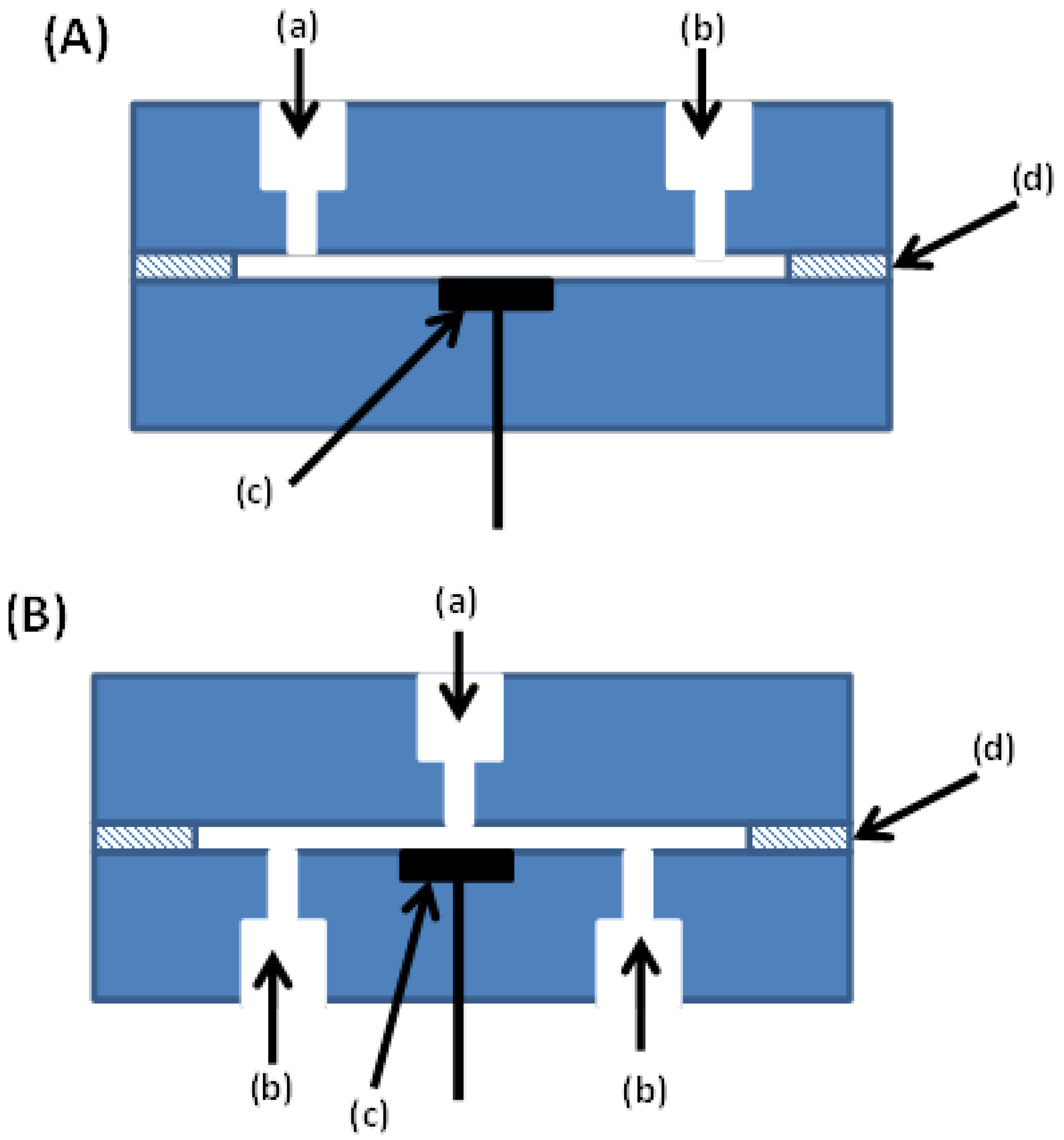
2.1. Thin Layer Cell Amperometric Detector
2.2. Wall-Jet Amperometric Detector
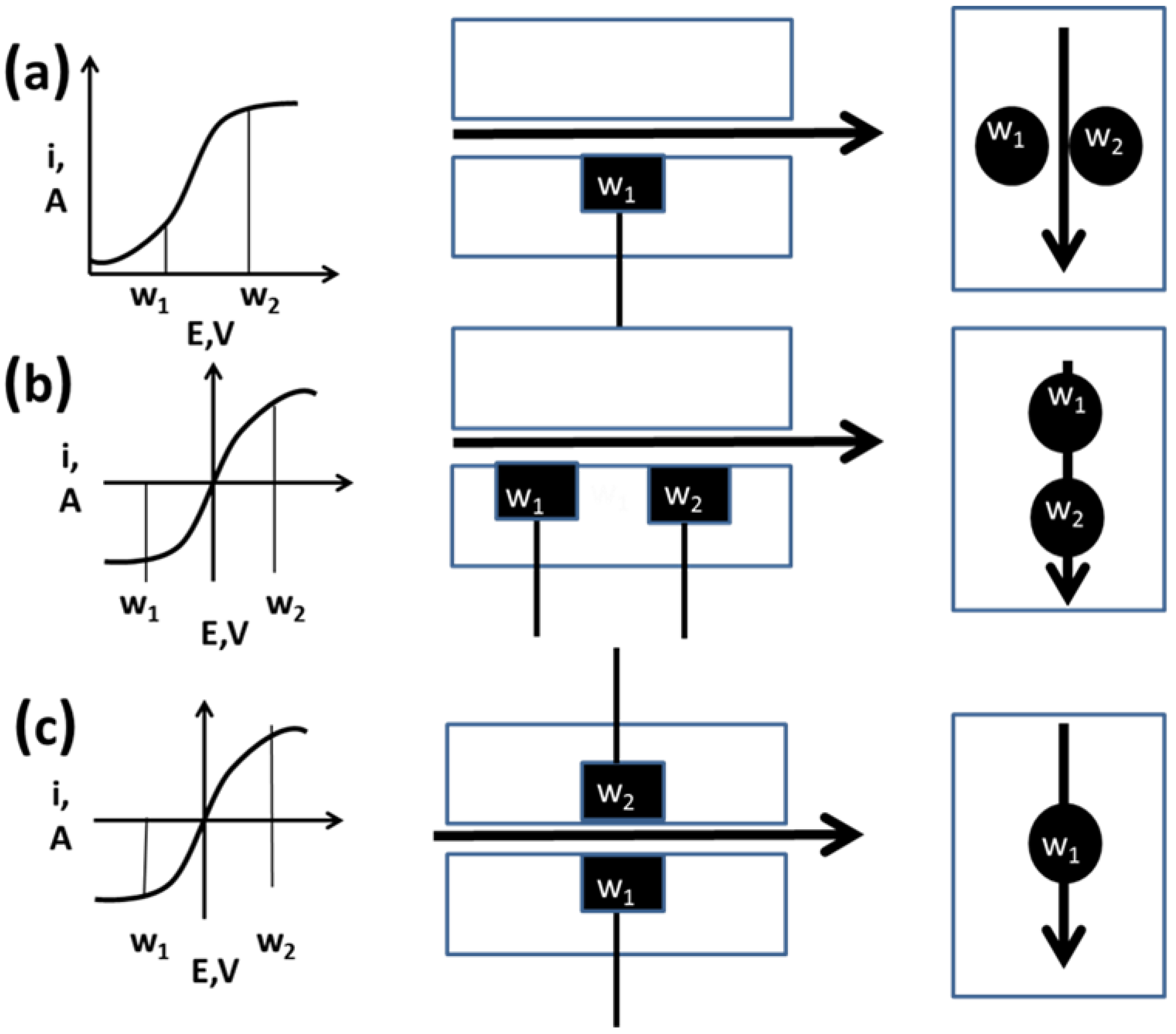
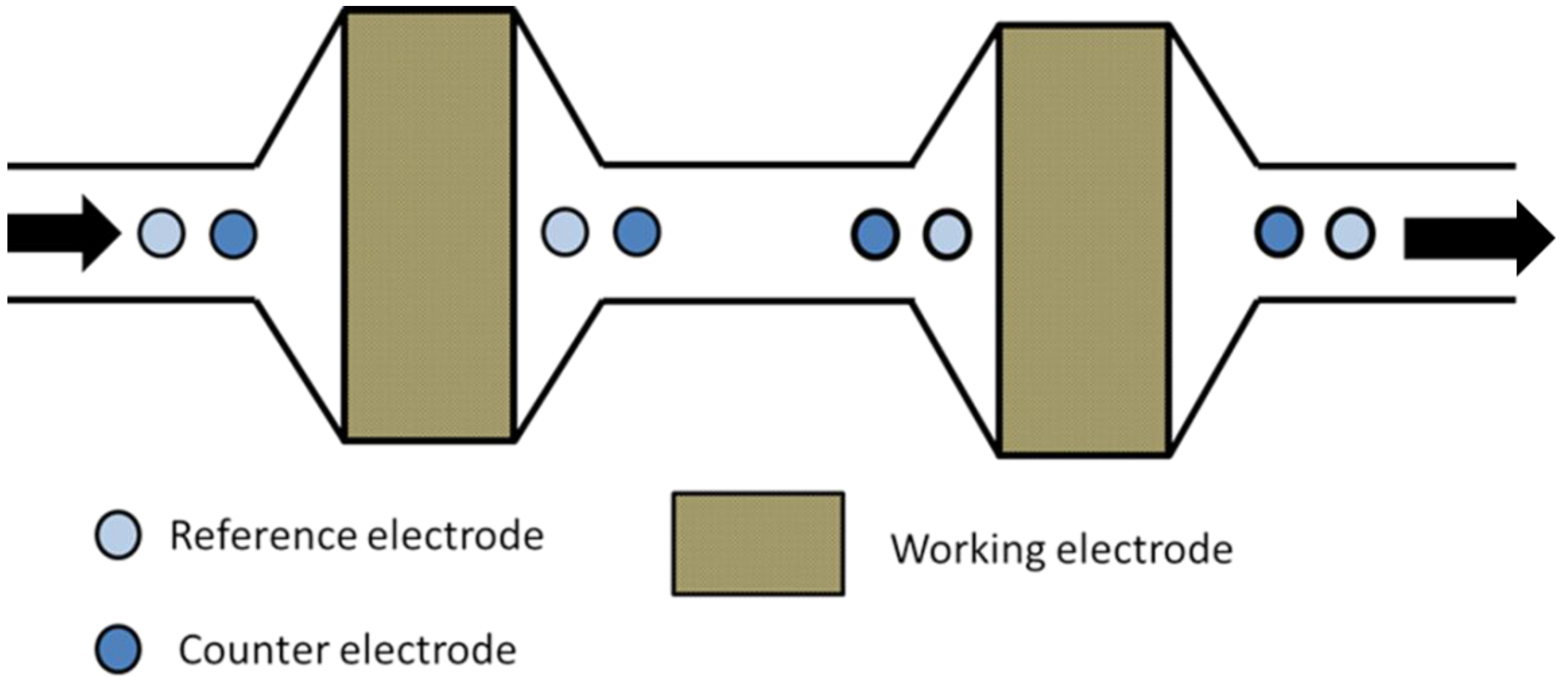
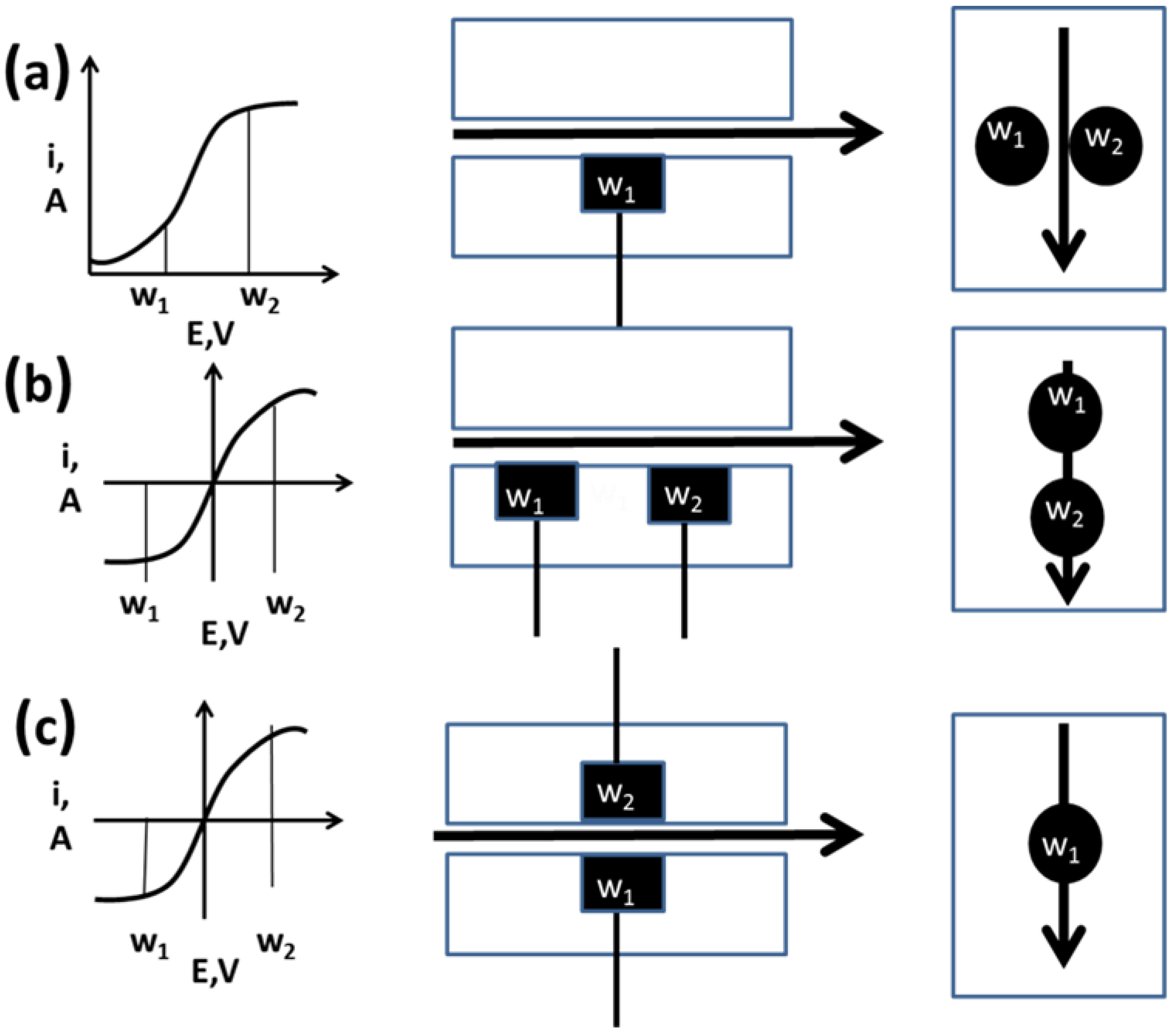
2.3. Multi-Electrode Amperometric Detectors
2.4. Pulse Amperometric Detection
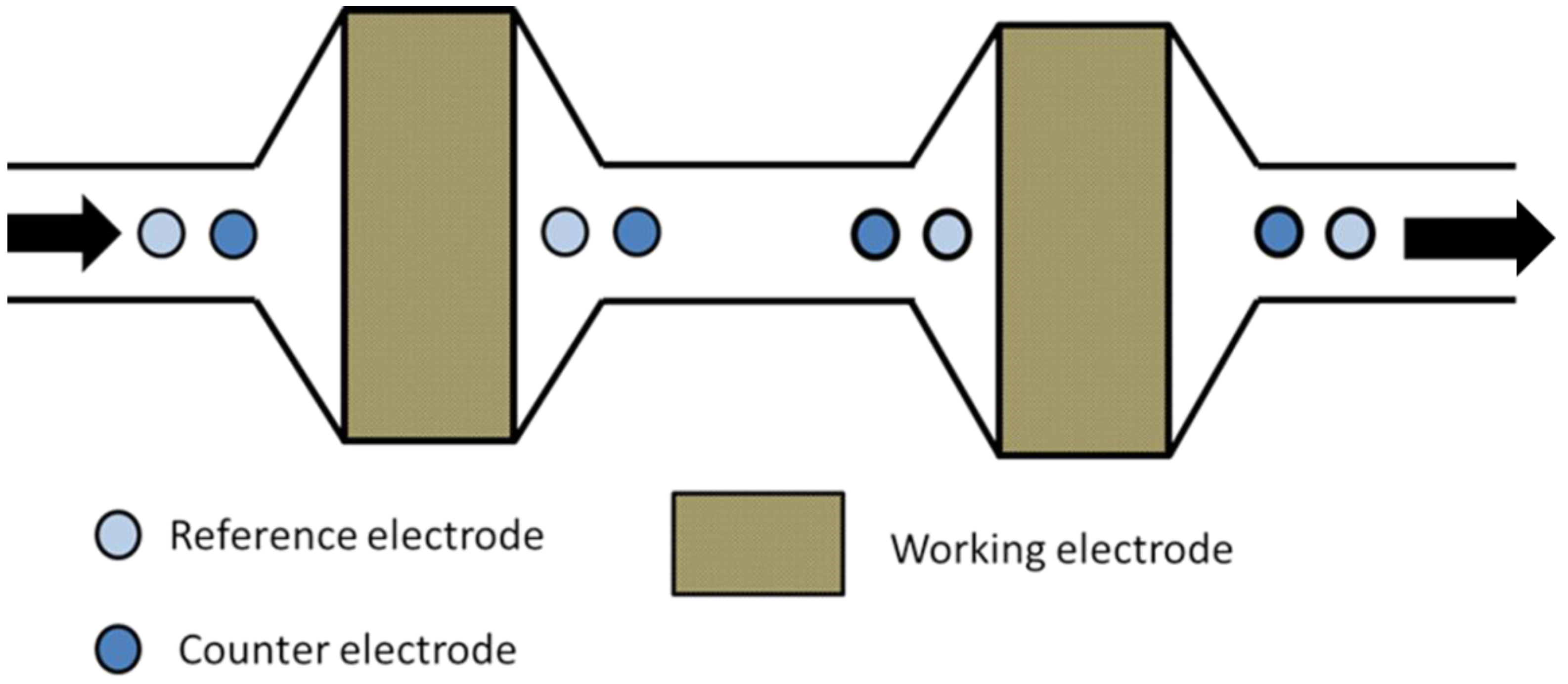
2.5. Coulometric Detectors
3. Applications
3.1. Cannabinoids
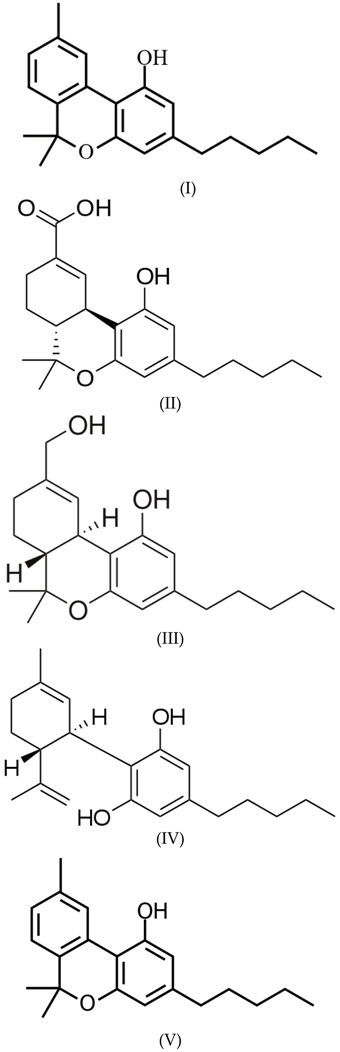
3.2. Ethanol
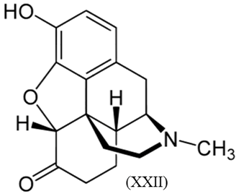
3.3. Alkaloids
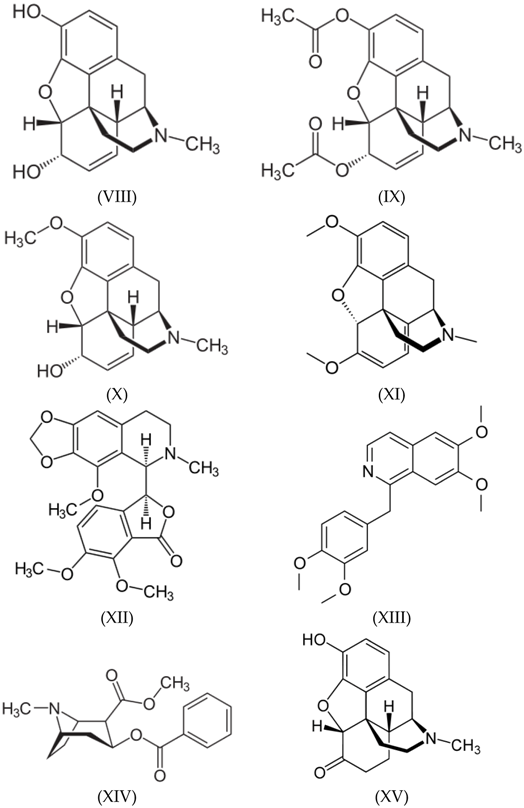
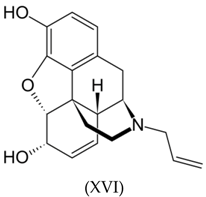
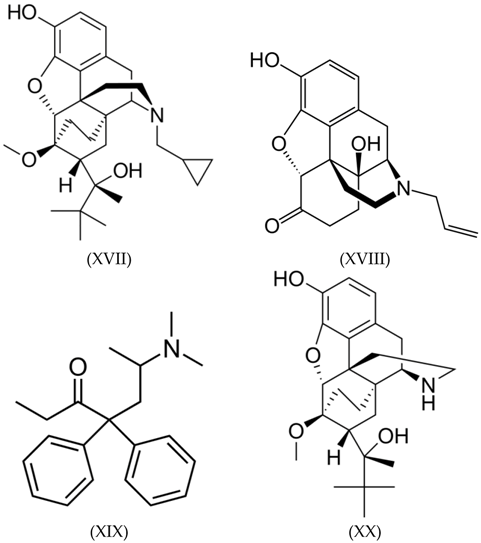
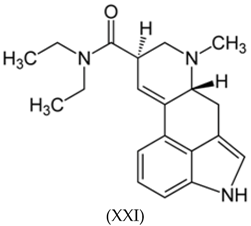
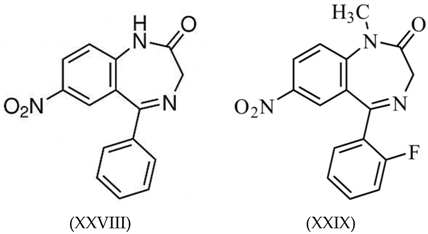
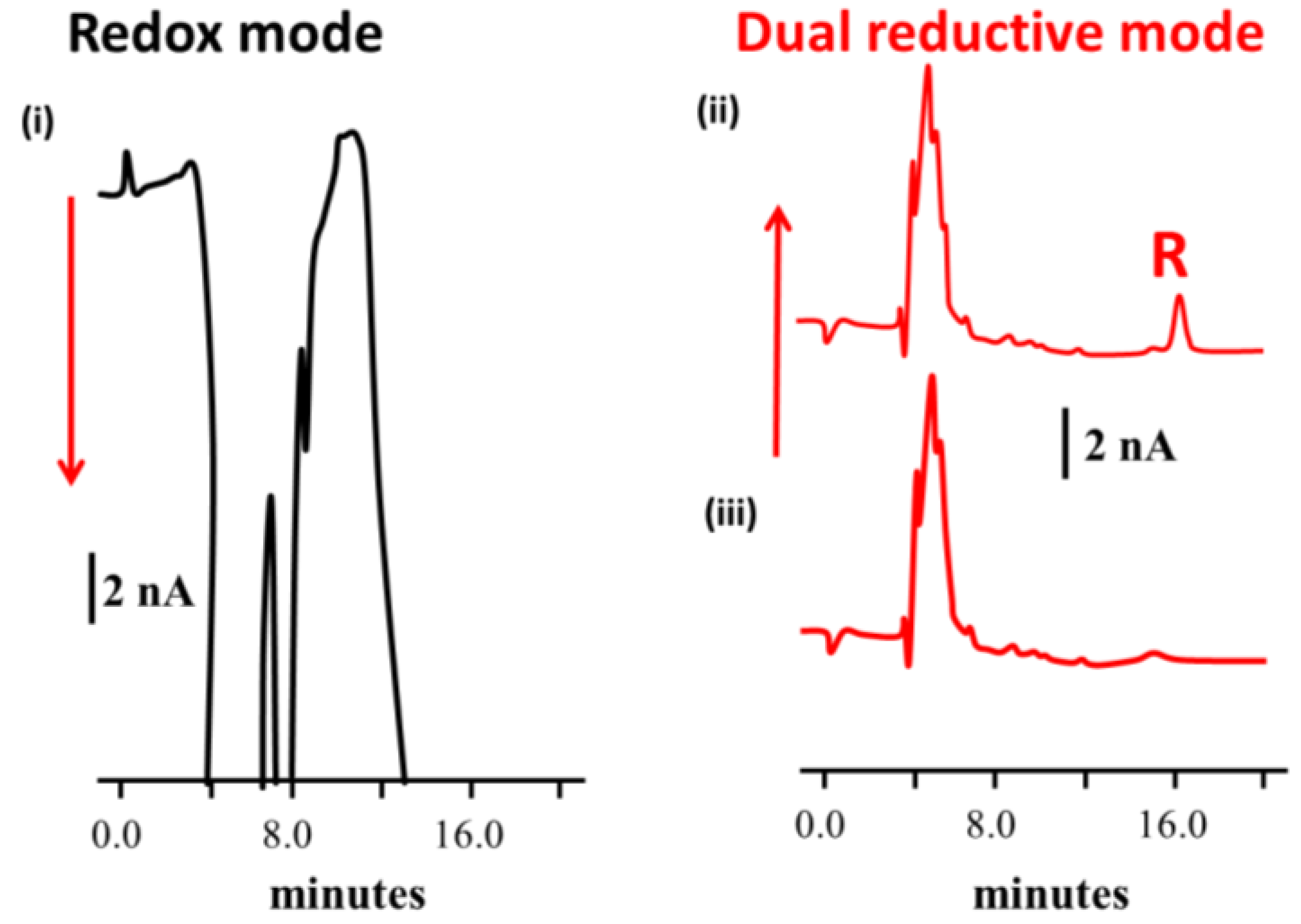
3.4. Benzodiazepines
3.5. Amphetamines
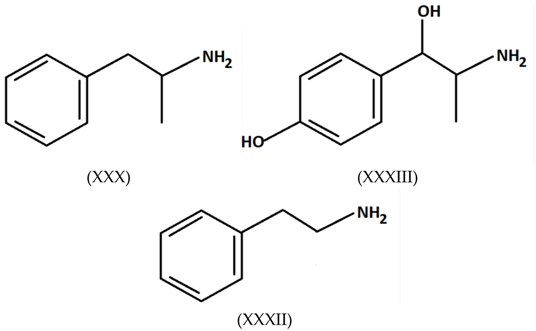
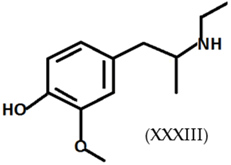
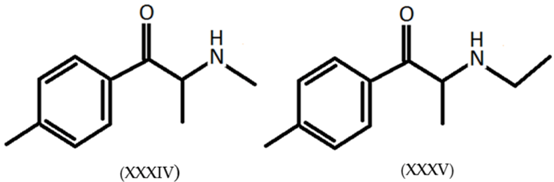
3.6. Legal Highs: Mephedrone and 4-Methylethcathinone
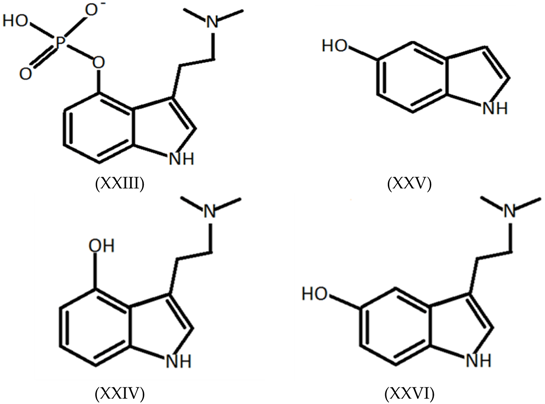
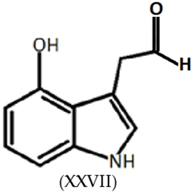
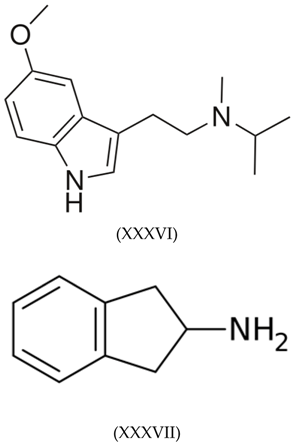
3.7. Tryptamines, Phenethylamines and Piperazines
4. Comparisons with Other Liquid Chromatographic Detection Systems
5. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| 2,5-DBA | 2,5-dihydroxybenzaldehyde |
| 4HIAA | 4-hydroxyindole-3-acetic acid |
| 5-MeO-MIPT | N-[2-(5-methoxy-1H-indol-3-yl)ethyl]-N-methylpropan-2-amine |
| Ag/AgCl | Silver/silver chloride |
| BAS | Bioanalytical Systems, Inc. |
| EtG | Ethyl glucuronide |
| GCE | Glassy carbon electrode |
| GC/MS | Gas chromatography mass spectrometry |
| HILIC | Hydrophilic interaction chromatography |
| HMEA | N-ethyl-4-hydroxy-3-methoxy-amphetamine |
| HPLC EA | High performance liquid chromatography electrochemical array detection |
| HPLC-UV | High performance liquid chromatography-ultra violet detection |
| IDU | Intravenous drug user |
| LC-DED | Liquid chromatography dual electrode detection |
| LC ED | Liquid chromatography electrochemical detection |
| LC/MS | Liquid chromatography mass spectrometry |
| LC-UV | Liquid chromatography ultraviolet detection |
| LSD | Lysergic acid diethylamide |
| MDE | Methylenedioxyethylamphetamine |
| MECD | Multichannel electrochemical detection |
| Na2EDTA | Disodium ethylenediaminetetraacetic acid |
| NRG-2 | 4-Methylethcathinone legal high marketed alone or in mixtures with other substituted cathinones |
| PBS | Phosphate buffered saline |
| PdH2 | Palladium-hydrogen reference electrode |
| PED | Pulsed electrochemical detection |
| PIA | Pulsed integrated amperometry |
| PHE | Phenylethylamine |
| S/N | Signal-to-noise ratio |
| SPE | Solid-phase extraction |
| THC | Tetrahydrocannabinol |
| THC–COOH | 11-nor-Δ9-tetrahydrocannabinol-9-carboxylic acid |
| TLC | Thin-layer cell |
References
- Johnson, M.W.; Garcia-Romeu, A.; Cosimano, M.P.; Griffiths, R.R. Pilot study of the 5-HT2AR agonist psilocybin in the treatment of tobacco addiction. J. Psychopharmacol. 2014, 28, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Frood, A. LSD helps to treat alcoholism. Retrospective analysis shows hallucinogenic drug helped problem drinkers. Nature 2012, 483. [Google Scholar] [CrossRef]
- Sewell, R.A.; Halpern, J.H.; Pope, H.G. Response of cluster headache to psilocybin and LSD. Neurology 2006, 66, 1920–1922. [Google Scholar] [CrossRef] [PubMed]
- Novotna, A.; Mares, J.; Ratcliffe, S.; Novakova, I.; Vachova, M.; Zapletalova, O.; Gasperini, C.; Pozzilli, C.; Cefaro, L.; Comi, G.; et al. A randomized, double-blind, placebo-controlled, parallel-group, enriched-design study of nabiximols (Sativex®), as add-on therapy, in subjects with refractory spasticity caused by multiple sclerosis. Eur. J. Neurol. 2011, 18, 1122–1131. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-W. Simultaneous Screening of 177 Drugs of Abuse in Urine Using Ultra-Performance Liquid Chromatography with Tandem Mass Spectrometry in Drug-Intoxicated Patients. Clin. Psychopharmacol. Neurosci. 2013, 11, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Saito, R.; Kikuchi, Y.; Iwasaki, Y.; Ito, R.; Nakazawa, H. Analysis of Drugs of Abuse in Biological Specimens. J. Health Sci. 2011, 57, 472–487. [Google Scholar] [CrossRef]
- Arora, B.; Velpandian, T.; Saxena, R.; Lalwani, S.; Dogra, T.D.; Ghose, S. Development and validation of an ESI-LC-MS/MS method for simultaneous identification and quantification of 24 analytes of forensic relevance in vitreous humour, whole blood and plasma. Drug Test. Anal. 2016, 8, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Xiang, P.; Shen, M.; Drummer, O.H. Review: Drug concentrations in hair and their relevance in drug facilitated crimes. J. Forensic Leg. Med. 2015, 36, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Tagliaro, F.; Franchi, D.; Dorizzi, R.; Marigo, M. High-performance liquid chromatographic determination of morphine in biological samples: An overview of separation methods and detection techniques. J. Chromatogr. A 1989, 488, 215–228. [Google Scholar] [CrossRef]
- Weber, S.G.; Purdy, W.C. Electrochemical Detectors in Liquid Chromatography. A Short Review of Detector Design. Ind. Eng. Chem. Prod. Res. Dev. 1981, 20, 593–598. [Google Scholar] [CrossRef]
- Flanagan, R.J.; Perrett, D.; Whelpton, R. Electrochemical Detection in HPLC: Analysis of Drugs and Poisons; Royal Society of Chemistry: Cambridge, UK, 2005. [Google Scholar]
- Kissinger, P.T. Electrochemical Detection in Liquid Chromatography and Flow Injection Analysis. In Laboratory Techniques in Electroanalytical Chemistry; Kissinger, P.T., Heineman, W.R., Eds.; Marcel Dekker: New York, NY, USA, 1984; pp. 611–635. [Google Scholar]
- Štulík, K.; Pacáková, V. Electroanalytical Measurements in Flowing Liquids; Ellis Horwood: Chichester, UK, 1987. [Google Scholar]
- Tóth, K.; Štulík, K.; Kutner, W.; Fehér, Z.; Lindner, E. Electrochemical Detection in Liquid Flow Analytical Techniques: Characterization and Classification, (IUPAC Technical Report). Pure Appl. Chem. 2004, 76, 1119–1138. [Google Scholar] [CrossRef]
- Trojanowicza, M. Recent developments in electrochemical flow detection—A review Part I. Flow analysis and capillary electrophoresis. Anal. Chim. Acta 2009, 653, 36–58. [Google Scholar] [CrossRef] [PubMed]
- Kissinger, P.T.; Refshauge, C.; Dreiling, R.; Adams, R.N. An Electrochemical Detector for Liquid Chromatography with Picogram Sensitivity. Anal. Lett. 1973, 6, 465–477. [Google Scholar] [CrossRef]
- Fleet, E.; Little, C.J. Design and Evaluation of Electrochemical Detectors for HPLC. J. Chromatogr. Sci. 1974, 72, 747–752. [Google Scholar] [CrossRef]
- Honeychurch, K.C. Design and Application of Liquid Chromatography Dual Electrode Detection. In Electrochemistry; Royal Society of Chemistry: Cambridge, UK, 2015; Volume 13, pp. 1–20. [Google Scholar]
- Chao, M.-H.; Huang, H.-J. A Bipotentiostat Based 4-Electrode Detection System for HPLC Analysis. J. Chin. Chem. Soc. 2001, 48, 763–768. [Google Scholar] [CrossRef]
- Honeychurch, K.C.; Smith, G.C.; Hart, J.P. Voltammetric Behavior of Nitrazepam and Its Determination in Serum Using Liquid Chromatography with Redox Mode Dual-Electrode Detection. Anal. Chem. 2006, 78, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Honeychurch, K.C.; Davidson, G.; Brown, E.; Hart, J.P. Novel reductive-reductive mode electrochemical detection of Rohypnol following liquid chromatography and its determination in coffee. Anal. Chim. Acta 2015, 853, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Honeychurch, K.C.; Hart, J.P. Voltammetric behaviour of p-nitrophenol and its trace determination in human urine by liquid chromatography with a dual reductive mode electrochemical detection system. Electroanalysis 2007, 19, 2176–2184. [Google Scholar] [CrossRef]
- Evans, O. On-line deoxygenation in reductive (and oxidative) amperometric detection: Environmental applications in the liquid chromatography of organic peroxides. Analyst 1999, 124, 1811–1816. [Google Scholar] [CrossRef]
- LaCourse, W.R. Pulsed Electrochemical Detection in High-Performance Liquid Chromatography; John Wiley & Sons: New York, NY, USA, 1997. [Google Scholar]
- Johnson, D.C.; Dobberpuhl, D.; Roberts, R.; Vanderberg, P. Pulsed amperometric detection of carbohydrates, amines, and sulfur species in ion chromatography: The current state of research. J. Chromatogr. 1993, 640, 79–96. [Google Scholar] [CrossRef]
- Jensen, M.B.; Johnson, D.C. Fast wave forms for pulsed electrochemical detection of glucose by incorporation of reductive desorption of oxidation products. Anal. Chem. 1997, 69, 1776–1781. [Google Scholar] [CrossRef] [PubMed]
- Martens, D.A.; Frankenberger, W.T., Jr. Determination of saccharides by high performance anion-exchange chromatography with pulsed amperometric detection. Chromatographia 1990, 29, 7–12. [Google Scholar] [CrossRef]
- Acworth, I.N.; Bowers, M. An Introduction to HPLC-Based Electrochemical Detection: From Single Electrode to Multi-Electrode Arrays. In Coulometric Electrode Array Detectors for HPLC (Progress in HPLC-HPCE), 6th ed.; Acworth, I.N., Naoi, M., Parvez, S., Parvez, H., Eds.; VSP Publications: Utrecht, The Netherlands, 1997; pp. 3–50. [Google Scholar]
- Svendsen, C.N. Tutorial review—Multi-electrode array detectors in high-performance liquid chromatography: A new dimension in electrochemical analysis. Analyst 1993, 118, 123–129. [Google Scholar] [CrossRef]
- Acworth, I.N.; Gamache, P. The coulometric electrode array for use in HPLC analysis. Part 1. Theory. Am. Lab. 1996, 5, 33–38. [Google Scholar]
- Acworth, I.N.; Waraska, J.; Gamache, P. The coulometric electrode array for use in HPLC analysis. Part 2: An application overview. Am. Lab. 1997, 11, 25–32. [Google Scholar]
- Raharjo, T.J.; Verpoorte, R. Methods for the Analysis of Cannabinoids in Biological Materials: A Review. Phytochem. Anal. 2004, 15, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, Y.; Sekine, H. Studies on Confirmation of Cannabis Use. I. Determination of the Cannabinoid Contents in Marijuana Cigarette, Tar, and Ash Using High Performance Liquid Chromatography with Electrochemical Detection. J. Anal. Toxicol. 1985, 9, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Harvey, D.J. Cannabinoids. In Mass Spectrometry, Clinical and Biomedical Applications; Springer: New York, NY, USA, 1992; pp. 207–257. [Google Scholar]
- Nyoni, E.C.; Sitaram, B.R.; Taylor, D.A. Determination of Δ9-tetrahydrocannabinol levels in brain tissue using high-performance liquid chromatography with electrochemical detection. J. Chromatogr. B 1996, 679, 79–84. [Google Scholar] [CrossRef]
- Bourquin, D.; Brenneisen, R. Confirmation of cannabis abuse by the determination of 11-nor-Δ9-tetrahydrocannabinol-9-carboxylic acid in urine with high performance liquid chromatography and electrochemical detection. J. Chromatogr. 1987, 414, 187–191. [Google Scholar] [CrossRef]
- ElSohly, M.A.; ElSohly, H.N.; Jones, A.B.; Dimson, P.A.; Wells, K.E. Analysis of the major metabolite of delta 9-tetrahydrocannabinol in urine II. A HPLC procedure. J. Anal. Toxicol. 1983, 7, 262–264. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, Y.; Sekine, H.; Cook, C.E. Confirmation of Cannabis Use II. Determination of Tetrahydrocannabinol Metabolites in Urine and Plasma by HPLC with ECD. J. Anal. Toxicol. 1989, 13, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, Y.; Sekine, H. Automatic extraction using the minicolumn liquid chromatography method for analysis of drugs of abuse in biological fluids. Chromatographia 1984, 19, 131–136. [Google Scholar] [CrossRef]
- Baker, T.S.; Harry, J.V.; Russell, J.W.; Myers, R.L. Rapid method for the GC/MS confirmation of 11-nor-Δ9-THC-9-COOH in urine. J. Anal. Toxicol. 1984, 8, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.H.; Broudy, M.I.; Fisher, L.M. Quantification of 9-Carboxy-11-nor-Δ9-tetrahydrocannabinol in Urine Using Brominated 9-Carboxy-11–nor-Δ9-tetrahydrocannabinol as the Internal Standard and High-Performance Liquid Chromatography with Electrochemical Detection. Biomed. Chromatogr. 1996, 10, 161–166. [Google Scholar] [CrossRef]
- Nakahara, Y.; Tanaka, K. Studies on discrimination of confiscated cannabis products by high performance liquid chromatography with electrochemical detector. Bull. Natl. Inst. Hygenic Sci. 1988, 106, 11–18. [Google Scholar]
- Kokubun, H.; Uezono, Y.; Matoba, M. Novel method of determination of Δ9-tetrahydrocannabinol (THC) in human serum by high-performance liquid chromatography with electrochemical detection. Gan Kagaku Ryoho 2014, 41, 471–473. [Google Scholar]
- Kaushik, R.; LaCourse, W.R.; Levine, B. Determination of ethyl glucuronide in urine using reversed-phase HPLC and pulsed electrochemical detection (Part II). Anal. Chim. Acta 2006, 556, 267–274. [Google Scholar] [CrossRef]
- Johnson, D.C.; LaCourse, W.R. Liquid Chromatography with Pulsed Electrochemical Detection at Gold and Platinum Electrodes. Anal. Chem. 1990, 62, 589A–597A. [Google Scholar] [CrossRef]
- Schwartz, R.S.; David, K.O. Liquid Chromatography of Opium Alkaloids, Heroin, Cocaine, and Related Compounds Using Electrochemical Detection. Anal. Chem. 1985, 57, 1362–1366. [Google Scholar] [CrossRef]
- Masui, M.; Sayo, H.; Tsuda, Y. Anodic oxidation of amines. Part I. Cyclic voltammetry of aliphatic amines at a stationary glassy-carbon electrode. J. Chem. Soc. B 1968, 9, 973–976. [Google Scholar] [CrossRef]
- Masui, M.; Sayo, H. Anodic oxidation of amines. Part II. Electrochemical dealkylation of aliphatic tertiary amines. J. Chem. Soc. B 1971, 1971. [Google Scholar] [CrossRef]
- Sayo, H.; Masui, M. Anodic-oxidation of amines part III. Cyclic voltammetry and controlled potential electrolysis of 4-dimethylaminoantipyrine (4-dimethylamino-2,3-dimethyl-1-phenyl-Δ-3-pyrazolin-5-one) in acetonitrile. J. Chem. Soc. Perkin Trans. 1973, 2, 1640–1645. [Google Scholar] [CrossRef]
- Mann, C.K. Cyclic Stationary Electrode Voltammetry of Some Aliphatic Amines. Anal. Chem. 1984, 36, 2424–2426. [Google Scholar] [CrossRef]
- Masui, M.; Ozaki, S. Anodic-oxidation of amines 5. Cyclic voltammetry and controlled potential electrolysis of ephedrine and related compounds in aqueous buffer solution. Chem. Pharm. Bull. 1978, 26, 2153–2159. [Google Scholar] [CrossRef]
- Sawyer, W.R.; Waterhouse, G.A.W.; Doedens, D.J.; Forney, R.B. Heroin, Morphine, and Hydromorphone Determination in Postmortem Material by High Performance Liquid Chromatography. J. Forensic Sci. 1988, 33, 1146–1155. [Google Scholar] [CrossRef] [PubMed]
- Zaromb, S.; Alcaraz, J.; Lawson, D.; Woo, C.S. Detection of airborne cocaine and heroin by high-throughput liquid-absorption preconcentration and liquid chromatography-electrochemical detection. J. Chromatogr. 1993, 643, 107–115. [Google Scholar] [CrossRef]
- Somaini, L.; Saracino, M.A.; Marcheselli, C.; Zanchini, S.; Gerra, G.; Raggi, M.A. Combined liquid chromatography-coulometric detection and microextraction by packed sorbent for the plasma analysis of long acting opioids in heroin addicted patients. Anal. Chim. Acta 2011, 702, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Huettl, P.; Koester, S.; Hoffer, L.; Gerhardt, G.A. Separation and Identification of Drugs of Abuse in Drug Cottons by High Performance Liquid Chromatography Coupled with Electrochemical Array Detectors. Electroanalysis 1999, 11, 313–319. [Google Scholar] [CrossRef]
- Ary, K.; Róna, K. LC determination of morphine and morphine glucuronides in human plasma by coulometric and UV detection. J. Pharm. Biomed. Anal. 2001, 26, 179–187. [Google Scholar] [CrossRef]
- Rashid, B.A.; Aherne, G.W.; Katmeh, M.F.; Kwasowski, P.; Stevenson, D. Determination of morphine in urine by solid-phase immunoextraction and high-performance liquid chromatography with electrochemical detection. J. Chromatogr. A 1998, 797, 245–250. [Google Scholar] [CrossRef]
- Xu, F.; Gao, M.; Wang, L.; Zhou, T.; Jin, L.; Jin, J. Amperometric determination of morphine on cobalt hexacyanoferrate modified electrode in rat brain microdialysates. Talanta 2002, 58, 427–432. [Google Scholar] [CrossRef]
- Guedes, A.G.P.; Papich, M.G.; Rude, E.P.; Rider, M.A. Pharmacokinetics and physiological effects of two intravenous infusion rates of morphine in conscious dogs. J. Vet. Pharmacol. Ther. 2007, 30, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Kukanich, B.; Lascelles, B.D.X.; Papich, M.G. Pharmacokinetics of morphine and plasma concentrations of morphine-6-glucuronide following morphine administration to dogs. J. Vet. Pharmacol. Ther. 2005, 28, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Aragon, C.L.; Read, M.R.; Gaynor, J.S.; Barnhart, M.D.; Wilson, D.; Papich, M.G. Pharmacokinetics of an immediate and extended release oral morphine formulation utilizing the spheroidal oral drug absorption system in dogs. J. Vet. Pharmacol. Ther. 2008, 32, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.P.; Fung, C.; Hanna, M. Serum morphine concentrations after buccal and intramuscular morphine administration. Br. J. Clin. Pharma. 1987, 24, 685–687. [Google Scholar] [CrossRef]
- Todd, R.D.; Muldoon, S.M.; Watson, R.L. Determination of morphine in cerebrospinal fluid and plasma by high-performance liquid chromatography with electrochemical detection. J. Chromatogr. 1982, 232, 101–110. [Google Scholar] [CrossRef]
- Jordan, P.H.; Hart, J.P. Voltammetric Behaviour of Morphine at a Glassy Carbon Electrode and Its Determination in Human Serum by Liquid Chromatography with Electrochemical Detection under Basic Conditions. Analyst 1991, 116, 991–996. [Google Scholar] [CrossRef] [PubMed]
- Oka, K.; Kantrowitz, J.D.; Spector, S. Isolation of morphine from toad skin (nonpeptide opiate/endogenous). Proc. Natl. Acad. Sci. USA 1985, 82, 1852–1854. [Google Scholar] [CrossRef] [PubMed]
- Lindenblatt, H.; Kramer, E.; Holzmann-Erens, P.; Gouzoulis-Mayfrank, E.; Kovar, K.-A. Quantitation of psilocin in human plasma by high-performance liquid chromatography and electrochemical detection: Comparison of liquid-liquid extraction with automated on-line solid-phase extraction. J. Chromatogr. B 1998, 709, 255–263. [Google Scholar] [CrossRef]
- Hasler, F.; Bourquin, D.; Brenneisen, R.; Bär, T.; Vollenweider, F.X. Determination of psilocin and 4-hydroxyindole-3-acetic acid in plasma by HPLC-ECD and pharmacokinetic profiles of oral and intravenous psilocybin in man. Pharm. Acta Helvetiae 1997, 72, 175–184. [Google Scholar] [CrossRef]
- Honeychurch, K.C.; Hart, J.P. Electrochemical detection of benzodiazepines, following liquid chromatography, for applications in pharmaceutical, biomedical and forensic investigations. Insci. J. Sens. 2014, 4, 1–18. [Google Scholar] [CrossRef]
- Santagati, N.A.; Ferrara, G.; Marrazzo, A.; Ronsisvalle, G. Simultaneous determination of amphetamine and one of its metabolites by HPLC with electrochemical detection. J. Pharm. Biomed. Anal. 2002, 30, 247–255. [Google Scholar] [CrossRef]
- Kramer, E.; Kovar, K.-A. On-line coupling of automated solid-phase extraction with highperformance liquid chromatography and electrochemical detection Quantitation of oxidizable drugs of abuse and their metabolites in plasma and urine. J. Chromatogr. B 1999, 731, 167–177. [Google Scholar] [CrossRef]
- Zuway, K.Y.; Smith, J.P.; Foster, C.W.; Kapur, N.; Banks, C.E.; Sutcliffe, O.B. Detection and quantification of new psychoactive substances (NPSs) within the evolved “legal high” product, NRG-2, using high performance liquid chromatography-amperometric detection (HPLC-AD). Analyst 2015, 140, 6283–6294. [Google Scholar] [CrossRef] [PubMed]
- Min, J.Z.; Yamashita, K.; Toyo’oka, T.; Inagaki, S.; Higashi, T.; Kikura-Hanajiri, R.; Goda, Y. Simultaneous and group determination methods for designated substances by HPLC with multi-channel electrochemical detection and their application to real samples. Biomed. Chromatogr. 2010, 24, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- Seger, C. Usage and limitations of liquid chromatography-tandem mass spectrometry (LC–MS/MS) in clinical routine laboratories. Wien. Med. Wochenschr. 2012, 162, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Hudson, J.; Hutchings, J.; Wagner, R.; Harper, C.; Friel, P. Validation of a Cannabiniod Quantitation Method Using an Agilent 6430 LC/MS/MS; Agilent Technologies Inc.: Santa Clara, CA, USA, 2013; Available online: https://www.agilent.com/cs/library/applications/5991-2554EN.pdf (accessed on 3 August 2016).
- Teles de Menezes, M.M.; Fernando de Andrade, J.; Firmino de Oliveira, M.; Tristão, H.M.; Aparecida Saczk, A.; Luiz Okumura, L. Analysis of δ9-THC in cosmetics by high performance liquid chromatography with UV-Vis detection. Braz. J. Anal. Chem. 2012, 2, 341–344. [Google Scholar]
- Liu, H.-C.; Lee, H.-T.; Hsu, Y.-C.; Huang, M.-H.; Liu, R.H.; Chen, T.-J.; Lin, D.-L. Direct Injection LC-MS-MS Analysis of Opiates, Methamphetamine, Buprenorphine, Methadone and Their Metabolites in Oral Fluid from Substitution Therapy Patients. J. Anal. Toxicol. 2015, 39, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Mostafavi, A.; Abedi, G.; Jamshidi, A.; Afzali, D.; Talebi, M. Development and validation of a HPLC method for the determination of buprenorphine hydrochloride, naloxone hydrochloride and noroxymorphone in a tablet formulation. Talanta 2009, 77, 1415–1419. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Shay, S.D.; Caraco, Y.; Wood, M.; Wood, A.J.J. Simultaneous determination of codeine and it seven metabolites in Plasma and urine by high-performance liquid chromatography with ultraviolet and electrochemical detection. J. Chromatogr. B 1998, 708, 185–193. [Google Scholar] [CrossRef]
- Moeller, M.R.; Steinmeyer, S.; Kraemer, T. Determination of drugs of abuse in blood. J. Chromatogr. B 1998, 713, 91–109. [Google Scholar] [CrossRef]
- Roda, E.; Lonati, D.; Buscaglia, E.; Papa, P.; Rocchi, L.; Locatelli, C.A.; Coccini, T. Evaluation of Two Different Screening ELISA Assays for Synthetic Cathinones (Mephedrone/Methcathinone and MDPV) with LC-MS Method in Intoxicated Patients. J. Clin. Toxicol. 2016, 6. [Google Scholar] [CrossRef]
- Rambabu, C.; Rao, S.V.; Ramu, G.; Babu, A.B. Quantitative Analysis of Mephedrone in Bulk and Pharmaceutical Formulations by Reverse Phase High Performance Liquid Chromatography. Rasӑyan J. Chem. 2010, 3, 796–799. [Google Scholar]
- Bogusz, M.J. Liquid chromatography-mass spectrometry as a routine method in forensic sciences: A proof of maturity. J. Chromatogr. B 2000, 748, 3–19. [Google Scholar] [CrossRef]
- Gambaro, V.; Roda, G.; Visconti, G.L.; Arnoldi, S.; Casagni, E.; Dell’Acqua, L.; Farè, F.; Paladino, E.; Rusconi, C.; Arioli, S.; et al. DNA-based taxonomic identification of basidiospores in hallucinogenic mushrooms cultivated in “grow-kits” seized by the police: LC-UV quali-quantitative determination of psilocybin and psilocin. J. Pharm. Biomed. Anal. 2016, 125, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Honeychurch, K.C.; Hart, J.P. Determination of Flunitrazepam and Nitrazepam in Beverage Samples by Liquid Chromatography with Dual Electrode Detection Using a Carbon Fibre Veil Electrode. J. Solid State Electr. 2008, 12, 1317–1324. [Google Scholar] [CrossRef]
- Borgesa, K.B.; Freire, E.F.; Martins, I.; Pereira Bastos de Siqueira, M.E. Simultaneous determination of multibenzodiazepines by HPLC/UV: Investigation of liquid-liquid and solid-phase extractions in human plasma. Talanta 2009, 78, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Glover, S.J.; Allen, K.R. Measurement of benzodiazepines in urine by liquid chromatography-tandem mass spectrometry: Confirmation of samples screened by immunoassay. Ann. Clin. Biochem. 2010, 47, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Hart, J.P.; McCalley, D.V. Determination of catecholamines in urine using hydrophilic interaction chromatography with electrochemical detection. J. Chromatogr. A 2011, 1218, 3854–3861. [Google Scholar] [CrossRef] [PubMed]
- Baumann, A.; Karst, U. Online electrochemistry/mass spectrometry in drug metabolism studies: Principles and applications. Expert Opin. Drug Metab. Toxicol. 2010, 6, 715–731. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
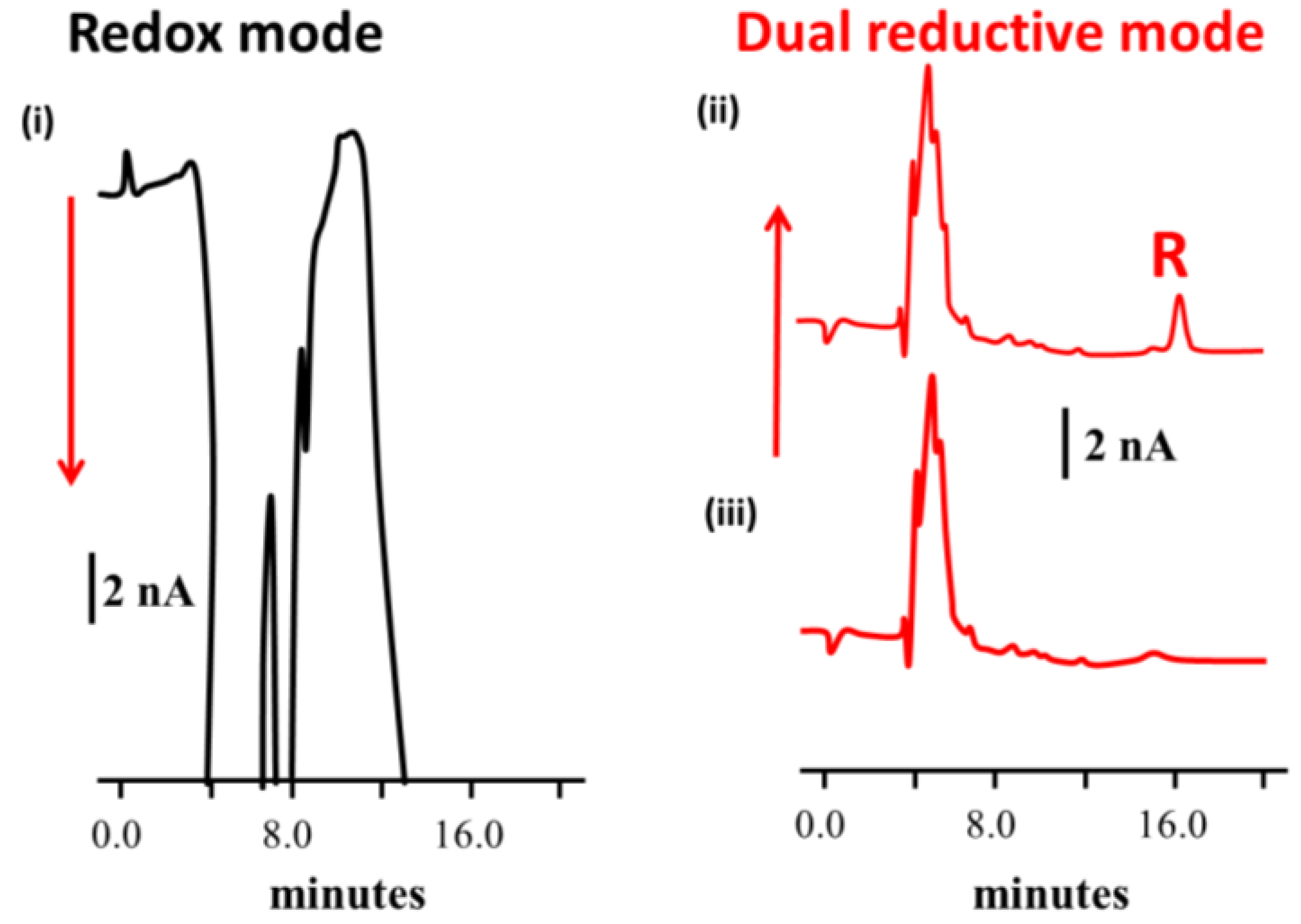{kind=link}
| LC ED Technique | Reference Electrode | Linear Range | Detection Limit | Comments | Ref. |
|---|---|---|---|---|---|
| Amperometric mode; +1.2 V | Ag/AgCl | 25–300 ng/mL | 5 ng/mL of urine (S/N = 5) | THC–COOH in urine | [36,37] |
| Amperometric mode; +1.2 V | Ag/AgCl | Up to 10 µg/mL | 1.5 ng on column | Δ9-tetrahydrocannablnol levels in brain tissue | [35] |
| Amperometric mode; +1.1 V | Ag/AgCl | 10–500 ng/mL | 0.5 ng/mL (S/N > 3) | THC and metabolites; THC–COOH) and 11-OH THC in rabbit and human urine | [38] |
| Amperometric mode; +0.85 V | Ag/AgCl | 0.012–0.20 µg/mL | THC–COOH is 0.012 µg/mL (limit of quantification) in human urine | Brominated 9-Carboxy-11-nor Δ9 tetrahydrocannabinol as internal Standard | [41] |
| Amperometric mode; +1.0 V | Ag/AgCl | 1–500 µg/mL | -- | Chemotaxonomical discrimination of confiscated cannabis. Studies made on stablity of cannabinoids in herbal cannabis. Mobile phase: CH3CN/CH3OH/0.02 N H2SO4. Benozic acid as internal standard | [42] |
| Amperometric mode; +0.40 V | Ag/AgCl | 10–100 ng/mL | Limit of quantification: 0.5 ng/mL (S/N = 3) | Blood THC levels in patients given the drug dronabinol | [43] |
| Amperometric mode; +1.2 V | Ag/AgCl | 5–500 ng/injection | 0.5 to 0.9 ng/injection for free cannabinoids and 1.2 to 2.5 ng/injection for cannabinoic acids (S/N > 4). | Cannabinoic contents in marijuana cigarettes and in tar and ash | [33] |
| Analyte | LC ED Technique | Reference Electrode | Linear Range | Detection Limit | Comments | Ref. |
|---|---|---|---|---|---|---|
| Cocaine and heroin | Amperometric mode; +1.0 V | Ag/AgCl | 25–300 ng/mL | ca. 1:1013 (v/v) | Airbourne concentrations of cocaine and heroin | [53] |
| Heroin, morphine and hydromorphone | Amperometric mode; +0.5 V | Ag/AgCl | l0 to 500 ng/mL (morphine), 62 to 1000 ng/mL (hydromorphone), and 250 to 2000 ng/mL (heroin) | For extracted sample was 0.5 ng/mL (morphine), 3.1 ng/mL (hydromorphone), and 12.5 ng/ mL (heroin) | Post-mortem samples of whole blood, urine, or vitreous humor | [52] |
| Morphine, heroin, codeine, thebaine, narcotine, papaverine and cocaine | Amperometric mode; +1.2 V | Ag/AgCl | Morphine base, 0.42–1.7 nM; heroin hydrochloride, 1.6–6.5 nM; cocaine hydrochloride, 3.1–12 nM. | 0.3 ng for morphine, 1 ng for heroin, and 2 ng for cocaine | Comparisum with LC UV detection made | [46] |
| Heroin, morphine, codeine and cocaine | 12 channel electrochemical array | PdH2 | -- | 4 pg/mL for morphine, 24 pg/mL for codeine, 444 pg/mL for heroin and 576 pg/mL for cocaine | Analysis of drug cottens | [55] |
| Morphine, morphine-3-glucuronide and morphine-6-glucuronide | LC coulometric DED in conjunction with UV detection | PdH2 | Morphine; 1–30 ng/mL | Morphine; 0.5 ng/mL | Morphine and its glucuronides extracted from human plasma by SPE | [56] |
| Buprenorphine, norbuprenorphine, naloxone and methadone | DED, screening −0.2 V detector +0.6 V | PdH2 | buprenorphine and norbuprenorphine, 3.0–1000.0 ng/mL for methadone and 0.13–10.0 ng/mL for naloxone. | 0.08 ng/mL for both buprenorphine and norbuprenorphine, 0.9 ng/mL for methadone and 0.04 ng/mL for naloxone | Plasma smaples from heroin addicts. Levosulpiride as an internal standard | [54] |
| Morphine | Amperometric mode; +0.60 V. 25 µL injection | Ag/AgCl | 1.0 × 10−6 M to 5.0 × 10−4 M | 5.0 × 10−7 M (S/N = 3). | Rat brain dialysates. | [58] |
| Morphine | LC coulometric DED | PdH2 | -- | 0.8 ng/mL | Buccal and intramuscular morphine adminstered to humans | [62] |
| Morphine | Amperometric mode; +0.45 V | Ag/AgCl | 1.2 × 10−12 to 4 × 10−10 M | 1.24 × 10−13 M | hydromorphone as an internal standard | [64] |
| Morphine | LC coulometric DED | PdH2 | -- | Limits of quantification: 25 ng/L morphine | Guard cell +750 mV; cell 1 +300 mV; cell 2 +450 mV | [60] |
| Morphine, morpine-6-glucronide | LC coulometric DED | PdH2 | -- | Limit of quantification for morphine; 7 ng/mL and morpine-6-glucronide; 4 ng/mL | Guard cell +750 mV; cell 1 +300 mV; cell 2 +450 mV | [59] |
| Morphine, morpine-6-glucronide | LC coulometric DED | PdH2 | -- | limits of quantification: morphine; 7.8 ng/mL; morpine-6-glucronide ng/mL | [61] | |
| Morphine | Amperometric mode; +0.75 V | Ag/AgCl | -- | -- | Detection of morphine in toad (Bufo marinus), rabbit and rat skin, bovine adrenal, cerebellum, cerebel cortex | [65] |
| Psilocybin | Amperometric mode; +0.650 V using 5-hydroxyindole or bufotenine as an internal standard | Ag/AgCl | 25–300 ng/mL | limit of quantitation; 10 ng/mL | Pooled human blood bank plasma and plasma obtained from seven volunteers (self-experimenting physicians) | [66] |
| Psilocin and 4-hydroxyindole-3-acetic acid | LC coulometric DED | PdH2 | -- | Limits of quantification of 0.8 ng/mL and 5.0 ng/mL for psilocin and 4HIAA | Column-switching. Analysis of human plasma | [67] |
| Analyte | LC ED Technique | Reference Electrode | Linear Range | Detection Limit | Comments | Ref. |
|---|---|---|---|---|---|---|
| Rohypnol (flunitrazepam) | Dual amperometric reductive-reductive mode; electrode 1; −2.4 V and electrode 2; −0.2 V | Stainless steel (generator cell); Ag/AgCl (detector cell) | 0.5–100 mg/mL | 20 ng/mL | Bovine and human serum | [20] |
| Amphetamine and 4-hydroxynorephedrine | Porous graphite electrode; +0.6 V. | PdH2 | 10–40 nM/mL | 50 ng/mL for each compound | Derivatisation with 2,5-dihydroxybenzaldehyde | [69] |
| Mephedrone and 4-methylethcathinone | Screen-printed carbon electrode; +1.4 V | Screen-printed Ag/AgCl | Mephedrone; 14.66 µg/mL; 4-methylethcathinone 9.35 µg/mL. | Samples of the ‘legal high’ NRG-2 analysed. | [71] | |
| Tryptamines, Phenethylamines and Piperazines | Multichannel electrochemical detection | PdH2 | Ranged from 17.1 pg to 117 ng indan-2-amine | 31 different drugs determined | [72] |
| Analyte | LC ED Detection Limit, ng/mL | Ref. | LC/MS Detection Limit, ng/mL | Ref. | LC/UV Detection Limit, ng/mL | Ref. |
|---|---|---|---|---|---|---|
| THC | 0.5 | [38] | 1.0 | [74] | 746 | [75] |
| methadone | 0.9 | [54] | 0.1 | [76] | ||
| buprenorphine | 0.08 | [54] | 5.0 | [76] | 20 | [77] |
| norbuprenorphine | 0.08 | [54] | 1.0 | [76] | ||
| morphine | 0.5 | [47] | 0.5 | [76] | 10 | [47] |
| codeine | 24 | [55] | 1.0 | [76] | 6.0 | [78] |
| amphetamine | <50 | [69] | 0.25 | [76] | 100 | [79] |
| mephedrone | 14,660 | [71] | 5.0 | [80] | 803 | [81] |
| 4-Methylethcathinone | 9350 | [71] | 5.0 | [80] | 2410 | [71] |
| psilocybin | 0.8 | [67] | 2.0 | [82] | 50 | [83] |
| Rohypnol | 20 | [84] | 0.2 | [82] | 30 | [85] |
| nitrazepam | 100 | [20] | 1.25 | [86] |
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Honeychurch, K. Review: The Application of Liquid Chromatography Electrochemical Detection for the Determination of Drugs of Abuse. Separations 2016, 3, 28. https://doi.org/10.3390/separations3040028
Honeychurch K. Review: The Application of Liquid Chromatography Electrochemical Detection for the Determination of Drugs of Abuse. Separations. 2016; 3(4):28. https://doi.org/10.3390/separations3040028
Chicago/Turabian StyleHoneychurch, Kevin. 2016. "Review: The Application of Liquid Chromatography Electrochemical Detection for the Determination of Drugs of Abuse" Separations 3, no. 4: 28. https://doi.org/10.3390/separations3040028
APA StyleHoneychurch, K. (2016). Review: The Application of Liquid Chromatography Electrochemical Detection for the Determination of Drugs of Abuse. Separations, 3(4), 28. https://doi.org/10.3390/separations3040028