
On the Extraction of Antibiotics from Shrimps Prior to Chromatographic Analysis



Abstract
:1. Introduction
2. Antibiotics
3. Trends in the Extraction of Antibiotics from Shrimps
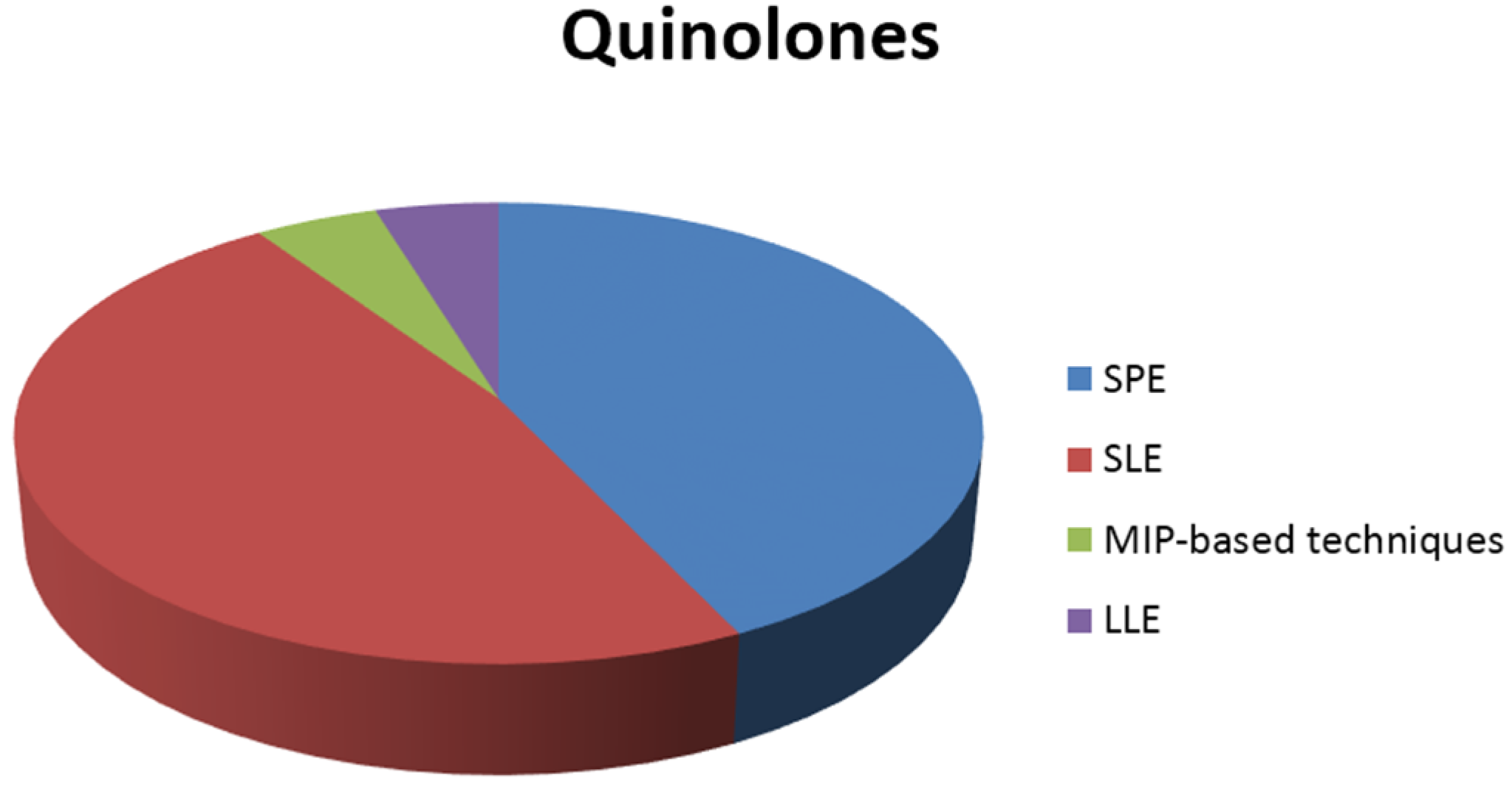
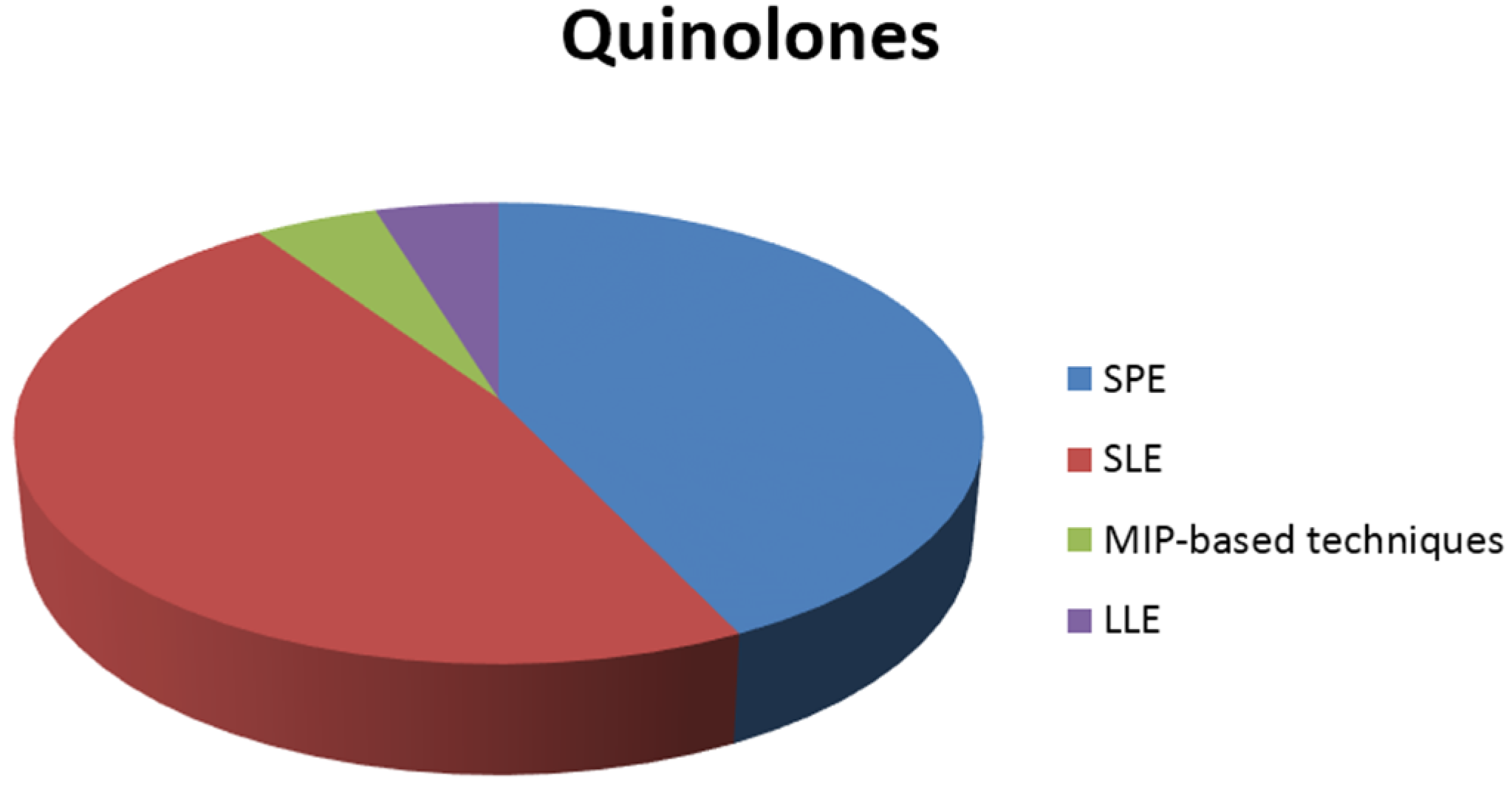
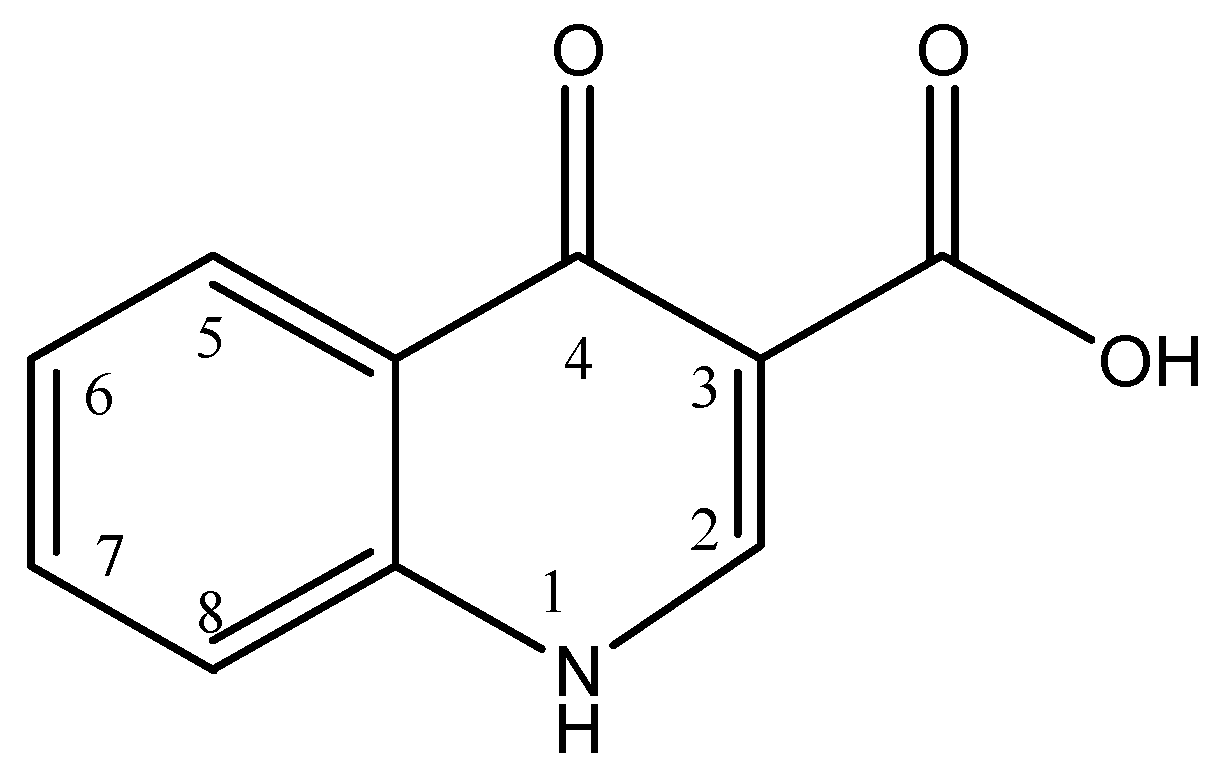
3.1. Extraction of Quinolones
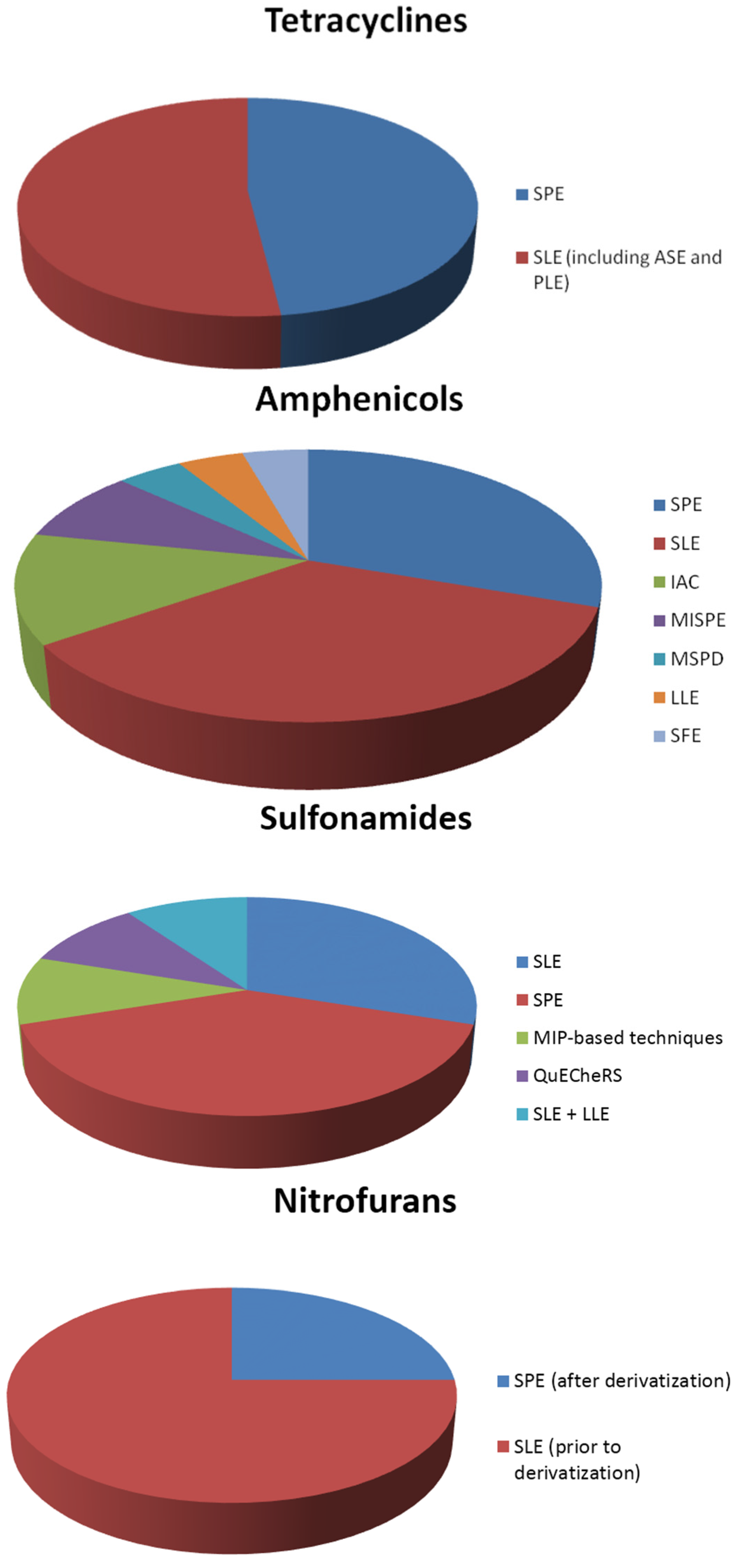
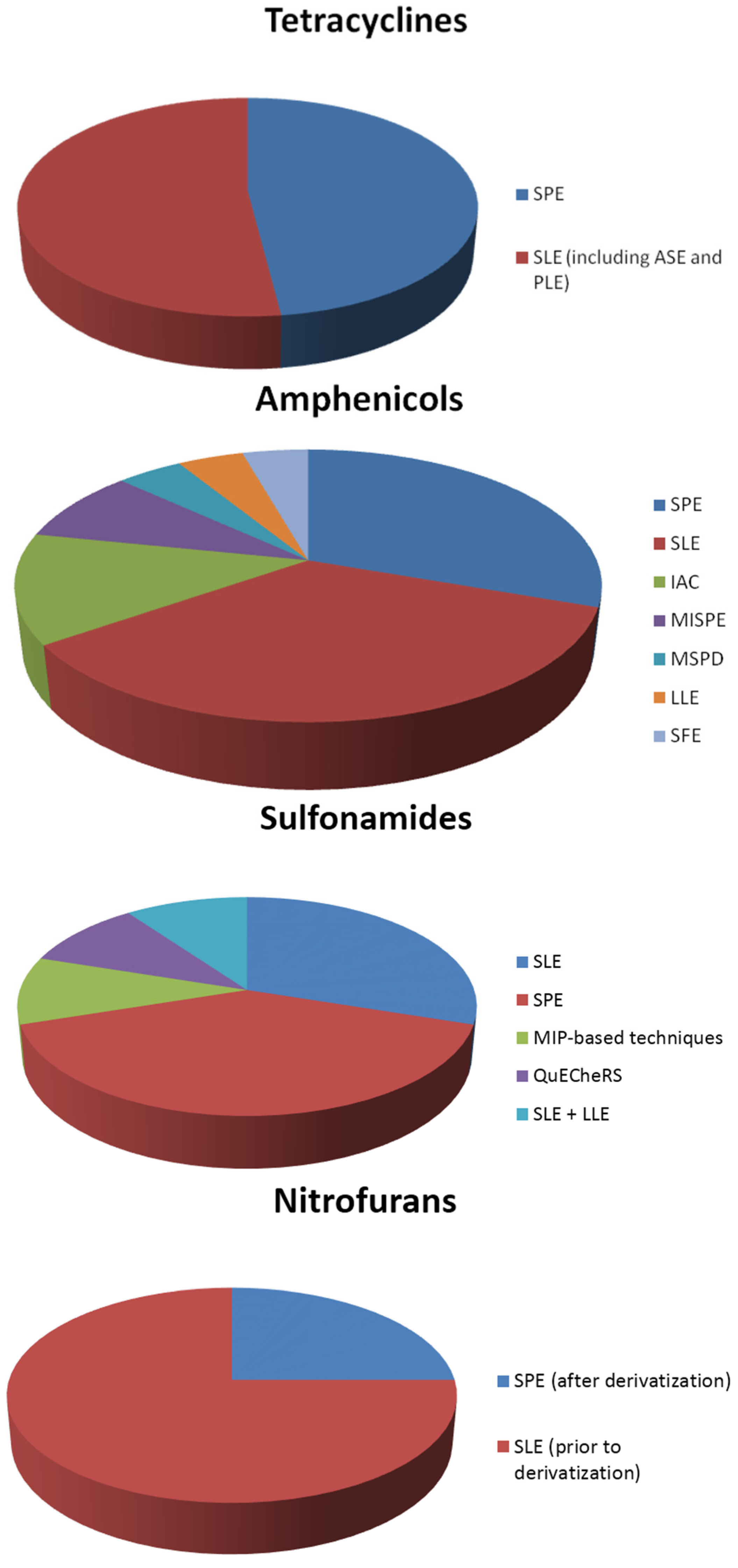
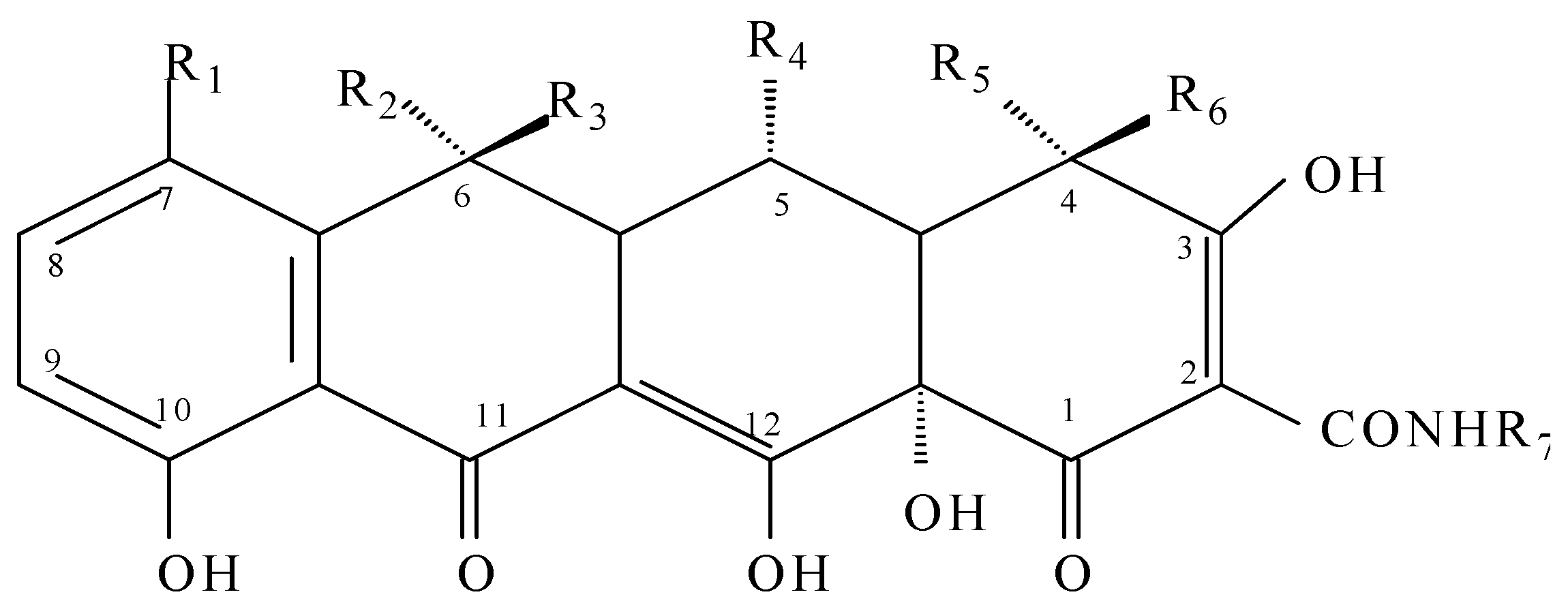
3.2. Extraction of Tetracyclines
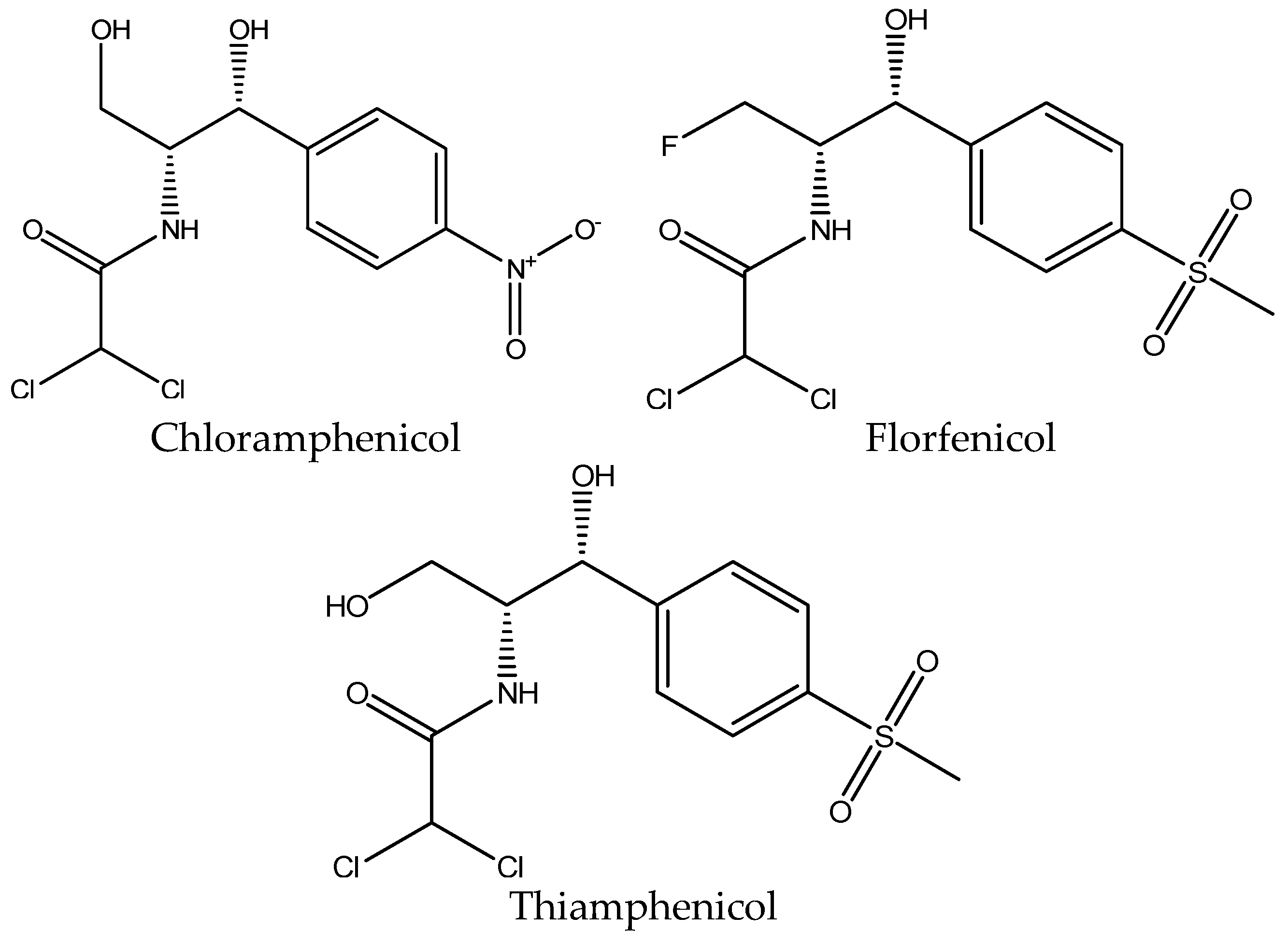
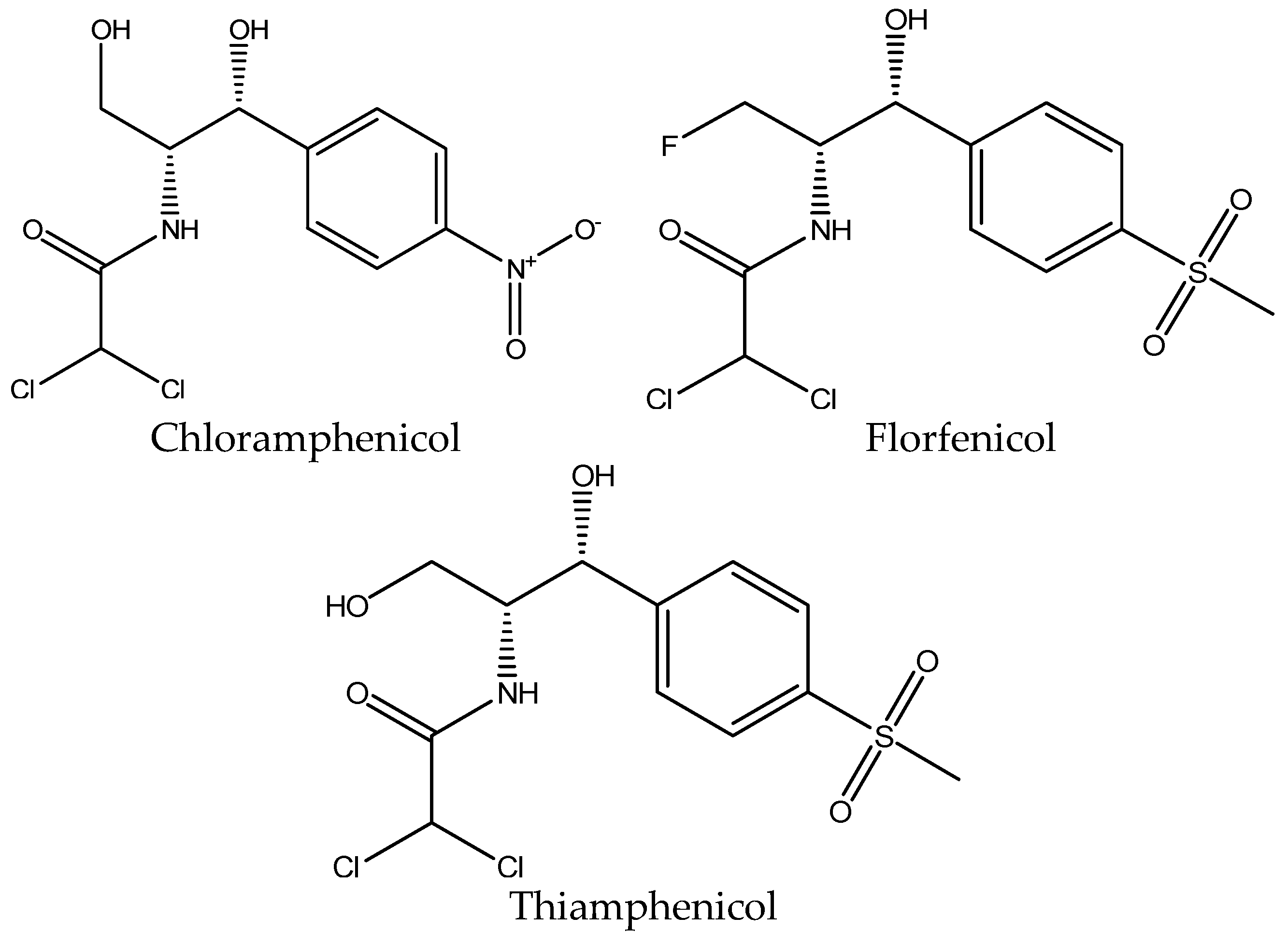
3.3. Extraction of Amphenicols
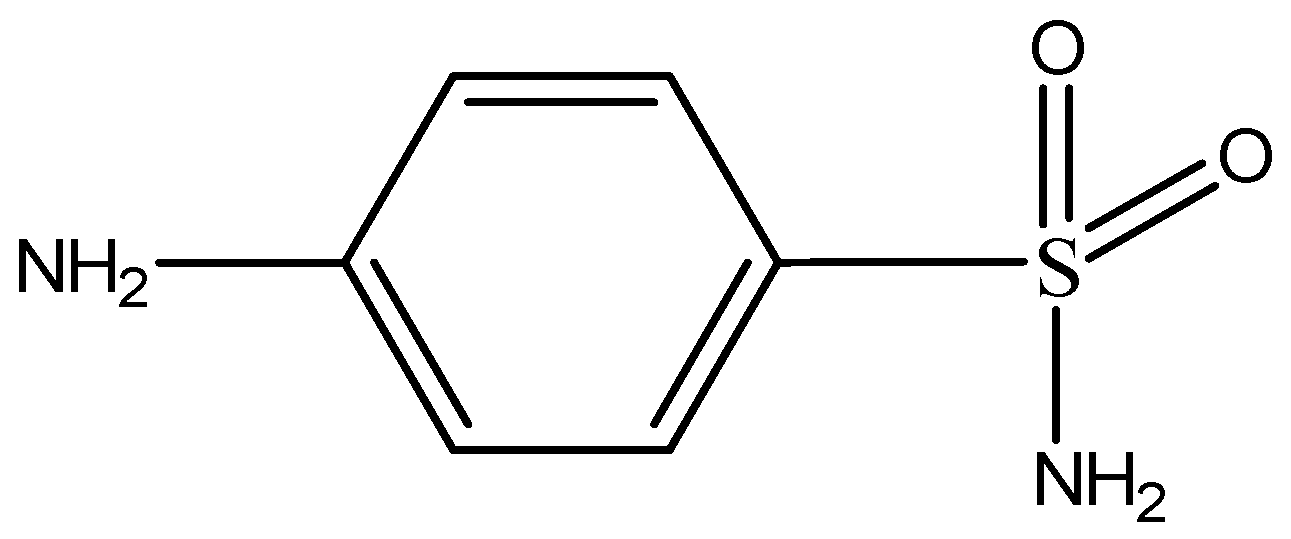
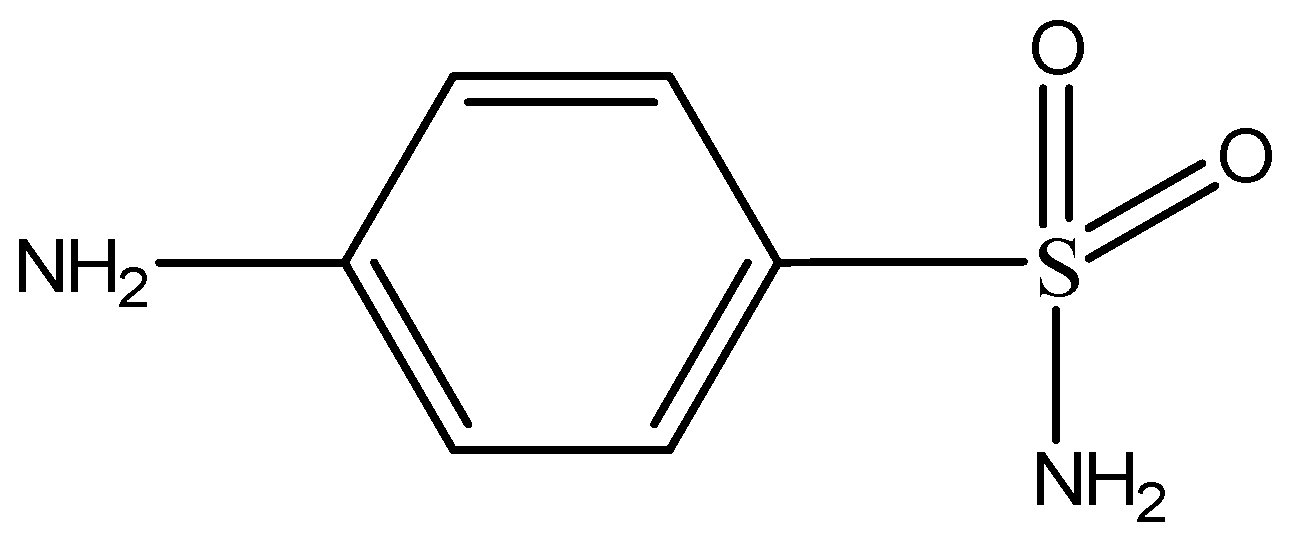
3.4. Extraction of Sulfonamides
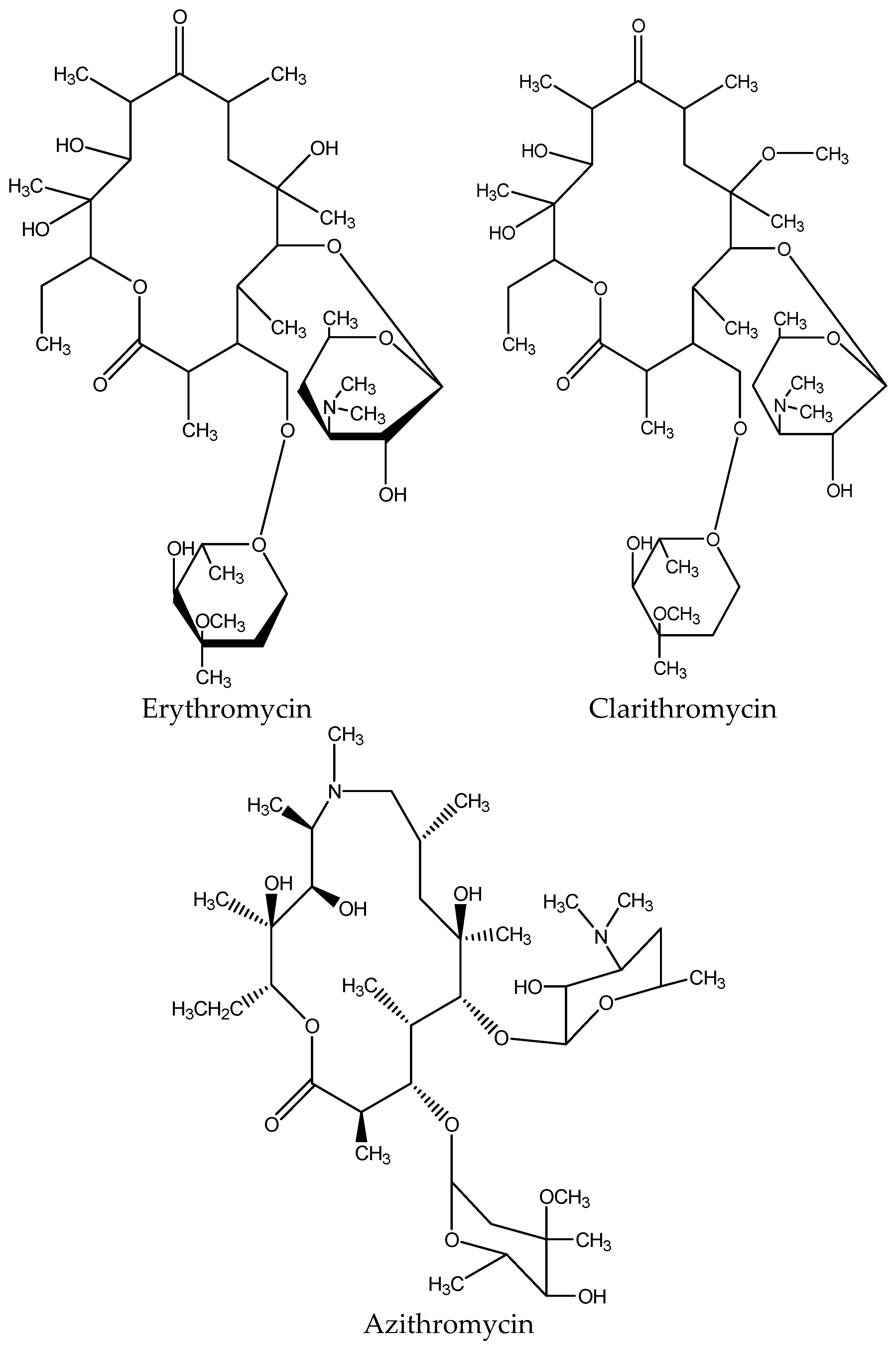
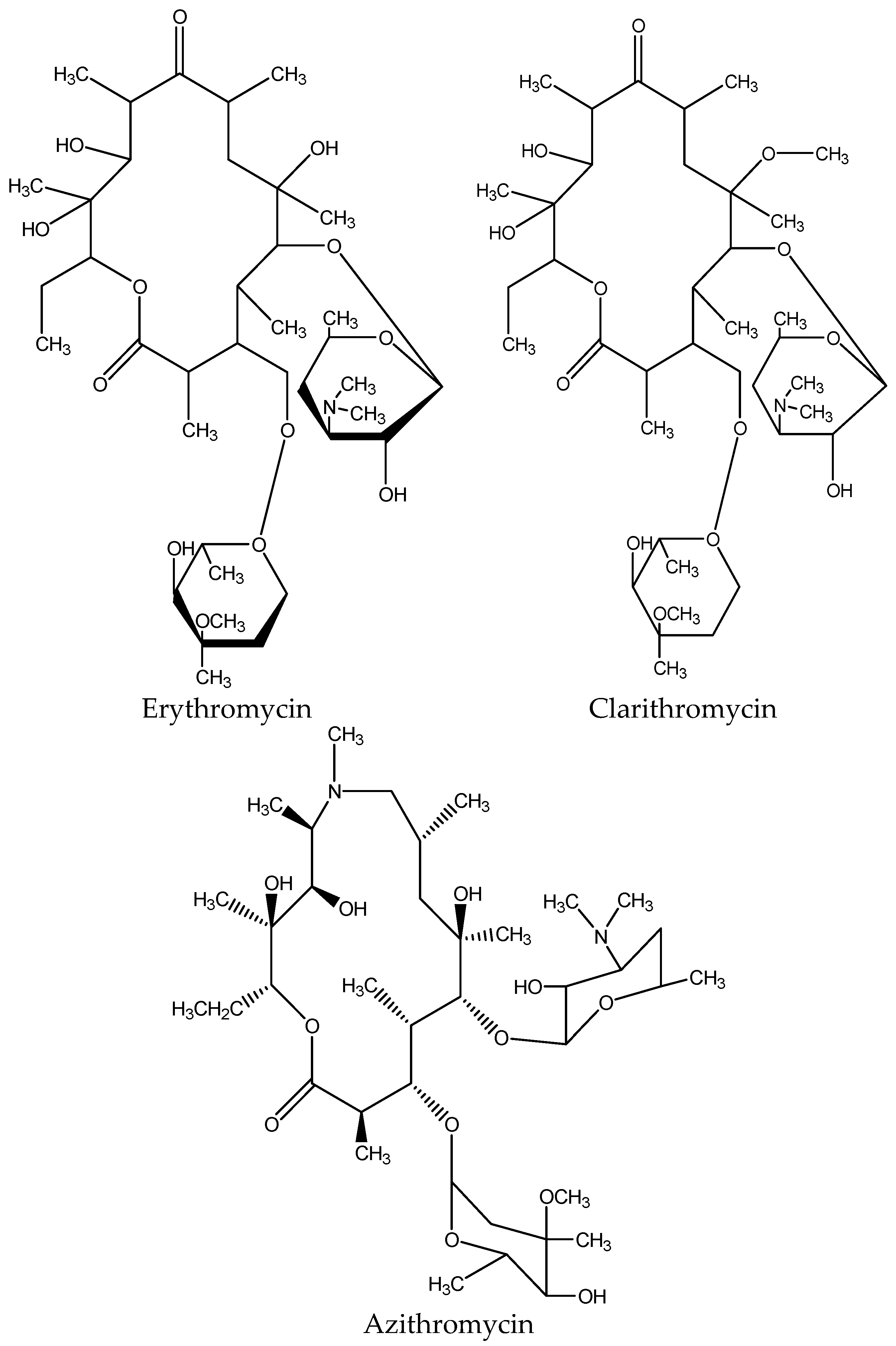
3.5. Extraction of Macrolides
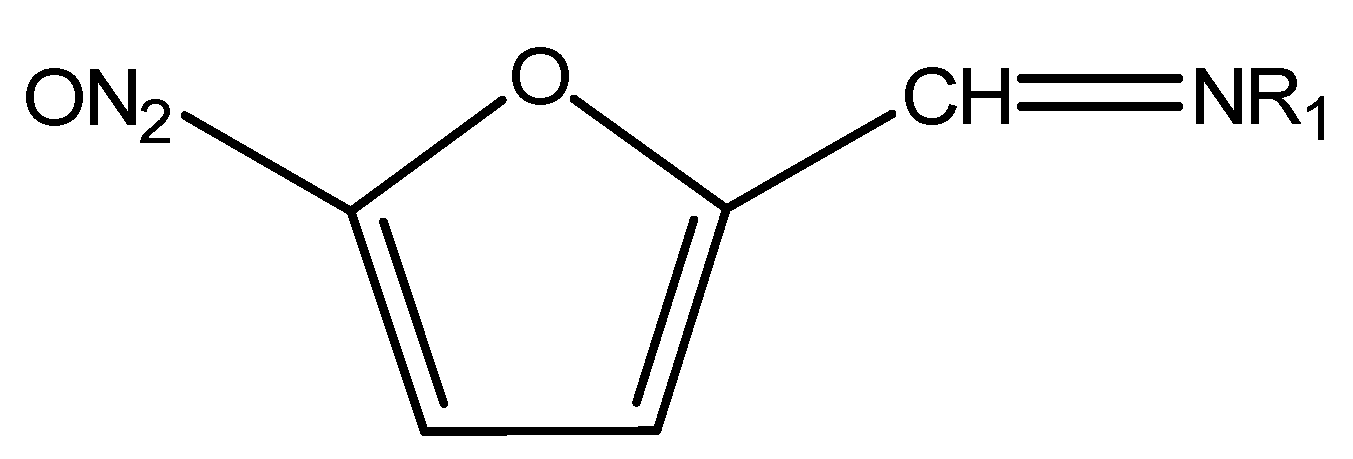
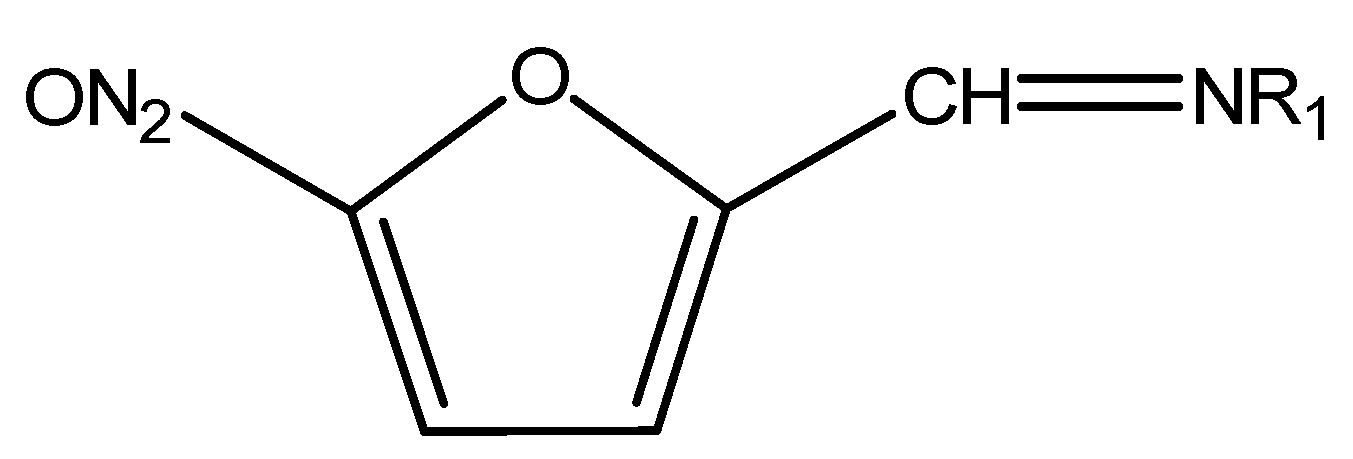
3.6. Extraction of Nitrofurans
3.7. Multi-Residue Methods
- (1)
- Spiked samples were extracted with 10 mL of acetonitrile containing 1% acetic acid with the addition of 4 g of anhydrous magnesium sulfate and 1.75 g of sodium chloride. 250 mg of primary-secondary amine and 750 mg of anhydrous magnesium sulfate were added to 5 mL of the acetonitrile supernatant. The final extract was evaporated to near dryness, reconstituted with 20% (v/v) methanol in water to a final volume of 2 mL.
- (2)
- Spiked samples were extracted with 6 mL of trifluoroacetic (20%, w/v), and the extract was evaporated to near dryness and reconstituted with 20% (v/v) methanol in water to a final volume of 0.5 mL.
- (3)
- Spiked shrimp samples were mixed with 2 g of aminopropyl (Bondesil-NH2), and the mixture was transferred to a SPE cartridge. The antibiotics were eluted twice with 5 mL of acetonitrile, and the eluates were evaporated to near dryness and reconstituted with 20% (v/v) methanol in water to a final volume of 1 mL.
- (4)
- Spiked shrimp samples were mixed with 3 mL of sulfuric acid 0.17 M, 0.158 g of sodium tungstate and 12 mL of acetonitrile in order to precipitate the proteins. An SPE cleanup step followed, with a C18 cartridge preconditioned with 5 mL of methanol and 5 mL of water. The analytes were eluted with 1 mL of acetonitrile/water (30:70 v/v), 2 × 2 mL of ethyl acetate were added to the eluate, and the organic extracts were evaporated to near dryness and with 20% (v/v) methanol in water to a final volume of 1 mL.
- (5)
- In order to precipitate the proteins, spiked shrimp samples were mixed with 100 mL of 0.2% of metaphosphoric acid in acetonitrile, the mixture was filtered through a 0.45 μm filter, and the extract was evaporated to a final volume of 30 mL. A SPE cleanup step followed with an Oasis HLB SPE cartridge, and the analytes were eluted with 5 mL of acetonitrile. The eluate was evaporated to near dryness and reconstituted with 20% (v/v) methanol in water to a final volume of 1 mL.
4. Conclusions
Author Contributions
Conflicts of Interest
Abbreviations
| ACN | Acetonitrile |
| AHD | 1-Aminohydatoin |
| AMOZ | 3-Amino-5-morpholino-methyl-1,3-Oxazolidinone |
| AOZ | 3-Amino-2-Oxazolidinone |
| APCI | Atmospheric Pressure Chemical Ionization |
| AQ | Acidic Quinolones |
| ASE | Accelerated Solvent Extraction |
| AVE | Average |
| AZM | Azithromycin |
| BC | Benzalkonium Chloride |
| BDD | Boron Doped Diamond |
| BG | Brilliant Green |
| BSA | N,O-Bis(trimethylsilyl)acetamide |
| CAP | Chloramphenicol |
| CCα | Decision Limit |
| CCβ | Detection Capability |
| CIN | Cinoxacin |
| CIP | Ciprofloxacin |
| CLM | Clarithromycin |
| CTC | Chlortetracycline |
| CV | Crystal Violet Cation |
| CWP | Coordinating Working Party |
| CZE | Capillary Zone Electrophoresis |
| d5-Cap | d5-Chloramphenicol |
| DC | Doxycycline |
| DES-CIP | Desethylene Ciprofloxacin |
| DI water | Deionized Water |
| DIF | Difloxacin |
| DMC | Demeclocycline |
| DNA | Deoxyribonucleic Acid |
| EDTA | Ethylenediaminetetraacetic Acid |
| ELAN | Elandomycin |
| ENR | Enrofloxacin |
| EQ | Ethoxyquin |
| ERY | Erythromycin |
| EU | European Union |
| FAO | Food and Agriculture Organization |
| FAO | Food and Agriculture Organization of The United Nations |
| FFC | Florfenicol |
| FLU | Flumequine |
| FQ | Fluoroquinolones |
| GC | Gas Chromatography |
| GC/MS-MS | Gas Chromatography-Mass Spectrometry |
| GC/NCI/MS | Gas Chromatography-Negative Chemical Ionization-Mass Spectrometry |
| GV | Gentian Violet |
| HLB | Hydrophilic-Lipophilic Balance |
| HPLC | High-Performance Liquid Chromatography |
| HPLC-CE | High-Performance Liquid Chromatography Cation-Exchange |
| HPLC-CL | High-Performance Liquid Chromatography-Chemiluminescenece Detection |
| HPLC-FLD | High-Performance Liquid Chromatography-Fluorescence Detection |
| HPLC-UV | High-Performance Liquid Chromatography-Ultraviolet Detection |
| IAC | Immunoaffinity Column |
| IS | Internal Standard |
| LC dye metabolites | Leuco Dye Metabolites |
| LC-ESI-MS/MS | Liquid Chromatography-Electrospray Ionization-Mass Spectrometry |
| LC-FLD | Liquid Chromatography-Fluorescence Detection |
| LC-FLD-MS | Liquid Chromatography-Fluorescence-Mass Spectrometry |
| LC-MS/MS | Liquid Chromatography-Tandem Mass Spectrometry |
| LC-TOFMS | Liquid Chromatography-Time-Of-Flight Mass Spectrometry |
| LC-UV | Liquid Chromatography-Ultraviolet Detection |
| LCV | Leucocrystal Violet |
| LDTD-MS/MS | Laser Diode Thermal Desorption-Mass Spectrometry |
| LGV | Leucogentian Violet |
| L-L partition | Liquid-Liquid Partition |
| LLE | Liquid-Liquid Extraction |
| LMG | Leucomalachite Green |
| LOD | Limit of Detection |
| LOME | Lomefloxacin |
| LOQ | Limit of Quantification |
| MARB | Marbofloxacin |
| MBZ | Mebendazole |
| MCX | Mixed Mode Cation Exchange |
| MDL | Method Detection Limit |
| MeCN | Acetonitrile |
| MeOH | Methanol |
| MG | Malachite Green Cation |
| MIP | Molecularly Imprinted Polymer |
| MISPE | Molecularly Imprinted Solid Phase Extraction |
| MNC | Minocycline |
| MQCA | 3-Methyl-quinoxaline-2-carboxylic Acid |
| MRLs | Maximum Residue Levels |
| MSPD | Matrix Solid Phase Dispersion |
| MT | Methyltestosterone |
| MTC | Methacycline |
| NAL | Nalidixic Acid |
| NBA | Nitrobenzaldehyde |
| Ni-DIA electrode | Nickel-Implanted Boron-Doped Diamond Thin Film Electrode |
| NIP | Non-Molecularly Imprinted Polymer |
| NOR | Norfloxacin |
| OFL | Ofloxacin |
| ORB | Orbifloxacin |
| OTC | Oxytetracycline |
| OXO | Oxolinic Acid |
| PABA | Para-Aminobenzoic Acid |
| PEF | Perfloxacin |
| PLE | Pressurized Liquid Extraction |
| PSA | Primary–Secondary Amine |
| PTFE | Polytetrafluoroethylene |
| p-TSA | p-Toluenesulfonic Acid Monohydrate |
| QCA | Quinoxaline-2-Carboxylic Acid |
| QuEChERS | Quick, Easy, Cheap, Effective, Rugged, and Safe |
| RNA | Ribonucleic Acid |
| RXM | Roxythromycin |
| SAR | Sarafloxacin |
| SAs | Sulfonamides |
| SCPD | Sulfachloropyridazine |
| SCPZ | Sulfachloropyrazine |
| SDB-RPS | Polystyrenedivinylbenzene-Reverse Phase Sorbent |
| SDM | Sulfadimethoxine |
| SDM | Sufladimethoxine |
| SDMP | Sulfadimethoxypyrimidine |
| SDX | Sulfadoxine |
| SDX | Sulfadoxine |
| SDZ | Sulfadiazine |
| SEM | Semicarbazide |
| SFE | Supercritical Fluid Extraction |
| SG | Sulfaguanidine |
| SLE | Solid Liquid Extraction |
| SMD | Sulfamethoxydiazine |
| SME | Sulfameter |
| SMM | Sulfamonomethoxine |
| SMP | Sulfamethoxypyridazine |
| SMR | Sulfamerazine |
| SMT | Sulfamethazine |
| SMTZ | Sulfamethizole |
| SMX | Sulfamethoxazole |
| SMZ | Sulfamethazine |
| SMZ-13C6 | Sulfamethazine-13C6 |
| SN | Sulfanilamide |
| SPD | Sulfapyridine |
| SPE | Solid Phase Extraction |
| SPZ | Sulfaphenazole |
| SQX | Sulfaquinoxaline |
| SSZ | Sulfisoxazole |
| SSZ | Sulfisoxazole |
| STZ | Sulfathiazole |
| Sylon BFT | {N,O-Bis(Trimethylsily) Trifluoroacetamide[BSTFA]-Trimethylchlorosilane [TMCS], 99 + 1} |
| TC | Tetracycline |
| TCA | Trichloroacetic Acid |
| TCs | Tetracyclines |
| THI | Thiamphenicol |
| TIM | Tilmicocin |
| TMPD | N,N,N′,N′-Tetramethyl-P-Phenylenediamine dihydrochloride |
| TOLSa | Toltrazurisulfone |
| TPM | Triphenylmethane Dyes |
| TRI | Trimethoprim |
| UPLC-MS/MS | Ultra-Performance Liquid Chromatography-Mass Spectrometry |
| UV | Ultra Violet |
References
- Food and Agriculture Organization of the United Nations. Available online: http://www.fao.org/fishery/cwp/handbook/j/en (accessed on 9 December 2015).
- Cole, D.W.; Cole, R.; Gaydos, S.J.; Gray, J.; Hyland, G.; Jacques, M.L.; Powell-Dunford, N.; Sawhney, C.; Au, W.W. Aquaculture: Environmental, Toxicological, and Health Issues. Int. J. Hyg. Environ. Health 2009, 212, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Sapkota, A.; Sapkota, A.R.; Kucharski, M.; Burke, J.; McKenzie, S.; Walker, P.; Lawrence, R. Aquaculture Practices and Potential Human Health Risks: Current Knowledge and Future Priorities. Environ. Int. 2008, 34, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- Gillett, R. Global Study of Shrimp Fisheries; FAO: Rome, Italy, 2008; Volume 475. [Google Scholar]
- Rigos, G.; Troisi, G.M. Antibacterial Agents in Mediterranean Finfish Farming: A Synopsis of Drug Pharmacokinetics in Important Euryhaline Fish Species and Possible Environmental Implications. Rev. Fish Biol. Fish. 2005, 15, 53–73. [Google Scholar] [CrossRef]
- Defoirdt, T.; Sorgeloos, P.; Bossier, P. Alternatives to Antibiotics for the Control of Bacterial Disease in Aquaculture. Curr. Opin. Microbiol. 2011, 14, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Bermúdez-Almada, M.C.; Espinosa-Plascencia, A. The Use of Antibiotics in Shrimp Farming. In Health and Environment in Aquaculture; Carvalho, E., Ed.; InTech: Rijeka, Croatia, 2010; pp. 199–214. [Google Scholar]
- Mo, W.Y.; Chen, Z.; Leung, H.M.; Leung, A.O.W. Application of Veterinary Antibiotics in China’s Aquaculture Industry and Their Potential Human Health Risks. Environ. Sci. Pollut. Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Samanidou, V.F.; Evaggelopoulou, E.N. Analytical Strategies to Determine Antibiotic Residues in Fish. J. Sep. Sci. 2007, 30, 2549–2569. [Google Scholar] [CrossRef] [PubMed]
- Finch, R.G.; Greenwood, D.; Whitley, R.J.; Norrby, S.R. Antibiotic and Chemotherapy, 9th ed.; Elsevier: Amsterdam, The Netherlands, 2010. [Google Scholar]
- Wagman, A.S.; Wentland, M.P. Quinolone Antibacterial Agents. In Comprehensive Medicinal Chemistry II; Elsevier: Amsterdam, The Netherlands, 2007; pp. 567–596. [Google Scholar]
- Sousa, J.; Alves, G.; Abrantes, J.; Fortuna, A.; Falcão, A. Analytical Methods for Determination of New Fluoroquinolones in Biological Matrices and Pharmaceutical Formulations by Liquid Chromatography: A Review. Anal. Bioanal. Chem. 2012, 403, 93–129. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Hao, H.; Dai, M.; Liu, Z.; Yuan, Z. Antibacterial Action of Quinolones: From Target to Network. Eur. J. Med. Chem. 2013, 66, 555–562. [Google Scholar] [CrossRef] [PubMed]
- EUR-Lex. Available online: http://eur-lex.europa.eu/legal-content/EN (accessed on 9 December 2015).
- Rang, H.P.; Ritter, J.M.; Flower, R.J.; Henderson, G. RANG & DALE’S Pharmacology, 8th ed.; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Nelson, M.L.; Ismail, M.Y. The Antibiotic and NonantibioticTetracyclines. In Comprehensive Medicinal Chemistry II; Elsevier: Amsterdam, The Netherlands, 2007; pp. 597–628. [Google Scholar]
- Nicolaou, K.C.; Chen, J.S.; Edmonds, D.J.; Estrada, A.A. Recent Advances in the Chemistry and Biology of Naturally Occurring Antibiotics. Angew. Chem. Int. Ed. 2010, 48, 660–719. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Available online: http://www.fda.gov/AnimalVeterinary/DevelopmentApprovalProcess/Aquaculture/ucm132954.htm (accessed on 9 December 2015).
- Hauser, A.R. Antibiotics Basics for Clinicians: The ABCs of Choosing the Right Antibacterial Agent, 2nd ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007. [Google Scholar]
- Manivasagan, P.; Venkatesan, J.; Sivakumar, K.; Kim, S.K. Marine Actinobacterial Metabolites: Current Status and Future Perspectives. Microbiol. Res. 2013, 168, 311–332. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, T.; Dougherty, T.J.; Magee, T.V. Macrolide Antibiotics. In Comprehensive Medicinal Chemistry II; Elsevier: Amsterdam, The Netherlands, 2007; pp. 520–539. [Google Scholar]
- Aronson, J.K. Meyler’s Side Effects of Drugs: The International Encyclopedia of Adverse Drug Reactions and Interactions; Elsevier: Amsterdam, The Netherlands, 2006; pp. 2248–2249. [Google Scholar]
- Dost, K.; Jones, D.C.; Davidson, G. Determination of sulfomamides by packed column supercritical fluid chromatography with atmospheric pressure chemical ionization mass spectrometric detection. Analyst 2000, 125, 1243–1247. [Google Scholar] [CrossRef] [PubMed]
- FAO/WHO. Evaluation of Certain Veterinary Drug Residues in Food; 40th Report of the Joint FAO/WHO Expert Committee on Food; World Health Organization: Geneva, Switzerland, 1993. [Google Scholar]
- Garrido-Frenich, A.; Plaza-Bolanos, P.; Aguilera-Luiz, M.M.; Martinez-Vidal, J.L. Recent advances in the analysis of veterinary drugs and growth promoting agents by chromatographic techniques. In Chromatography Types, Techniques and Methods; Quintin, T.J., Ed.; Nova Science Publishers, Inc.: New York, NY, USA, 2009; pp. 1–102. [Google Scholar]
- Delatour, T.; Gremaud, E.; Mottier, P.; Richoz, J.; Vera, F.A.; Stadler, R.H. Preparation of stable isotope-labeled 2-nitrobenzaldehyde derivatives of four metabolites of nitrofuran antibiotics and their comprehensive characterization by UV, MS, and NMR techniques. J. Agric. Food Chem. 2003, 51, 6371–6379. [Google Scholar] [CrossRef] [PubMed]
- Tarbin, J.A.; Potter, R.A.; Stolker, A.A.M.; Berendsen, B. Single-residue quantitative and confirmatory methods. In Chemical Analysis of Antibiotic Residues in Food; Wiley: Hoboken, NJ, USA, 2011; pp. 227–262. [Google Scholar]
- Sriket, P.; Benjakul, S.; Visessanguan, W.; Kijroongrojana, K. Comparative Studies on Chemical Composition and Thermal Properties of Black Tiger Shrimp (Penaeus monodon) and White Shrimp (Penaeus vannamei) Meats. Food Chem. 2007, 103, 1199–1207. [Google Scholar] [CrossRef]
- Danyi, S.; Widart, J.; Douny, C.; Dang, P.K.; Baiwir, D.; Wang, N.; Tu, H.T.; Tung, V.T.; Phuong, N.T.; Kestemont, P.; et al. Determination and Kinetics of Enrofloxacin and Ciprofloxacin in Tra Catfish (Pangasianodon Hypophthalmus) and Giant Freshwater Prawn (Macrobrachium Rosenbergii) Using a Liquid Chromatography/mass Spectrometry Method. J. Vet. Pharmacol. Ther. 2011, 34, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Pearce, J.N.; Burns, B.G.; van de Riet, J.M.; Casey, M.D.; Potter, R.A. Determination of Fluoroquinolones in Aquaculture Products by Ultra-Performance Liquid Chromatography-Tandem Mass Spectrometry (UPLC-MS/MS). Food Addit. Contam. A Chem. Anal. Control Expo. Risk Assess. 2009, 26, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Wan, G.H.; Cui, H.; Pan, Y.L.; Zheng, P.; Liu, L.J. Determination of Quinolones Residues in Prawn Using High-Performance Liquid Chromatography with Ce(IV)-Ru(bpy)32+-HNO3 Chemiluminescence Detection. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2006, 843, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Poapolathep, A.; Jermnak, U.; Chareonsan, A.; Sakulthaew, C.; Klangkaew, N.; Sukasem, T.; Kumagai, S. Dispositions and Residue Depletion of Enrofloxacin and Its Metabolite Ciprofloxacin in Muscle Tissue of Giant Freshwater Prawns (Macrobrachium Rosenbergii). J. Vet. Pharmacol. Ther. 2009, 32, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Takeda, N.; Gotoh, M.; Matsuoka, T. Rapid Screening Method for Quinolone Residues in Livestock and Fishery Products Using Immobilised Metal Chelate Affinity Chromatographic Clean-up and Liquid Chromatography-Fluorescence Detection. Food Addit.Contam. A Chem. Anal. Control Expo. Risk Assess. 2011, 28, 1168–1174. [Google Scholar] [CrossRef] [PubMed]
- Schröder, U.; MacHetzki, A. Determination of Flumequine, Nalidixic Acid and Oxolinic Acid in Shrimps by High-Performance Liquid Chromatography with Fluorescence Detection. Eur. Food Res. Technol. 2007, 225, 627–634. [Google Scholar] [CrossRef]
- Karbiwnyk, C.M.; Carr, L.E.; Turnipseed, S.B.; Andersen, W.C.; Miller, K.E. Determination of Quinolone Residues in Shrimp Using Liquid Chromatography with Fluorescence Detection and Residue Confirmation by Mass Spectrometry. Anal. Chim. Acta 2007, 596, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Lohne, J.J.; Andersen, W.C.; Clark, S.B.; Turnipseed, S.B.; Madson, M.R. Laser Diode Thermal Desorption Mass Spectrometry for the Analysis of Quinolone Antibiotic Residues in Aquacultured Seafood. Rapid Commun. Mass Spectrom. 2012, 26, 2854–2864. [Google Scholar] [CrossRef] [PubMed]
- Costi, E.M.; Sicilia, M.D.; Rubio, S. Supramolecular Solvents in Solid Sample Microextractions: Application to the Determination of Residues of Oxolinic Acid and Flumequine in Fish and Shellfish. J. Chromatogr. A 2010, 1217, 1447–1454. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Dai, J.; Meng, M.; Song, Z.; Pan, J.; Yan, Y.; Li, C. Surface Molecularly Imprinted Polymers Based on Yeast Prepared by Atom Transfer Radical Emulsion Polymerization for Selective Recognition of Ciprofloxacin from Aqueous Medium. J. Appl. Polym. Sci. 2014. [Google Scholar] [CrossRef]
- Andersen, W.C.; Roybal, J.E.; Gonzales, S.A.; Turnipseed, S.B.; Pfenning, A.P.; Kuck, L.R. Determination of Tetracycline Residues in Shrimp and Whole Milk Using Liquid Chromatography with Ultraviolet Detection and Residue Confirmation by Mass Spectrometry. Anal. Chim. Acta 2005, 529, 145–150. [Google Scholar] [CrossRef]
- Treetepvijit, S.; Preechaworapun, A.; Praphairaksit, N.; Chuanuwatanakul, S.; Einaga, Y.; Chailapakul, O. Use of Nickel Implanted Boron-Doped Diamond Thin Film Electrode Coupled to HPLC System for the Determination of Tetracyclines. Talanta 2006, 68, 1329–1335. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, P.; Kumar, N.A.; Prasad, R.H.; Krishnamoorthy, K.B.; Prasath, K.H.; Soumya, V. LC-MS/MS Analysis of Tetracycline Antibiotics in Prawns (Penaeus monodon) from South India Coastal Region. JOPR J. Pharm. Res. 2012, 6, 48–52. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, H.; Yang, S.; Hu, Q.; Cheng, H.; Liu, H.; Qiu, Y. High-Performance Liquid Chromatography Using Pressurized Liquid Extraction for the Determination of Seven Tetracyclines in Egg, Fish and Shrimp. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2013, 917–918, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Wu, Y.; Li, Y.; Li, L. Determination of the Residues of 3-Methyl-Quinoxaline-2-Carboxylic Acid and Quinoxaline-2-Carboxylic Acid in Animal Origin Foods by High Performance Liquid Chromatography-Tandem Mass Spectrometry. Chin. J. Chromatogr. 2013, 30, 660–664. [Google Scholar] [CrossRef]
- Schneider, M.J.; Vazquez-Moreno, L.; Bermudez-Almada, M.C.; Guardado, R.B.; Ortega-Nieblas, M. Multiresidue Determination of Fluoroquinolones in Shrimp by Liquid Chromatography-Fluorescence-Mass Spectrometry. J. AOAC Int. 2005, 88, 1160–1166. [Google Scholar] [PubMed]
- Dufresne, G.; Fouquet, A.; Forsyth, D.; Tittlemier, S.A. Multiresidue Determination of Quinolone and Fluoroquinolone Antibiotics in Fish and Shrimp by Liquid Chromatography/tandem Mass Spectrometry. J. AOAC Int. 2007, 90, 604–612. [Google Scholar] [PubMed]
- Chonan, T.; Fujimoto, T.; Inoue, M.; Tazawa, T.; Ogawa, H. Multiresidue Determination of Quinolones in Animal and Fishery Products by HPLC. J. Food Hyg. Soc. Jpn. 2008, 49, 244–248. [Google Scholar] [CrossRef]
- Sun, Y.; Xu, X.; Zhu, X.; Mou, Z.; Ge, Y.; Wu, S.; Du, N. Optimization of Sample Preparation for the Determination of Fluoroquinolone Antibiotics Residues in Aquatic Products by UPLC-MS/MS. J. Harbin Inst. Technol. 2013, 45, 52–57. [Google Scholar]
- Chang, C.S.; Wang, W.H.; Tsai, C.E. Simultaneous Determination of 18 Quinolone Residues in Marine and Livestock Products by Liquid Chromatography/tandem Mass Spectrometry. J. Food Drug Anal. 2010, 18, 87–97. [Google Scholar]
- Chang, C.S.; Wang, W.H.; Tsai, C.E. Simultaneous Determination of Eleven Quinolones Antibacterial Residues in Marine Products and Animal Tissues by Liquid Chromatography with Fluorescence Detection. J. Food Drug Anal. 2008, 16, 87–96. [Google Scholar]
- Zhao, S.; Jiang, H.; Li, X.; Mi, T.; Li, C.; Shen, J. Simultaneous Determination of Trace Levels of 10 Quinolones in Swine, Chicken, and Shrimp Muscle Tissues Using HPLC with Programmable Fluorescence Detection. J. Agric. Food Chem. 2007, 55, 3829–3834. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, S.R.R.; Donato, J.L.; de Nucci, G.; Reyes, F.G.R. A High-Throughput Method for Determining Chloramphenicol Residues in Poultry, Egg, Shrimp, Fish, Swine and Bovine Using LC-ESI-MS/MS. J. Sep. Sci. 2009, 32, 4012–4019. [Google Scholar] [CrossRef] [PubMed]
- Douny, C.; Widart, J.; de Pauw, E.; Maghuin-Rogister, G.; Scippo, M.L. Determination of Chloramphenicol in Honey, Shrimp, and Poultry Meat with Liquid Chromatography-Mass Spectrometry: Validation of the Method According to Commission Decision 2002/657/EC. Food Anal. Methods 2013, 6, 1458–1465. [Google Scholar] [CrossRef]
- Tyagi, A.; Vernekar, P.; Karunasagar, I.; Karunasagar, I. Determination of Chloramphenicol in Shrimp by Liquid Chromatography-Electrospray Ionization Tandem Mass Spectrometry (LC-ESI-MS-MS). Food Addit. Contam. A Chem. Anal. Control Expo. Risk Assess. 2008, 25, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Barreto, F.; Ribeiro, C.; Hoff, R.B.; Costa, T.D. Determination and Confirmation of Chloramphenicol in Honey, Fish and Prawns by Liquid Chromatography-tandem Mass Spectrometry with Minimum Sample Preparation: Validation according to 2002/657/EC Directive. Food Addit.Contam. A 2012, 29, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Polzer, J.; Hackenberg, R.; Stachel, C.; Gowik, P. Determination of Chloramphenicol Residues in Crustaceans: Preparation and Evaluation of a Proficiency Test in Germany. Food Addit. Contam. 2006, 23, 1132–1140. [Google Scholar] [CrossRef] [PubMed]
- Ashwin, H.M.; Stead, S.L.; Taylor, J.C.; Startin, J.R.; Richmond, S.F.; Homer, V.; Bigwood, T.; Sharman, M. Development and Validation of Screening and Confirmatory Methods for the Detection of Chloramphenicol and Chloramphenicol Glucuronide Using SPR Biosensor and Liquid Chromatography-tandem Mass Spectrometry. Anal. Chim. Acta 2005, 529, 103–108. [Google Scholar] [CrossRef]
- Shi, X.; Wu, A.; Zheng, S.; Li, R.; Zhang, D. Molecularly Imprinted Polymer Microspheres for Solid-Phase Extraction of Chloramphenicol Residues in Foods. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 850, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Zhu, F.; Chen, D.; Wei, H.; Pan, Y.; Wang, X.; Liu, Z.; Huang, L.; Wang, Y.; Yuan, Z. Evaluation of Matrix Solid-Phase Dispersion (MSPD) Extraction for Multi-Fenicols Determination in Shrimp and Fish by Liquid Chromatography-Electrospray Ionisation Tandem Mass Spectrometry. Food Chem. 2014, 150, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.L.; Lee, R.J.; Lee, M.R. Supercritical Fluid Extraction in Situ Derivatization for Simultaneous Determination of Chloramphenicol, Florfenicol and Thiamphenicol in Shrimp. Food Chem. 2010, 121, 797–802. [Google Scholar] [CrossRef]
- Stidl, R.; Cichna-Markl, M. Sample Clean-up by Sol-Gel Immunoaffinity Chromatography for Determination of Chloramphenicol in Shrimp. J. Sol Gel Sci. Technol. 2007, 41, 175–183. [Google Scholar] [CrossRef]
- Samanidou, V.F.; Makrygianni, E.A. Ultrasound-Assisted Matrix Solid Phase Dispersion for the HPLC-DAD Analysis of Amphenicols in Shrimps. Sample Prep. 2015, 2, 66–73. [Google Scholar] [CrossRef]
- Shi, X.; Meng, Y.; Liu, J.; Sun, A.; Li, D.; Yao, C.; Lu, Y.; Chen, J. Group-selective molecularly imprinted polymer solid phase extraction for the simultaneous determination of six sulfonamides in aquaculture products. J.Chromatogr. B 2011, 879, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Won, S.Y.; Lee, C.H.; Chang, H.S.; Kim, S.O.; Lee, S.H.; Kim, D.S. Monitoring of 14 sulfonamide antibiotic residues in marine products using HPLC-PDA and LC-MS/MS. Food Control 2011, 22, 1101–1107. [Google Scholar] [CrossRef]
- Gehring, T.A.; Griffin, B.; Williams, R.; Geiseker, C.; Rushing, L.G.; Siitonen, P.H. Multiresidue determination of sulfonamides in edible catfish, shrimp and salmon tissues by high-performance liquid chromatography with postcolumn derivatization and fluorescence detection. J. Chromatogr. B 2006, 840, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Sangjarusvichai, H.; Dungchai, W.; Siangproh, W.; Chailapakul, O. Rapid separation and highly sensitive detection methodology for sulfonamides in shrimp using a monolithic column coupled with BDD amperometric detection. Talanta 2009, 79, 1036–1041. [Google Scholar] [CrossRef] [PubMed]
- Done, H.Y.; Halden, R.U. Reconnaissance of 47 antibiotics and associated microbial risks in seafood sold in the United States. J. Hazard. Mater. 2015, 282, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Thammasoontaree, N.; Rattanarat, P.; Ruecha, N.; Siangproh, W.; Rodthongkum, N.; Chailapakul, O. Ultra-performance liquid chromatography coupled with graphene/polyaniline nanocomposite modified electrode for the determination of sulfonamide residues. Talanta 2014, 123, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Dickson, L.C. Performance characterization of a quantitative liquid chromatography-tandem mass spectrometric method for 12 macrolide and lincosamide antibiotics in salmon, shrimp and tilapia. J. Chromatogr. B 2014, 967, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, T.; Jin, H.; Jing, T.; Song, B.; Zhou, Y.; Mei, S.; Lee, Y. Rapid and selective extraction of multiple macrolide antibiotics in foodstuff samples based on magnetic molecularly imprinted polymers. Talanta 2015, 137, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Alarcón-Flores, M.I.; Romero-González, R.; Vidal, J.L.M.; Frenich, A.G. Multiclass determination of phytochemicals in vegetables and fruits by ultra high performance liquid chromatography coupled to tandem mass spectrometry. Food Chem. 2013, 141, 1120–1129. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhou, R.; Zhao, Y.F.; Wu, Y.N. Analysis of Chloramphenicol in Shrimps by Gas Chromatography Tandem Mass Spectrometry. Wei Sheng Yan Jiu 2005, 34, 584–587. [Google Scholar] [PubMed]
- Shi, X.; Song, S.; Sun, A.; Liu, J.; Li, D.; Chen, J. Characterisation and Application of Molecularly Imprinted Polymers for Group-Selective Recognition of Antibiotics in Food Samples. Analyst 2012, 137, 3381–3389. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Shen, J.; Zhang, S.; Jiang, H.; Sun, Z. Determination of Chloramphenicol Residue in Fish and Shrimp Tissues by Gas Chromatography with a Microcell Electron Capture Detector. J. AOAC Int. 2005, 88, 57–60. [Google Scholar] [PubMed]
- Zhang, Q.J.; Peng, T.; Chen, D.D.; Xie, J.; Wang, X.; Wang, G.M.; Nie, C.M. Determination of Chloramphenicol Residues in Aquatic Products Using Immunoaffinity Column Cleanup and High Performance Liquid Chromatography with Ultraviolet Detection. J. AOAC Int. 2013, 96, 897–901. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.Y.; Chung, Y.J.; Shih, Y.C.; Hwang, D.F. Determination of Chloramphenicol Residues in Foods by Liquid Chromatography-Electrospray Tandem Mass Spectrometry. Taiwan. J. Agric. Chem. Food Sci. 2010, 48, 239–247. [Google Scholar]
- Mackie, J.; Marley, E.; Donnelly, C. Immunoaffinity Column Cleanup with LC/MS/MS for the Determination of Chloramphenicol in Honey and Prawns: Single-Laboratory Validation. J. AOAC Int. 2013, 96, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Sun, F.; Li, J.; Cheng, L.; Shen, J. Simultaneous Determination of Florfenicol and Florfenicol Amine in Fish, Shrimp, and Swine Muscle by Gas Chromatography with a Microcell Electron Capture Detector. J. AOAC Int. 2006, 89, 1437–1441. [Google Scholar] [PubMed]
- Peng, T.; Li, S.; Chu, X.; Cai, Y.; Li, C. Simultaneous Determination of Residues of Chloramphenicol, Thiamphenicol and Florfenicol in Shrimp by High Performance Liquid Chromatography-Tandem Mass Spectrometry. Fenxi Huaxue 2005, 33, 463–466. [Google Scholar]
- Tao, X.; Huang, H.; Liao, J.; Gao, P.; Huang, G.; Zeng, D. Simultaneous Determination of Residues of Chloramphenicol, Thiamphnicol, Florfenicol and Florfenicol Amine in Shrimp Muscle and Pork by High Performance Liquid Chromatography-Tandem Mass Spectrometry. J. Chin. Inst. Food Sci. Technol. 2014, 14, 232–238. [Google Scholar]
- Fernando, R.; Munasinghe, D.M.S.; Gunasena, A.R.C.; Abeynayake, P. Determination of nitrofuran metabolites in shrimp muscle tissue by liquid chromatography-photo diode array detection. Food Control 2015, in press. [Google Scholar] [CrossRef]
- Douny, C.; Widart, J.; de Pauw, E.; Silvestre, F.; Kestemont, P.; Tu, H.T.; Phuong, N.T.; Maghuin-Rogister, G.; Scippo, M.L. Development of an analytical method to detect metabolites of nitrofurans: Application to the study of furazolidone elimination in Vietnamese black tiger shrimp (Penaeus monodon). Aquaculture 2013, 376–379, 54–58. [Google Scholar] [CrossRef]
- Hossain, M.B.; Ahmed, S.; Rahman, M.F.; Kamaruzzam, B.Y.; Jalal, K.C.A.; Amin, S.M.N. Method Development and Validation of Nitrofuran Metabolites in Shrimp by Liquid Chromatographic Mass Spectrometric System. J. Biol Sci. 2013, 13, 33–37. [Google Scholar]
- Storey, J.M.; Clark, S.B.; Johnson, A.S.; Andersen, W.C.; Turnipseed, S.B.; Lohne, J.J.; Burger, R.J.; Ayres, P.R.; Carr, J.R.; Madson, M.R. Analysis of Sulfonamides, Trimethoprim, Fluoroquinolones, Quinolones, Triphenylmethane Dyes and Methyltestosterone in Fish and Shrimp Using Liquid Chromatography-Mass Spectrometry. J. Chromatogr. B 2014, 972, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Villar-Pulido, M.; Gilbert-López, B.; García-Reyes, J.F.; Martos, N.R.; Molina-Díaz, A. Multiclass Detection and Quantitation of Antibiotics and Veterinary Drugs in Shrimps by Fast Liquid Chromatography Time-of-Flight Mass Spectrometry. Talanta 2011, 85, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, T.; Nagano, C.; Kanda, M.; Hayashi, H.; Hashimoto, T.; Kanai, S.; Matsushima, Y.; Tateishi, Y.; Sasamoto, T.; Takano, I. Single-Laboratory Validation Study of Rapid Analysis Method for Multi-Class Veterinary Drugs in Milk, Fish and Shellfish by LC-MS/MS. ShokuhinEiseigakuZasshi 2013, 54, 335–344. [Google Scholar] [CrossRef]
- Wang, N.; Su, M.; Liang, S.; Sun, H. Sensitive Residue Analysis of Quinolones and Sulfonamides in Aquatic Product by Capillary Zone Electrophoresis Using Large-Volume Sample Stacking with Polarity Switching Combined with Accelerated Solvent Extraction. Food Anal. Methods 2015, 9, 1020–1028. [Google Scholar] [CrossRef]
- Li, H.; Kijak, P.J.; Turnipseed, S.B.; Cui, W. Analysis of veterinary drug residues in shrimp: A multi-class method by liquid chromatography-quadrupole ion trap mass spectrometry. J. Chromatogr. B 2006, 836, 22–38. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Kijak, P.J. Development of a Quantitative Multiclass/multiresidue Method for 21 Veterinary Drugs in Shrimp. J. AOAC Int. 2011, 94, 394–406. [Google Scholar] [PubMed]
- Kanda, M.; Nakajima, T.; Hayashi, H.; Hashimoto, T.; Kanai, S.; Nagano, C.; Matsushima, Y.; Tateishi, Y.; Yoshikawa, S.; Tsuruoka, Y.; et al. Multi-Residue Determination of Polar Veterinary Drugs in Livestock and Fishery Products by Liquid Chromatography/Tandem Mass Spectrometry. J. AOAC Int. 2015, 98, 230–247. [Google Scholar] [CrossRef] [PubMed]
- An, H.; Parrales, L.; Wang, K.; Cain, T.; Hollins, R.; Forrest, D.; Liao, B.; Paek, H.C.; Sram, J. Quantitative Analysis of Nitrofuran Metabolites and Chloramphenicol in Shrimp Using Acetonitrile Extraction and Liquid Chromatograph-Tandem Mass Spectrometric Detection: A Single Laboratory Validation. J. AOAC Int. 2015, 98, 602–608. [Google Scholar] [PubMed]
- Wang, Z.; Leng, K.; Sun, W.; Ning, J.; Zhai, Y. Simultaneous Determination of 33 Quinolone and Sulfonamide Residues in Eels and Shrimps by High Performance Liquid Chromatography-Tandem Mass Spectrometry. Chin. J. Chromatogr. 2009, 27, 138–143. [Google Scholar]
- El-Demerdash, A.; Song, F.; Reel, R.K.; Hillegas, J.; Smith, R.E. Simultaneous Determination of Nitrofuran Metabolites and Chloramphenicol in Shrimp with a Single Extraction and LC-MS/MS Analysis. J. AOAC Int. 2015, 98, 595–601. [Google Scholar] [PubMed]
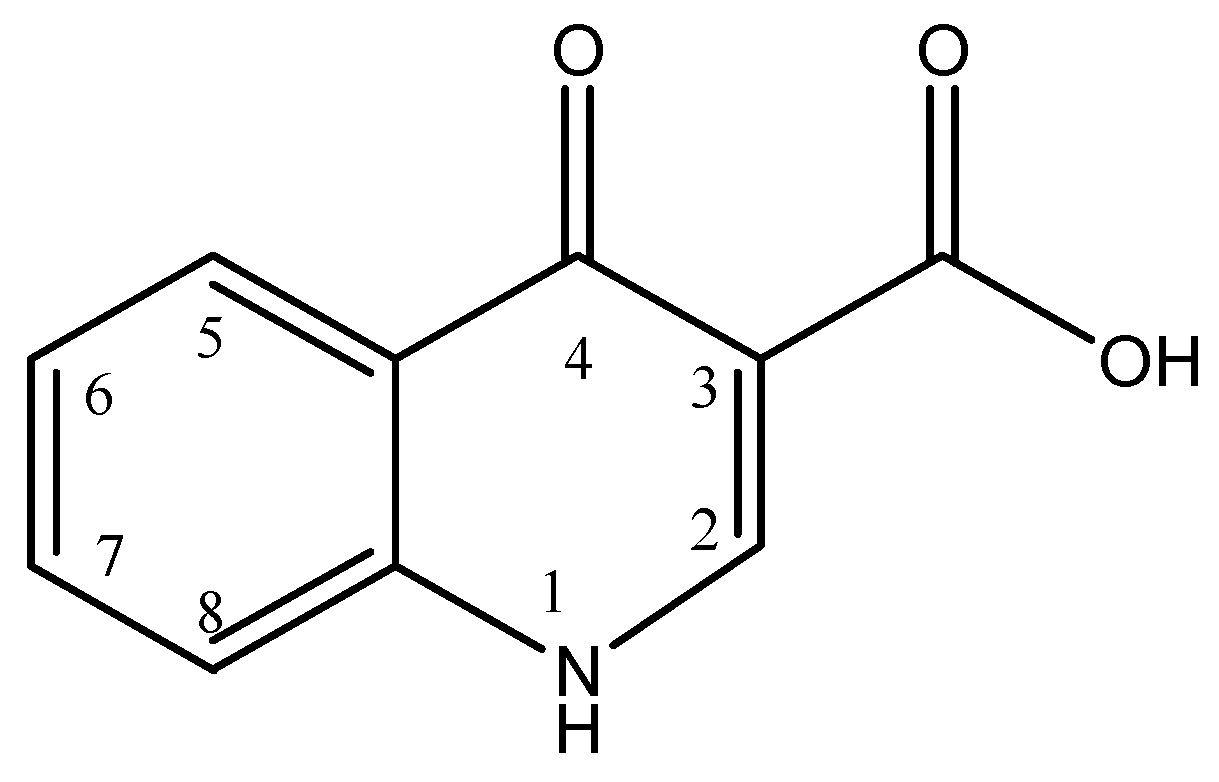

| Compound | R1 | R2 | R3 | R4 | R5 | R6 | R7 | |
|---|---|---|---|---|---|---|---|---|
| TC | Tetracycline | H | CH3 | OH | H | N(CH3)2 | H | H |
| OTC | Oxytetracycline | H | CH3 | OH | OH | N(CH3)2 | H | H |
| CTC | Chlortetracycline | Cl | CH3 | OH | H | N(CH3)2 | H | H |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Analytes | Sample | Sample Preparation | Analytical Technique | LOD-LOQ, CCα-CCβ | Recovery (%) |
|---|---|---|---|---|---|---|
| [29] | ENR and CIP | fish and prawn | Homogenized prawn tissue (1 g), spiked (100 μL IS, 3 μg/mL), extraction (10 mL ACN, vortex (1 min), shaker (15 min), centrifuged (10 min, 3700 g). Supernatant evaporated (dryness, 37 °C), add ammonium acetate buffer, vortex (15 s), sonicated (15 min). Purification (SPE cartridges, SDB-RPS), conditioned (successively 2 × 1 mL MeOH, 2 × 1 mL water and 2 × 1 mL ammonium acetate buffer), sample loaded, cartridge dried (centrifugation, 5 min, 3700 g), eluted (4 mL elution solution). Evaporation to dryness, 37 °C, reconstituted (300 μL formic acid), vortex (15 s), filtration (0.2 μm). | LC-MS/MS | LOD (μg/kg): ENR: 4, CIP: 3 | ENR: 94.0–106.0, CIP: 97.0–103.0 |
| LOQ (μg/kg): ENR: 14, CIP: 10 | ||||||
| CCα (μg/kg): ENR and CIP: 111 | ||||||
| [30] | CIP, DAN, ENR and SAR | salmon, shrimp and tilapia | Tissue (4 g), mixed with acidic ACN (16 mL), CH2Cl2 added (to 25 mL), rotated (10 min), centrifuged (10 min, 2000 rpm), supernatant (10 mL) removed, evaporated (45 °C, N2, to 2 mL). SPE column preconditioned (2 mL ACN), samples rinsed (2 × 2 mL ACN), passed through column, add 1 mL ACN. Eluent collected, evaporated (dryness, 45 °C, N2), residue reconstituted (vortexing, 200 μL ACN, deionized water 800 μL), hexane (1 mL) added, vortexed, centrifuged (1000 rpm, 5 min), aqueous layer filtered (0.2 μm syringe filter). | UPLC-MS/MS | LOD (ng/g): CIP: 0.13, DAN: 0.14, ENR: 0.19, SAR: 0.14 | CIP: 63.0–117.0, DAN: 71.0–87.0, ENR: 72.0–92.0, SAR: 95.0–125.0 |
| LOQ (ng/g): CIP: 0.4, DAN: 0.43, ENR: 0.56, SAR: 0.41 | ||||||
| [31] | OFL, NOR, CIP and LOME | prawn | Sample (5 g) dissolved (30 mL, 1% acetic acid ethanol solution), homogenized, centrifuged (4500 rpm, 5 min). Sample poured into SPE cartridge, adsorbed quinolones washed (10 mL MeOH, water, MeOH in order, 2.0 mL/min), quinolones absorbed eluted (10 mL, 25% ammonia MeOH, 1.5 mL/min). Eluent evaporated (N2, 35 °C), dissolved (mobile phase), filtered (0.45 μm filter). | HPLC-CL | LOD (ng/mL): OFL: 0.43, NOR: 0.36, CIP: 0.40, LOME: 2.4 | OFL: 90.3–101.4, NOR: 89.5–107.8, NOR: 88.0–107.2, LOME: 94.4–106.0 (prawn sample with shell) |
| LOQ (ng/mL): - | OFL: 88.3–99.8, NOR: 95.9–109.4, NOR: 91.2–107.0, LOME: 88.9–103.4 (peeled prawn sample without shell) | |||||
| [32] | ENR and CIP | prawns | Prawn muscle (1 g) extracted (5 mL MeOH: CH3COOH, 98:2 v/v), vortexed (2 min), sonication (10 min), centrifuged (1968 g, 10 min), pellet tissue extracted, evaporated (to 2 mL, 50 °C, N2), filtration, water: CH3COOH (10 mL, 98:2 v/v) added. Sample applied to SPE cartridge (C18, 500 mg, 6 mL), washed (6 mL water: phosphoric acid 1 M, 3:2 v/v), cartridges preconditioned and equilibrated (6 mL MeOH and 6 mL Milli-Q water), eluted (6 mL MeOH: phosphoric acid 1 M, 9:1 v/v and 4 mL MeOH). Eluate evaporated (dryness, N2, 50 °C), residue re-dissolved (1 mL Tris buffer, pH 9.1), filtered (syringe filter 0.45 μm, nylon). | HPLC-FLD | LOD (μg/g): ENR: 0.015, CIP: 0.025 | ENR: 88.43 (ave.), CIP: 80.41 (ave.) |
| LOQ (μg/g): - | ||||||
| [33] | 9 FQ: MARB, OFL, NOR, CIP, ENR, DAN, ORB, DIF and SAR and 3 AQ OXO, NAL and FLU | muscle (cattle, swine and chicken), liver (chicken), raw fish (shrimp and salmon), egg (chicken), and processed food (ham, sausage and fish sausage) | Sample (2 g), ACN/MeOH (20 mL, 1:1) added, homogenized (20 s), centrifuged (2500 g, 5 min), supernatant diluted (×2 with MeOH). Diluted extract (1 mL) added to the Fe3+ IMAC cartridge, allowed to pass, cartridge washed (1 mL MeOH and water), eluted (0.5 mL McIlvaine-EDTA-NaCl buffer). | LC-FLD | LOQ (mg/kg): DAN: 0.8, SAR: 6.5, ORB, DIF, OXO, FLU: 1.5, NOR, OFL, CIP, ENR: 2.5, NAL: 3, MARB: 50 | MARB: 88.2–89.5, OFL: 76.6–87.5, NOR: 85.4–86.4, CIP: 86.1–91.8, ENR: 88.2–90.0, DAN: 98.7–103.5, ORB: 87.3–88.7, DIF: 86.8–92.3, SAR: 81.1–81.8, OXO: 81.6–84.2, NAL: 87.7–94.9, FLU: 83.1–85.3 |
| [34] | FLU, NAL and OXO | shrimps | Samples spiked (100 μL IS), extraction (5 mL ACN), homogenized, centrifuged (10 min, 4000× g, 20 °C), supernatants transferred, re-extraction. ACN extracts combined, ammonia solution (2 mL, 0.1 mol/L) and n-hexane (2 mL) added, vortexed, centrifuged, n-hexane supernatant removed, procedure subsequently repeated (without ammonia). ACN extract evaporated (water-bath, 45 °C, vacuum), HCl (6 mL 0.1 mol/L) and ethyl acetate (6 mL) added, vortexed, centrifuged, extraction twice replicated. Acetate supernatants combined, water-bath, 45 °C, vacuum, evaporation to dryness, 40 °C, N2), dissolved (300 μL MeOH, ultrasonic bath). | HPLC-FLD | LOD (μg/kg): FLU: -, NAL: 6.9, OXO: - | OXO: 73.3–84.5, NAL: 80.4–90.4, FLU: 79.2–88.3 |
| CCα (μg/kg) FLU: 559.7, NAL: 10.3, OXO: 110.1 | ||||||
| CCβ (μg/kg) FLU: 610.9, NAL: 13, OXO: 117.3 | ||||||
| [35] | OXO, FLU and NAL | shrimp | Sample (2.0 g), fortification, vortexed (10 s), equilibrated (15 min), ethyl acetate (12 mL) and anhydrous sodium sulfate (2 g) added, shaken (1 min), centrifuged (1500 rpm, 10 min, 4 °C), supernatant transferred, evaporated (50–55 °C, N2), sample re-extracted (additional 12 mL ethyl acetate, as above). Supernatant added to original, evaporated (dryness or until oily residue), residue re-dissolved (2 mL, 0.2% formic acid), vortex (30 s), hexane (2 mL) added, mixed, centrifuged (5 min, 2600 rpm, 4 °C), hexane layer discarded (aspiration), aqueous liquid filtered (0.45 μm glass microfiber syringe filter). | LC-FLD, LC-MS/MS | LC-FL: | OXO: 88.0–97.7 (ave. 92.6), FLU: 77.1–80.7 (ave. 79.3), NAL: 78.6–81.7 (ave. 79.8) |
| MDL (ng/g): OXO: 3, FLU: 2.7, NAL: 2.3 | ||||||
| LOQ (ng/g): OXO: 9, FLU: 8.1, NAL: 6.9 | ||||||
| [36] | FLU, OXO and NAL | catfish, shrimp, and salmon | Sample (2.5 g) fortified, equilibrated (15 min), vortex-mixed (30 s, 5 mL 1% acetic acid), ACN (10 mL) and NaCl (2 g) added, shaken (5 min, 2500 rpm), centrifuged (10,000 rpm, 5 min, 4 °C), organic layer, transferred, evaporated (55 °C, dryness, N2,5 psi first 5 min, then 30–35 min at 12–15 psi). Reconstitution solution (2.5 mL) added to dried extracts, sonicated (1 min), vortex-mixed (30 s), centrifuged (10 min, 17,250 g, 4 °C), supernatants passed through filters (0.2 μm nylon syringe). Aliquots of each sample (3 mL) spotted into individual wells (96-well LazWell microtiter plate), evaporate (dryness, room temperature). | LDTD-MS/MS | MDL (ng/g): FLU: 1.7, OXO: 2.6, NAL: 4.4 | FLU: 79.0–88.0 (ave.), OXO: 91.0–95.0 (ave.), NAL: 100.0–101.0 (ave.) |
| LOQ (ng/g): FLU: 7.9, OXO: 17.3, NAL: 7.0 | ||||||
| [37] | FLU and OXO | fish and shellfish (salmon, sea trout, sea bass, gilt-head bream, megrim and prawns) | Sample (200 mg) and supramolecular solvent (400 μL) mixed, micro PTFE-coated bar introduced, vortex-shaken (2500 rpm, 15 min), thermostated (15 °C), centrifuged (15,000 rpm, 15 min). | LC-FLD | CCα (g/kg): OXO: 104, FLU: 611 | FLU: 100.0–102.0, OXO: 100.0–101.4 |
| CCβ (g/kg): OXO: 109, FLU: 622 (salmon) | ||||||
| [38] | CIP | shrimp | Sample (5 g) homogenated, dispersed in trichloroacetic acid aqueous solution (15%, w/v), stirred (2 h, 398 K), centrifugation and filtration, extraction solution collected and spiked. yeast@MIPs or yeast@NIPs (5 mg) dispersed in spiked samples (10 mL), incubated (6 h, 298 K). yeast@MIPs or yeast@NIPs collected (centrifugal filtration), washed (10 mL MeOH-acetic acid solution, 59:1 v/v), extracts dried (N2, 298 K), residues redissolved (0.4 mL MeOH). | HPLC | - | 86.4 |
| [43] | MQCA and QCA | porcine, chicken (muscles and livers), fish and shrimp (muscles) | Samples deproteinated (5% metaphosphoric acid in 10% MeOH), LLE, cleanup (solid phase extraction, mixed mode anion-exchange columns). | LC-MS/MS | LOQ (μg/kg): MQCA and QCA: 0.1 | MQCA and QCA: 62.4–118.0 |
| [44] | DES-CIP, NOR, CIP, DAN, ENR, ORB, SAR, and DIF | shrimp | Shrimp tissue extracted (ammoniacal ACN), extract defatted, evaporated, dissolution (basic phosphate buffer). | LC-FLD-MS | LOD (ng/g): - | 75.0–92.0 |
| LOQ (ng/g): 0.1–1 | ||||||
| [45] | 8 FQ: (NOR, OFL, DAN, CIP, DES-CIP, ENR, SAR and DIF) and 4 AQ (OXO, FLU, NAL | salmon, trout, and shrimp | Drugs extracted with mixture of ethanol and 1% acetic acid, diluted (aqueous HCl), defatted (hexane), cation-exchange solid phase extraction. | LC-MS/MS | LOD (ng/g): | 57.0–96.0 |
| QNs: 0.1, FQNs: 0.4 | ||||||
| [46] | CIP, DAN, DIF, ENR, FLU, MARB, NAL, NOR, OFL, ORB, OXO and SAR | muscle, liver, chicken eggs, milk, prawn and rainbow trout | Sample extracted (ACN-water, 95:5), 1/5 of filtered extract diluted (water, keep ACN ratio at ca. 60%), passed through C18 mini-column, eluate evaporated (dryness), residues dissolved (MeOH-water, 30:70, v/v) | HPLC-FLD | LOD (μg/g): - | CIP: 75.7, DAN: 96.5, DIF: 77.9, ENR: 97.3, FLU: 75.7, MARB: 80.7, NAL: 71.0, NOR: 74.4, OFL: 96.1, ORB: 74.3, OXO: 70.2, SAR: 72.3 |
| LOQ (μg/g): 0.005 | ||||||
| [47] | 6 FQ | fish, shrimp and crab | Sample extraction (acid ACN), defatted (n-hexane, water removed (Na2SO4). | UPLC-MS/MS | LOD (μg/kg): 0.1 (all) | 76.9–95.9 |
| LOQ (μg/kg): 0.2 (all) | ||||||
| [48] | 12 FQ (MARB, NOR, ENR, CIP, DES-CIP, LOME, DAN, SAR, DIF, OFL, ORB and ENO) and 6 AQ (OXO, NAL, FLU, CIN, piromidic acid and pipemidic acid) | milk, chicken, pork, fish and shrimp | Extraction with ACN-1% HCOOH, diluted (10% ACN), defatted (hexane). | LC-MS/MS | CCα (ng/g): 0.18–0.68 | - |
| CCβ (ng/g): 0.24–0.96 (all) | ||||||
| [49] | MARB, NOR, CIP, LOME, DAN, ENR, SAR, DIF, OXO, NAL and FLU | chicken, pork, fish and shrimp | Extraction with 0.3% metaphosphoric acid and ACN (1:1, v/v), cleanup (HLB cartridge). | HPLC-FLD | LOD (ng/g): - | 71.7–105.3 |
| LOQ (ng/g): 5.0–28.0 | ||||||
| [50] | MARB, CIP, NOR, LOME, DAN, ENR, SAR, DIF, OXO and FLU | swine, chicken, and shrimp tissues | Samples (≤2.0 g) and small volume of organic reagent (≤4.6 mL) of a nonchlorinated solvent. | HPLC-FLD | LOQ (ng/g): - | 72.8–106.8 |
| LOQ (ng/g): 0.3–1 (all) |
| Reference | Analytes | Sample | Sample Preparation | Analytical Technique | LOD-LOQ, CCα-CCβ | Recovery (%) |
|---|---|---|---|---|---|---|
| [39] | TC, OTC, and CTC | shrimp and whole milk | Shrimp extracted (1–1.5 g sodium chloride, 10 mL succinic acid, tissue disruptor, blending 20–30 s), tissue disruptor rinsed (2 × 3 mL succinic acid), centrifuged (5 min, 4000 rpm, 4 °C), supernatant decanted (25 mL depth filter, into centrifuge tube with 1–1.5 g alumina and 5 mL, 0.01 M oxalic acid), re-extracted (10 mL succinic acid), blending (tissue disruptor), centrifuged (5 min, 4000 rpm, 4 °C), supernatant decanted (depth filter), filter washed (4 mL water), extracts shaken (10 s), centrifuged (5 min, 4000 rpm, 4 °C).(HLB) SPE columns (6 mL, 200 mg) conditioned (sequentially 4 mL MeOH, 4 mL water and 4 mL succinic acid, 4 mL succinic acid above column bed), extracts applied (flow rate 45 drops/min), column washed (4 mL water), dried (5 min, full vacuum), eluted (2.0 mL MeOH), extracts evaporated (dryness, 60 °C, N2, 15 min), ACN (1 mL) added , residue dissolved (2.0 mL, 0.1% HCOOH), vortexed. | LC-UV, LC-MS/MS | LOD (ng/g): - | OTC: 82.9 (ave.), TC: 93.2 (ave), CTC: 76.8 (ave) |
| LOQ (ng/g): 50 (all, ave.) | ||||||
| [40] | OTC, TC, CTC and DC | shrimp | Sample (2.50 g), Na2EDTA-McIlvaine buffer (12.5 mL, pH 4) added, blended (30 s), shaken (10 min), centrifuged (30 min, 3500 rps). Supernatant loaded into SPE cartridge, activated (10 mL MeOH and 10 mL Milli-Q water), washed (10 mL Milli-Q water), eluted (10 mL MeOH), solvent removed at room temp., residues filtered (0.45 μm PTFE filter). | Ni-DIA electrode, HPLC/Ni-implanted electrode, HPLC/Ni-DIA electrode | Ni-DIA electrode: LOD (g/mL):OTC: 0.1, TC: 0.1, CTC: 0.5, DC: 0.5, LOQ (g/mL): - | Ni-implanted electrode: OTC: 84.8–102.5, TC:85.9–97.0, CTC: 91.48–97.9, DC: 88.4–103.7 |
| Ni-DIA electrode OTC: 83.3–96.5, TC: 88.4–96.9, CTC: 86.0–93.3, DC: 90.6–102.0 | ||||||
| [41] | TC, OTC, CTC and DC | prawns | Sample, homogenized, HPLC grade MeOH added, centrifuged (15 min, 3000 rpm), supernatant evaporated (dryness), dissolved (mobile phase, 0.1% formic acid in MeOH), filtered (0.22 μm membrane filter). | LC-MS/MS | LOD (ng/g): TC: 11, OTC: 12, CTC: 20, DC: 13 | TC: 91.0–98.0, OTC: 81.0–99.0, CTC: 84.0–101.0, DC: 80.0–85.0 |
| LOQ (ng/g): TC: 19, OTC: 20, CTC: 20, DC: 20 | ||||||
| [42] | OTC, TC, CTC, MNC, MTC, DMC and DC | egg, fish and shrimp | Extraction with Dionex accelerated solvent extractor 200 using MeOH and 1 mmol/L TCA at 80 °C/85 bar/pH 4.0. Sample (5 g) mixed (5 g Na2EDTA-washed sand), packed in extraction cell (circular glass microfiber filters, 1.98 cm, above and below the packing). Resulting extracts diluted, evaporated (dryness, 40 °C, N2), residue dissolved (1 mL mobile phase), vortexed, filtered (0.22 μm nylon Millipore chromatographic filter). | HPLC | CCα (μg/kg): MNC: 5.5, OTC: 101.8, TC: 102.2, DMC: 6.5, CTC: 106.8, MTC: 8.8, DC: 13.0 | OTC: 80.5–101.8, TC: 81.5–85.4, CTC: 78.7–85.7, DC: 83.4–89.1, DMC: 82.1–85.3, MNC: 80.4–99.7, MTC: 81.4–86.2 |
| CCβ (μg/kg): MNC:7.8, OTC:104.2, TC: 104.4, DMC:8.6, CTC:108.1, MTC: 10.5, DC: 15.3 |
| References | Analytes | Sample | Sample Preparation | Analytical Technique | LOD-LOQ, CCα-CCβ | Recovery (%) |
|---|---|---|---|---|---|---|
| [51] | CAP | fish, shrimp, poultry, eggs, bovine and swine | Samples homogenized, IS and phosphate extraction solution (4 mL) added, ultrasonic bath (15 min), centrifugation (3000 g, 10 min).Supernatant transferred, 4.5 mL ethyl acetate added, vortexed (1 min), centrifuged (3000 g, 10 min). Organic layer transferred, evaporated (45 °C, N2). Residue re-suspended (300 μL MeOH water, 50:50 v/v), vortexed (20 s). | LC-ESI-MS/MS | LOQ (ng/g): 0.03, LOQ (ng/g): 0.1 | 98.3–100.0 |
| [52] | CAP | honey, shrimp, and poultry meat | Sample (1 g), spiked (50 μL IS, 10 ng/mL), sodium sulfate anhydrous (1 g) and ethyl acetate (7 mL) added, shaken (20 min), centrifuged (15 min, 2000 rpm). Supernatant transferred, ethyl acetate (7 mL) added to sediment, both supernatants combined, evaporated (dryness, N2, 40 °C).ACN (1 mL) added on residues, evaporated (N2, 40 °C), re-suspended (500 μL MeOH/H2O, 10:90), vortexing (15 s), filtered | LC-ESI–MS/MS | CCα (μg/kg): 0.03, CCβ (μg/kg): 0.04 | - |
| [53] | CAP | shrimp | Homogenized shrimp meat (3.0 g), water (2 mL) added, spiked (5d-CAP IS, final concentration 0.5 mg/kg), ethyl acetate (5 mL), shaken (10 min, 100 rpm), vortexed (30 s), centrifugation (3000 g, 10 min). Extract evaporated (dryness, N2, water bath, 45 °C). Residue dissolved (1 mL, 1 M ammonium acetate and 4 mL petroleum ether), vortexed (60 s), centrifuged (5 min, 3000 g), upper phase discarded, isooctane (2 mL) added, vortexed (30 s), centrifuged (3000 g, 10 min), upper phase discarded, ethyl acetate (3 mL), vortexing (60 s), centrifugation (3000 g, 5 min). Organic layer collected, reduced (dryness, N2, water bath, 45 °C. Residue dissolved (0.5 mL hexane:CCl4, 1:1 v/v), mixed (0.7 mL HPLC-grade CAN-water, 1:1 v/v), vortexed (30 s), centrifuged (4000 g, 6 min), upper phase filtered (0.22 μm PVDF syringe filter). | LC-ESI-MS-MS | CCβ (mg/kg): 0.057, CCβ (mg/kg): 0.098 | 95.88–96.96 |
| [54] | CAP | honey, fish and prawns | Homogenized tissue (1.0 g) weighed, spiked (50 μL d5-CAP IS, 6 ng/mL), vortexed (30 s), allowed (20 min), ACN added (5 mL), vortexed (15 s), shaken (20 min, 180 rpm), centrifuged (5 min, 4000 rpm). Supernatant transferred; chloroform added (5 mL), vortexed (15–20 s), agitation and centrifugation, centrifugation (5 min, 2000 rpm), chloroform layer discarded. ACN phase evaporated (dryness, N2, water bath at 40–45 °C). Residue re-constituted (1 mL of mobile phase, water–ACN, 90:10 v/v). | LC-ESI-MS/MS | CCα (mg/kg): 0.04, CCβ (mg/kg): 0.06, LOD (mg/kg): 0.02, LOQ (mg/kg): 0.06 | 85.5–115.6 (all) |
| [55] | CAP | shrimp, crayfish and prawns | Freeze-dried samples (2 g), reconstituted (water, original sample 10 g, IS (d5-CAP) added, extraction (ACN/4% NaCl, 1:1 v/v, 20 mL), homogenized, centrifuged (4000 rpm, 15 min). Supernatant separated, de-fatted (2 × 10 mL n-hexane), ethyl acetate (7 mL) added, vortexed, supernatant transferred, extraction repeated. Combined supernatants evaporated (dryness), re-dissolved (3 mL of water/ACN, 95:5 v/v). Re-dissolved sample applied to C18 cartridge (500 mg, Separtis). Cartridge preconditioned (10 mL MeOH and 10 mL water/ ACN, 95:5 v/v), sample eluted (3 mL water/ACN, 45:55 v/v), ethyl acetate (4 mL) added to eluted sample, vortexed, extraction repeated. Combined extracts evaporated (dryness, N2), residue re-dissolved (1 mL of acetone/toluene, 20:80 v/v), applied onto a Silica cartridge (1 g). Cartridge preconditioned (6 mL of acetone/ toluene, 20:80 v/v), washed (2 ×3 mL acetone/toluene 20:80 v/v), eluted (6 mL acetone/ toluene, 70:30 v/v), extract evaporated (dryness), derivatization mixture (50 μL, BSA/n-heptane, 1:1 v/v) added, react (45 min, 60 °C). | GC/NCI/MS | CCα (mg/kg): 0.074, CCβ (mg/kg): 0.087 | 95.0 (all) |
| [56] | CAP and CAP glucuronide | honey and prawns | Sample (3 g) homogenized, 7 mL ACN added, centrifugation (10 min, 3900 G, 4 °C). Supernatant applied to cartridge (10 mL Chem-Elut, 5 min), elution (15 mL and 10 mL CH2Cl2), evaporation (dryness, 45–50 °C), residue re-suspended (5 mL hexane:ethyl acetate, 50:50 v:v), extract loaded (pre-conditioned SPE cartridge, 500 mg, 3 cc). SPE cartridge washed (3 mL ethyl acetate), eluting (3 mL ethyl acetate:MeOH, 50:50 v/v), evaporation (dryness, N2, 45–50 °C), dissolved (300 μL HPLC grade water). | Biacore Q biosensor, LC–MS/MS | Biosensor: CCα (μg/kg): 0.04, CCβ (μg/kg): 0.17 | - |
| LC-MS/MS: CCα (μg/kg): 0.09, CCβ (μg/kg): 0.17 | ||||||
| [57] | CAP | milk and shrimp | Samples (10 g), spiked, placed statically (15 min), phosphate buffer (40 mL, 0.05 mol/L, pH 7.0) added, vortexed (2 min), sonicated (15 min), centrifuged (1.4 × 103 g, 10 min). Supernatant transferred, to precipitate proteins TCA in water (3 mL, 15%) added, vortexed (2 min), centrifugation (1.4 × 103 g, 10 min), filtered (microfilters, 0.45 μm). Eluent samples from MISPE cartridges evaporated (dryness, N2), re-dissolved (mobile phase). | HPLC-UV | - | 84.9–89.0 |
| [58] | CAP, THI, FFC and FFC amine | shrimp and fish | Sample (1 g) placed into a mortar, blended (2 g, C18 material-dispersion adsorbent, 5 min) with a pestle, homogeneous mixture transferred in glass column (300 × 15 mm i.d.) with degreased cotton packed at the bottom/top of the sample, column tightly compressed. Extraction solvent mixture ethyl acetate-ACN-25% NH4OH (10/88/2, v/v/v, 5 mL) used for elution, eluate dried (N2, 50 °C), residue reconstituted (1.0 mL 5% MeOH in 0.1% formic acid 5 mmol/L CH3COONH4). | LC–MS/MS | CCα (μg/kg): FFC: 0.01, FFC amine: 0.05, THI: 0.07, CAP: 0.01 | 84.0–98.8 |
| CCβ (μg/kg): FFC: 0.05, FFC amine: 0.11, THI: 0.13, CAP: 0.04 | ||||||
| [59] | CAP, FFC and THI | shrimp | Sample (0.5 g), 500 μL working standard solution added, homogenized, dehydrated (0.5 g sea sand and 1 g anhydrous Na2SO4) in a glass mortar. Dry mixture placed into the SFE chamber, modifier introduced, chamber closed and attached to SFE system. Extracted substances collected in situ (silylation in a glass tube), tube filled with solvent containing derivatization reagent (20 mL, Sylon BFT), placed in a column oven. After extraction, solvent evaporated (dryness, N2), residue resolved (200 μL ethyl acetate). | NCI–GC/MS | LOD (pg/g): CAP: 8.7, FFC: 15.8 and THI:17.4 | CAP: 92.0, FFC: 87.0, THI: 85.0 |
| [60] | CAP | shrimp | Sample (10 g) homogenized, extracted (×2, ethyl acetate), acetate extracts evaporated, residue dissolved (15 mL salting out solution), fatty components removed (n-hexane), extraction (ethyl acetate), extract evaporated, residue dissolved (5 mL ACN-water, 10:90 v/v). Solution pumped through sol-gel filter column (flow-rate 0.5 mL/min) on-line coupled to immunoaffinity column (containing 0.67 mg anti-CAP antibodies). Flushing (10 mL ACN-water, 10:90 v/v, 0.5 mL/min), filter column removed, IAC washed (10 mL ACN-water, 10:90 v/v), eluting CAP (10 mL ACN-water, 40:60 v/v, 1.0 mL/min). Eluate extracted (2 × 3 mL ethyl acetate), combined extracts dried (2 g Na2SO4), centrifugation (1580 g, 5 min), ethyl acetate phase decanted, evaporated (N2), residue dissolved (1 mL ACN-water, 10:90 v/v). | HPLC-UV | LOD (ng/g): 1.8, LOQ (ng/g): - | 68.0 |
| [61] | CAP, THI and FFC | shrimp | Matrix solid phase dispersion: SPE cartridge conditioned (2 mL MeOH and 2 mL water), frits and sorbent removed, sorbent placed in a beaker with 0.5 g homogenized shrimp (spiked with 400 μL of mixture of three antibiotics), blending, sonicated (10 min). SPE cartridges repacked (one frit at the bottom, then sorbent/spiked sample, second frit on top, compressed with glass stirring rod), cartridge washed (1 mL ultra pure water), sequential elution (1 mL ACN and then 1 mL MeOH).Evaporation (dryness, water bath, 40 °C, under stream of nitrogen), dry residue dissolved (400 μL aqueous solution of lamotrigine, 10 ng/mL), filtration (syringe filter, 0.2 μm). | HPLC | LOQ (μg/kg): 20 (all), CCα (μg/kg): THI: 58.8, FFC: 1030.8, CAP: 59.2, CCβ (μg/kg): THI: 64.6, FFC: 1046.8, CAP: 63.8 | THI: 81.3–114.5, FFC: 72.0–103.3, CAP: 89.1–120.6 |
| [71] | CAP | shrimp tissue | Samples spiked (isotopically labeled internal standard, d5-Cap), ethyl acetate extraction, defatted (hexane), cleanup (SPE C18). Elute evaporated, derivatized with Sylon BFT. | GC/MS-MS | LOQ (ng/g): 0.3 | 95.0–111.0 (ave.) |
| [72] | CAP, THI, FFC and FFC amine | shrimp | MISPE | LC | LOD (μg/kg): CAP: 0.016, THI: 0.093, FFC: 0.102, FFC amine: 0.029, LOQ (μg/kg): - | 92.4- 98.8 (all) |
| [73] | CAP | fish and shrimp | Samples extracted (ethyl acetate), defatted (hexane), derivatized (Sylon BFT). | GC-MS | LOD (ng/g): 0.04 (all), LOQ (ng/g): 0.1 (all) | 69.9–86.3 (all) |
| [74] | CAP | aquatic products | Samples extracted with (ethyl acetate-NH4OH, 98:2 v/v), cleanup (IAC). | HPLC-UV, HPLC-MS/MS | LOD (μg/kg): -, LOQ (μg/kg): 0.25 (all) | 92.0–97.3 (ave.) |
| [75] | CAP | chicken meat, fish meat, shrimp meat and honey | Analytes extracted (ethylacetate), defatted (n-hexane, LLE). | LC-MS/MS | - | 76.2 |
| [76] | CAP | honey and prawns | Sample shaken with buffer, centrifuged, applied to IAC | LC-MS/MS | LOD (μg/kg): 0.05, LOQ (μg/kg): - | 84.0%–108.0% |
| [77] | FFC and FFC amine | fish, shrimp, and swine muscle | Samples extracted, defatted (hexane), cleaned (SPE, Oasis MCX cartridges), eluate evaporated (dryness), derivatized. | GC-microcell electron capture detector | LOD (ng/g): FFC: 0.5, FFC amine: 1 (all), LOQ (ng/g): - | 94.1–103.4 (ave.) |
| [78] | CAP, THI and FFC | shrimp | Samples extracted (basic ethyl acetate), extracts defatted (L-L partition), cleaned (C18 SPE cartridge). | LC-MS/MS | LOD (ng/g): CAP and THI: 0.01, FFC: 0.05, LOQ (ng/g): - | CAP: 73.9–96.0, THI: 78.6–99.5, FFC: 74.9–103.7 |
| [79] | CAP, THI, FFC and FFC amine | shrimp muscle and pork | Samples extracted with 2% basic ethyl acetate, L-L partition, SPE. | HPLC-MS/MS | LOD (μg/kg): CAP: 0.001, THI: 0.020, FFC: 0.002, FFC amine: 0.003 (all), LOQ (μg/kg): - | 78.17–99.86 |
| Reference | Analytes | Sample | Sample Preparation | Analytical Technique | LOD, LOQ, CCα, CCβ | Recovery % |
|---|---|---|---|---|---|---|
| [62] | SDZ, SMR, SME, SMZ, SMX, SDM | Fish, Shrimp | Fish and shrimp samples (5.0 g), spiked at 3 levels, MISPE extraction, 1% acetic acid (10.0 mL), vortexed, sonicated (5 min), centrifuged (5.0 × 103 g, 5 min), supernatant applied to MISPE/NISPE cartridges, washed (5% ACN 1.0 mL in water—1% acetic acid), elution (3 mL MeOH/acetic acid 9/1 v/v), evaporation with N2 at 40 °C, dissolved with 0.5 mL 28% ACN in water, filtration (0.22 μm) | HPLC-UV | LOD (μg/kg): 8.4–10.9 | 85.5%–106.1% |
| LOQ (μg/kg): 22.4–27.7 | ||||||
| [63] | SDZ, STZ, SMZ, SMX, SMP, SCPD, SDM, SMM, SPZ, SDX, SSZ, SCP, SMT, SQX | flatfish, jacopever, sea bream, common eel, blue crab, shrimp, abalone | Sample (1 g), ACN (5 mL), homogenized (1 min), extraction × 2 -sonication (10 min), centrifugation (4500× g, 10 min), supernatant collected, add 100 mg C18, homogenization (30 s), powder dispersed, centrifugation (4500× g, 10 min), evaporation with N2 to dryness, dissolved with KH2PO4 (5 mM, 1 mL), filtration (0.45 μm) | HPLC-PDA Confirmation with LC-MS/MS | LOD (μg/kg): 3–6 LOQ (μg/kg): 9–18 | 51.8%–89.7% |
| [64] | SN, SDZ, STZ, SPD, SMR, SMZ, SMTZ, SMP, SCP, SMM, SDX, SMX, SDM, SQX | catfish, shrimp, salmon | Samples (10 g), 0.2% CH3COOH-MeOH-ACN (10 mL, 85:10:5), homogenized (30 s, 20000 rpm), ACN (90 mL), shaker (10 min), centrifugation (10 min), ACN (30 mL) for extraction, shaker, centrifugation, CH2Cl2 (60 mL), shake (3 min), leave 15 min, bottom layer to flask with boiling chips, extraction repeated, concentration to 2–3 mL, CH2Cl2:acetone (60:40, 5 mL). SPE on SCX cartridges preconditioned (2.5 mL acetone, 2.5 mL 0.2% acetic acid, 2.5 mL acetone), elution with acetone-0.4 M CH3COONH4 (50:50 v/v, 5 mL), evaporation with N2 to 2 mL. | HPLC-FLD | LOQ (ng/g): 1 | 67.3%–90.5%. |
| [65] | SG, SDZ, SMT, SMM, SMX, SDM, SQX | Shrimp | Homogenizes shrimp (2 g), Na2EDTA-Mcllaine’s buffer (10 mL), mixed, vortexed (5 min), sonicated, centrifuged (3500 rpm, 10 min), SPE with Oasis HLB (200 mg, conditioned with 5 mL Milli-Q water, 5 mL Na2EDTA-Mcllvaine buffer solution), elution with MeOH (7 mL), evaporation with N2, reconstituted with mobile phase (10 mL), filtered (0.45 μm). | HPLC-EC (BBD amperometric detection) | LOD (ng/mL): 1.2–3.4 | (Spiked samples 1.5, 5, 10 μg·g−1) 81.7 to 97.5% |
| LOQ (ng/mL): 4.1–11.3 | ||||||
| [67] | SDZ, SMR, SG, SSZ, SDM, SMM, SDX, SMX | Shrimp | Homogenized shrimp (2 g), Na2EDTA—Mcllvaine buffer, vortexed (5 min), sonicated, centrifuged (3500 rpm, 10 min), SPE Microcolumn VertipakTM HCP (conditioned with MeOH, Milli-Q water, Na2EDTA-Mcllvaine buffer), supernatant (10 mL), supernatant (10 mL) loaded, elution with MeOH (7 mL), filtration (0.20 μm pore size). | UPLC-ECD | LOD (ng/mL): 1.162–2.900 | - |
| LOQ (ng/mL): 3.336–20.425 ng·mL−1 |
| Reference | Analytes | Sample | Sample Preparation | Analytical Technique | LOD, LOQ, CCα, CCβ | Recovery % |
|---|---|---|---|---|---|---|
| [68] | Macrolides—(erythromycin, tylosin, josamycin, spiromicyn, neospiromycin, tilmicosin, gamithromycin, tildipirosin and oleandomycin) Lincosamides—Lincomycin, Pirlimycin, Clindamycin | Salmon, Shrimp, Tilapia | Homogenized sample (5 g), extraction with ACN (10 mL), water (1 mL), shaker (700 rpm), centrifugation (5 min, 400× g RCF), re-extraction with ACN, phosphate buffer (3 mL), shaker, centrifugation, the two supernatants centrifuged again (5 min, 6100× g RCF), SPE on Bond-Elut cartridge (pre-conditioned with water, 12% ACN), elution with methanolic CH3COONH4 (750 μL × 2), fat removal with water and hexane, vortexed, centrifuged (5 min, 1000× g RCF), evaporation with N2 to volume < 0.75 mL, methanolic CH3COONH4 (50 μL), mixed, MeOH to volume 1 mL, centrifugation (15 min, 2130× g, 5 °C). | LC-MS/MS | LOD (μg/kg): 0.5 | 47%–99% |
| [69] | Macrolides—ERY, ELAN, AZM, CLM, TIM, RXM, Quinolones—CIP, SPFX Amphenicols—CAP, TAP | Pork, Fish, Shrimp | Spiked samples, NaOH for hydrolysis of lipids (500 μL), extraction with ACN (20 mL), vortexed (15 min), centrifuged (5 min, 7000 rpm), supernatant with MMIPs (100 mg) mixed, magnetically removed, washed with ACN:water, elution with (10 mL) MeOH/50 mM KH2PO4 (pH 8), evaporation to dryness, residue reconstituted with mL ACN/25 mM KH2PO4 | UPLC-UV | LOD (μg/kg): 0.015–0.2 LOQ (μg/kg): 0.075–0.5 | - |
| [70] | Nitrofurans—Furazolidone, furaltadone, nitrofurazone, nitrofurantoin | Shrimp | Homogenized samples (2.5 g), added HCl aqueous and 2-NBA for derivatization, incubating overnight, neutralized with di-sodium hydrogen phosphate and NaOH,centrifugation (5 min, 4000 rpm), supernatants SPE Oasis HLB (conditioning with ethyl acetate, MeOH, Milli-Q water), cartridge washed with water, elution with ethyl acetate (6 mL), evaporation to dryness, redissolved with mobile phase (1 mL), filtered (0.20 μm) | UHPLC-QqQ-MS/MS | LOD (mg/kg):0.5–0.8, LOQ (mg/kg): 1 | - |
| [80] | Nitrofurans—nitrofuran metabolites, AOZ, AMOZ, SEM and AHD, | Shrimp | Homogenized sample (5 g), washed with MeOH (20 mL) mixing, centrifugation (10 min, 2500 rpm), washing with MeOH and water, HCl (10 mL), derivatization with 2-NBA, incubated overnight, Na3PO4·12H2O solution added, neutralized with NaOH (2 M), extraction with ethyl acetate, evaporation to dryness, reconstituted with 500 μL reconstitution solvent, extracted tree times with hexane, filtration (0.45 μm) | HPLC-UV | LOD (mg/kg): 2 | 107%–115% |
| [81] | Nitrofurans—AOZ, AMOZ, SEM and AHD | Shrimps | Sample (1 g) spiked at 2 μg/Kg, hydrolysis with HCl (5 mL), derivatization with 50 μL 2-NBA, overnight incubation, neutralizing with NaOH and phosphate buffer, extraction with ethyl acetate, evaporation to dryness, dissolution with HPLC grade water, filtration. | LC-IDMS/MS | CCα (μg/kg): 0.08–0.20 CCβ (μg/kg):0.13–0.85 | - |
| [82] | Nitrofurans—furalizone—AOZ, furaltadone—AMOZ, nitrofurazone—SEM and nitrofurantoin—AHD | Shrimps | Homogenized sample (1 g) washed with methanol, centrifuged (4 min 4000 rpm) repeated, HCl and 2-NBA to the sample, incubated overnight, neutralized with NaOH, ethyl acetate added (4 mL), centrifuged, extraction again, supernatant evaporation near dryness, residue dissolved with methanol, filtrated (0.45 μm) analysis, AMOZ-d5 internal standard | LC-MS/MS | CCα (μg/kg): 0.12–0.23, CCβ (μg/kg): 0.21–0.38 | 88%–110% |
| Reference | Analytes | Sample | Sample Preparation | Analytical Technique | LOD, LOQ, CCα, CCβ | Recovery (%) |
|---|---|---|---|---|---|---|
| [83] | 26 veterinary drugs: 13 SAs TRI, 3 FQ, 3 AQ, 3 TPM, 2 LC dyes metabolites, 1 hormone | fish and other aquaculture products (tilapia, catfish, eel, pangasius, sablefish,swai, salmon, trout, and shrimp | Tissue (4.0 g ± 0.03 g) weighed, SMZ-13C solution (0.040 mL) added, EDTA-McIlvaine buffer (2.0 mL) added, mixed (10 s). ACN (10 mL), p-TSA (0.100 mL), TMPD solution (0.100 mL), NaCl (2.0 g) and ceramic homogenizer pellet added, shaken (5 min), centrifuged (6000 rpm, 5 °C, 5 min) Organic layer transferred, ACN (10 mL) added, shaken (5 min), centrifuged. ACN layers combined, evaporated (dryness, water bath, 50–55 °C, N2). Residue reconstituted (2.0 mL the dissolution solution), mixed (30 s), sonicator (5 min), centrifuged (10,000 rpm, 5 °C, 5 min). 0.5 mL portion filtered (0.2 μm PTFE syringe filter). | LC-MS/MS | - | 98.0–104.0 (all, ave.) |
| [84] | BC, EQ, LMG, MG, MBZ, SDZ, SDM, SMZ, SMTZ, SN, SPD, STZ,TRO | shrimp | Five different sample treatment methodologies were tested: ACN extraction followed by cleanup by QuEChERS. Sample (10 g) homogenized, ACN containing 1% acetic acid (10 mL) added, shaken (1 min), anhydrous MgSO4 (4 g) and NaCl (1.75 g) added, shaking repeated (1 min).Extract centrifuged (3700 rpm, 3 min), supernatant (5 mL) (ACN phase) transferred to centrifuge tube (with 250 mg PSA and 750 mg anhydrous MgSO4), shaken (20 s), centrifuged (3700 rpm, 3 min), extract (2 mL) evaporated (near dryness, reconstituted (20% v/v MeOH in water, to a final volume of 2 mL), filtered (0.45 μm PTFE filter). | LC-TOFMS | LOD (μg/kg): BC-C12: 0.6, EQ: 7.1, LMG: 0.6, MG: 0.06, MBZ: 0.1, SDZ: 4.5, SDM: 0.3, SMZ: 0.1, SMTZ: 0.8, SN: 3.5, SPD: 0.5, STZ: 2.9, TRI: 0.7 | BC-C12: 53.0, EQ: 53.0, LMG: 90.0, MG: 118.0, MBZ: 118.0, SDZ: 82.0, SDM: 85.0, SMZ: 114.0, SMTZ: 33.0, SN: 115.0, SPD: 109.0, STZ: 81.0, TRI: 87.0 |
| ||||||
| ||||||
| ||||||
| ||||||
| [85] | 43 multi-class veterinary drugs (sulfonamides, quinolones, coccidiostats and antiparasites) | milk, fish and shellfish (salmon, tiger shrimp, red sea bream and bastard halibut) | Sample (5 g), working standard solution (50 μL or 500 mL) added, waiting (>30 min), water (5 mL) and HCOOH in ACN (15 mL, 1 vol. %, Method A) or ACN alone (15 mL, 1 vol. %, Method B) added, homogenized, magnesium sulfate (4 g), trisodium citrate dehydrate(1.5 g) and NaCl (2 g) added, shaken (1 min), centrifuged (1800× g, 10 min), supernatant transferred, dilution extraction solvent, portion of the solution transferred to a microtube, centrifuged (16,000× g, 10 min). | LC-MS/MS | LOQ (μg/kg): 1–10 (all) | 48.5–113.6 (all) (Method A) 11.1–116.5 (all) (Method B) |
| [86] | LOME, ENO, SAR, ENR, SDZ, SMD, SDMP | shrimp and sardine | Sample (5 g) and diatomite (1.5 g) mixed, transferred into extraction cell (ACN extraction solvent). Extraction conditions: oven temperature 60 °C, 3 min heat-up time, pressure 10.3 MPa, two static cycles, static time 5 min, flush volume 40% of extraction cell volume. Extract purged (pressurized N2, 90 s), evaporated (dryness, N2, 45 °C), residue dissolved (MeOH, to 1 mL), filtered (0.45 μm). | CZE | LOD (μg/mL): LOME: 0.025, ENO: 0.033, SAR: 0.025, ENR: 0.020, SDZ: 0.013, SMD: 0.013, SDMP: 0.013 | LOME: 88.9–94.8, ENO: 88.0–93.1, SAR: 87.6–95.7, ENR: 88.0–93.2, SDZ: 88.7–91.0, SMD: 86.7–90.0, SDMP: 85.4–88.8 |
| LOQ (μg/mL): LOME: 0.08, ENO: 0.10, SAR: 0.08, ENR: 0.07, SDZ: 0.04, SMD: 0.04, SDMP: 0.04 | ||||||
| [87] | SDZ, SMR, SMZ, SCP, SDM, SQX, ENR, SAR, DIF, OXO, NAL, FLU, LMV, LVG, MG, GV, OTC, TOLSa | Shrimp | blended shrimp (2 g), 100 μL standard, TCA & NH2OH-HCl added, homogenized, vortex (10 min), centrifugation (4000 rcf, 4 °C, 15 min), supernatant into solution sodium succinate (2.5 mL) and NaOH (10N, 280 μL)—pH 3.6 ± 0.1, Oasis HLB cartridge pre-conditioned (washed 3 mL ammonium formate buffer, 3 mL Milli- Q water, dried for 2 min), elution with MeOH (2 mL) CH3CN/MeOH 1:1 v/v (1 mL), to the elute ammonium formate buffer (20 mM, pH 3.9), EDTA (50 μL, 0.1 M), ascorbic acid (1 mg/mL in MeOH), evaporation with N2 till 0.8 mL, aliquot of 1:1 water/AC N added to fill 1 mL, centrifugation (14000 rpm, 10 min), analysis of the middle portion (~0.8 mL) | LC-ion trap-MS | 10 ng/g for SQX | 40%–90% |
| [88] | 21 veterinary drugs: SAs (SDZ, SMR, SMZ, SCP, SDM, SQX), TCs (OTC, TC, CTC), FQ (NOR, CIP, ENR, SAR, DIF, FLU, OXO,NAL) and cationic dyes (MG, GV, LMG, and LGV) | shrimp | Sample (2 g) extracted (×2, 2 different pH values), supernatant diluted (aqueous internal standard), online SPE automated sample cleanup. | HPLC-MS/MS | - | - |
| [89] | FQ, TCs, macrolides, lincosamides, SAs and others | livestock and fishery products | Extraction with two solutions of different polarity: highly polar compounds extracted with Na2EDTA-McIlvaine's buffer (pH 7.0) and medium polar compounds were extracted with ACN containing 0.1% HCOOH. Cleanup with SPE polymer cartridge, first extracted solution applied to the cartridge (highly polar compounds retained), second extracted solution applied to the same cartridge, both highly and medium polar compounds eluted. | LC-MS/MS | LOQ (μg/kg): 0.1–5 | - |
| [90] | CAP and nitrofuran metabolites | shrimp | Extraction steps: neutralization of hydrolysates, addition of ACN for extraction, salting out of organic phase from the ACN-aqueous mixture | LC-MS/MS | - | 98.6–109.2 (all) |
| [91] | 33 FQ and SAs | eels and shrimps | Sample extracted with acidified ACN, cleaned-up (hexane), concentrated (evaporator). | HPLC-MS/MS | LOD (μg/kg): 1.0, LOQ (μg/kg): 2.0 | 66.0–123.0 |
| [92] | AMOZ, AOZ, AHD, SEM CAP | shrimp | single extraction procedure | LC-MS/MS | LOD (ng/g): nitrofuran metabolites: 0.5, CAP: 0.3 | - |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samanidou, V.; Bitas, D.; Charitonos, S.; Papadoyannis, I. On the Extraction of Antibiotics from Shrimps Prior to Chromatographic Analysis. Separations 2016, 3, 8. https://doi.org/10.3390/chromatography3010008
Samanidou V, Bitas D, Charitonos S, Papadoyannis I. On the Extraction of Antibiotics from Shrimps Prior to Chromatographic Analysis. Separations. 2016; 3(1):8. https://doi.org/10.3390/chromatography3010008
Chicago/Turabian StyleSamanidou, Victoria, Dimitrios Bitas, Stamatia Charitonos, and Ioannis Papadoyannis. 2016. "On the Extraction of Antibiotics from Shrimps Prior to Chromatographic Analysis" Separations 3, no. 1: 8. https://doi.org/10.3390/chromatography3010008
APA StyleSamanidou, V., Bitas, D., Charitonos, S., & Papadoyannis, I. (2016). On the Extraction of Antibiotics from Shrimps Prior to Chromatographic Analysis. Separations, 3(1), 8. https://doi.org/10.3390/chromatography3010008