Abstract
The efficient separation of light hydrocarbons, particularly alkanes from their isomers (C5–C6), represents a significant and energy-intensive challenge for the petrochemical industry. Metal-Organic Frameworks (MOFs) offer promising solutions due to their exceptional porosity, surface area, and, crucially, their structural and chemical tunability. This study employs advanced computational methods, including Grand Canonical Monte Carlo (GCMC) simulations and Molecular Dynamics (MD), to systematically investigate the adsorption and separation of pentane isomers (n-pentane, isopentane, and neopentane) in the UiO-66 MOF family. Specifically, the impact of organic linker functionalization with -H (parent), -NH2, -CH3, and -COOH groups on adsorption isotherms, isosteric heats, and competitive behavior in mixtures is evaluated. The analysis provides a molecular-level view of host-guest and guest-guest interactions, elucidating the recognition and selectivity mechanisms governing the separation of these C5 isomers and the potential for engineering MOF materials for this application.
1. Introduction
The separation of linear alkanes from their branched isomers is an important operation in the petrochemical industry, with direct implications for the efficiency of processes such as catalytic cracking and reforming, as well as for the quality of final products like gasoline [1]. Specifically, enrichment in branched isomers within the C5–C6 fractions is crucial for increasing the fuel’s octane number (RON), which improves engine performance and reduces self-ignition or knocking [1,2]. This improvement in fuel quality not only impacts combustion efficiency but also has environmental consequences related to emissions [3,4].
However, achieving this separation is very difficult and costly using conventional technologies. Fractional distillation requires high energy consumption due to the minimal differences in boiling points between isomers. On the other hand, molecular sieving using zeolites, such as Zeolite 5A, although effective for separating n-alkanes, often shows limitations in capacity or in its ability to discriminate between different branched isomers [3,5]. The inefficiency and high energy cost of these traditional methods [6,7] drive the search for more efficient and sustainable alternative separation materials and processes.
In this context, Metal-Organic Frameworks (MOFs) have emerged as a promising class of adsorbent materials [8,9]. These porous crystalline solids, constructed through the self-assembly of metal nodes or clusters and organic linkers, possess unique characteristics such as ultra-high surface areas and, fundamentally, structural and chemical tunability [10]. The ability to design the size, shape, and chemical environment of the pores allows for optimizing host-guest interactions to discriminate molecules with subtle differences in size, shape, and polarizability, as is the case with alkane isomers [11,12]. Separation can be based on size/shape exclusion mechanisms (entropic effects) or differences in adsorption affinity (enthalpic effects), or a combination of both [13].
Within the vast diversity of reported MOFs, the UiO-66 family, based on Zr6O4(OH)4 clusters and 1,4-benzenedicarboxylic acid (BDC) linkers, is particularly attractive. A key property that has attracted much attention is its exceptional thermal, chemical, and hydrolytic stability. Seminal studies emerging shortly after its original synthesis [14] demonstrated that the robust Zr6 secondary building units impart remarkable stability even under aqueous or acidic conditions. For example, Kandiah et al. showed that UiO-66 maintains crystallinity in water and acidic environments [15], an observation corroborated and extended in subsequent studies that also examined the stability of functionalized variants [16]. Complementarily, modulated synthesis methods have been shown to produce monodisperse and highly stable microcrystals [17], consolidating UiO-66 as one of the most robust MOF platforms available. This stability is fundamental to consider modifications for specific separation applications.
Another major thrust in the UiO-66 literature is the general impact of linker functionalization on adsorption and separation properties. Incorporating groups like -NH2, -NO2, -OH, -CH3, or -COOH onto the BDC linker has been widely explored to tailor the pore environment and chemical reactivity. For instance, introducing -OH and -NO2 groups has been shown to adjust adsorption capacities for small molecules like CO2 and CH4 [18], while other modifications can modulate hydrophilicity/hydrophobicity profiles, affecting both adsorption isotherms and selectivity [19]. A wide range of post-synthetic modification strategies also reinforces the concept that functionalization is an effective tool for tuning host-guest interactions in UiO-66 [20]. Collectively, these studies justify the use of functionalized UiO-66 systems to optimize separation performance.
Despite these advances, a detailed and comparative molecular-level understanding of how different functional groups selectively modulate the interactions and packing of alkane isomers within the UiO-66 structure, especially under competitive conditions, remains an active area of research. Pentane isomers (linear n-pentane, mono-branched isopentane, and di-branched, quasi-spherical neopentane) represent an excellent model system for studying these effects due to their distinct molecular shapes. It is particularly interesting to understand how contrasting functional groups—such as the electron-donating and H-bond acceptor/donor (-NH2), the hydrophobic and sterically relevant (-CH3), and the polar and potentially acidic (-COOH)—modulate adsorption and selectivity for these C5 isomers compared to the parent MOF (-H).
In this context, molecular simulation has established itself as an indispensable tool for investigating adsorption/separation phenomena in MOFs at the molecular scale [21]. Techniques like Grand Canonical Monte Carlo (GCMC) allow probing the atomistic details of adsorption processes and competition in flexible or rigid mixtures [13]. Recent studies combine experimentation and theoretical calculations (e.g., DFT) to elucidate adsorption behavior even in functionalized variants of UiO-66, demonstrating that simulation is essential not only for predicting thermodynamic properties but also for clarifying the role of molecular-level interactions and structural defects [22].
In the present work, we perform a systematic computational study of the adsorption and separation of pentane isomers (n-pentane, including its relevant conformers, isopentane, and neopentane) in UiO-66-H, UiO-66-NH2, UiO-66-CH3, and UiO-66-COOH at 298 . Using GCMC simulations, we calculate single-component and mixture adsorption isotherms, as well as isosteric heats of adsorption. We compare the performance of the four MOFs and analyze the predictive accuracy of the Ideal Adsorbed Solution Theory (IAST). The main objective is to elucidate the specific impact of each functional group in modulating affinity and selectivity for these C5 isomers, providing a fundamental molecular understanding of the separation mechanisms and guiding the future design of optimized MOF materials for this industrially relevant application.
2. Computational Methods
The adsorption behavior of pentane isomers in the UiO-66 variants was investigated using a combination of Grand Canonical Monte Carlo (GCMC) simulations. GCMC simulations were performed using our in-house code [23,24] in the ensemble. MD simulations for the conformational analysis of n-pentane were carried out with the LAMMPS package [25] in the canonical (NVT) ensemble at 300 .
2.1. Computational Models
2.1.1. Modeling of Adsorbates (Pentane Isomers)
To model the adsorbates, a united-atom (UA) representation was employed for the three considered pentane isomers: n-pentane (nP), isopentane (iP), and neopentane (neoP). This coarse-graining approach balances computational efficiency with accuracy in predicting adsorption behavior in nanoporous materials. Intermolecular interactions for the pentane isomers were described using the Transferable Potentials for Phase Equilibria United-Atom (TraPPE-UA) force field [26,27], a widely validated model for hydrocarbons.
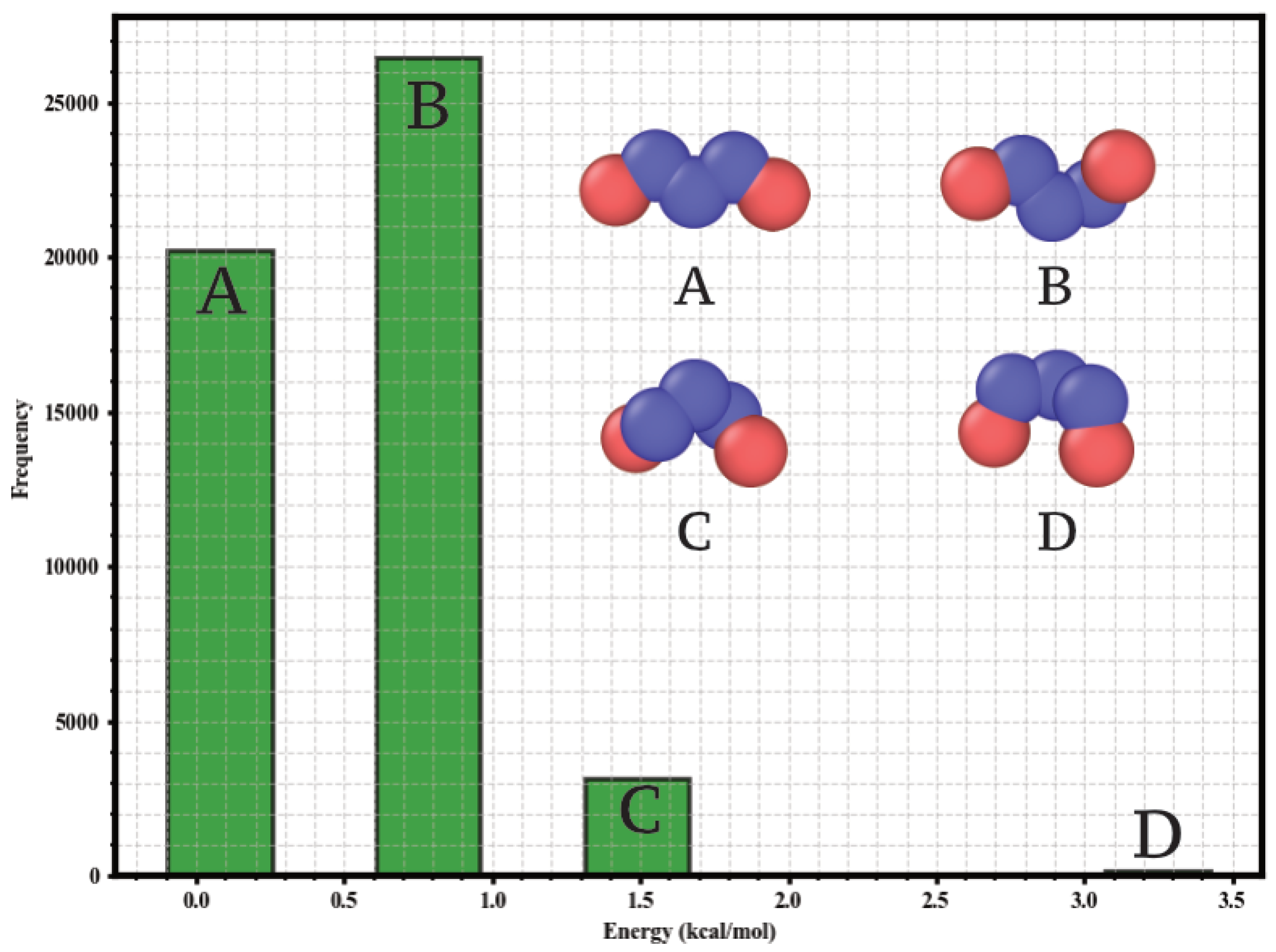
Given the conformational flexibility of n-pentane, an explicit analysis of its conformers was performed. MD simulations (NVT, 298 , 1 ns equilibration + 5 ns production) were conducted with LAMMPS using the TraPPE-UA force field and periodic boundary conditions (1 fs timestep, velocity-Verlet integrator, Nosé-Hoover thermostat) to sample the conformational space. From 50,000 configurations, a histogram of geometric minimization energies (Figure 1) was constructed, identifying four dominant populations corresponding to the tt, tg, gg+, and gg- conformers. These conformations agree with previous ab initio studies [28]. The equilibrium mole fractions determined at 298 and used in the GCMC simulations were 0.40 (tt), 0.53 (tg), 0.06 (gg+), and 0.01 (gg-).
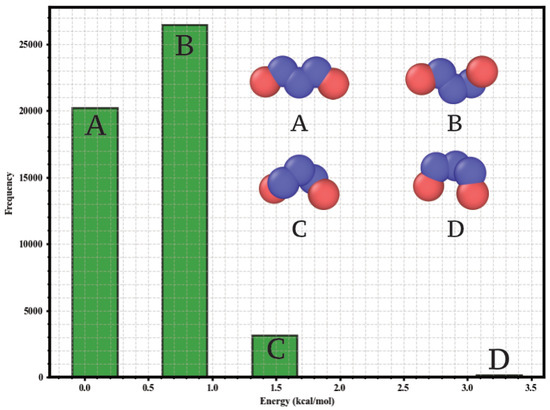
Figure 1.
Histogram of the geometric minimization energies for n-pentane conformers, constructed from 50,000 configurations sampled by MD at 298 K. Frequency of structures versus energy is shown. Superimposed are the united-atom model (CH3 and CH2 sites) and the representative structure for each conformational population.
2.1.2. Modeling of the MOF Network (UiO-66 Variants)
The crystal structures of the UiO-66 variants (-H, -NH2, -CH3, -COOH) were treated as rigid frameworks, with atomic positions fixed according to their reported/optimized crystallographic coordinates [29]. Interactions between framework atoms and adsorbates were modeled using the Lennard-Jones (LJ) potential. The LJ parameters for the framework atoms (C, H, O, N) were taken from the generic DREIDING force field [30], known for its transferability in MOFs. For the zirconium (Zr) atom, parameters from the Universal Force Field (UFF) [31] were used.
2.1.3. Interaction Parameters and Mixing Rules
All non-bonded interactions (fluid-fluid and fluid-framework) were modeled using the 12-6 LJ potential:
where r is the interatomic distance, is the potential well depth, and is the collision diameter. The and parameters for all TraPPE-UA pseudo-atoms and DREIDING/UFF framework atoms are summarized in Table 1. Cross-interactions between different atom types were calculated using the standard Lorentz–Berthelot mixing rules:

Table 1.
Lennard–Jones parameters ( in K, in Å) used in the simulations. Hydrocarbon pseudo-atoms (top block) follow TraPPE-UA; framework atoms (bottom block) follow DREIDING, except Zr † which uses UFF.
2.2. GCMC Simulation Details
GCMC simulations were performed in the ensemble, fixing the chemical potential (), system volume (V, corresponding to 2 × 2 × 2 unit cells of UiO-66), and temperature (). Periodic boundary conditions were applied in all three dimensions. Each simulation consisted of Monte Carlo steps for equilibration, followed by steps for production, during which ensemble averages were collected. A cutoff radius of Å was used for LJ interactions, with standard long-range tail corrections applied.
The Monte Carlo moves implemented included translation, insertion, and deletion of molecules. Their acceptance probabilities were defined by the Metropolis criteria as follows:
where the insertion bias term B is defined as:
Here, f is the fugacity of the bulk phase, V is the system volume, k is Boltzmann’s constant, and T is the temperature. represents the energy change resulting from the proposed move.
The adsorbed amount (q) was calculated as the average number of molecules in the ensemble, , per simulation cell. The average total potential energy of the system, , was calculated as the sum of all pairwise fluid-fluid () and fluid-framework () interactions:
where indices run over fluid particles and k over framework atoms.
2.3. Calculation of Isosteric Heat of Adsorption
The isosteric heat of adsorption () at a given loading was calculated from fluctuations in the number of particles (N) and the total potential energy (U) during the GCMC simulation in the ensemble, using the standard fluctuation formula [32]:
where R is the ideal gas constant, T is the absolute temperature, and angular brackets denote ensemble averages obtained during the production phase of the GCMC simulation. This method provides a direct measure of the average interaction strength between the adsorbate and the adsorbent at a specific loading.
3. Results
3.1. Single-Component Adsorption of Pentane Isomers
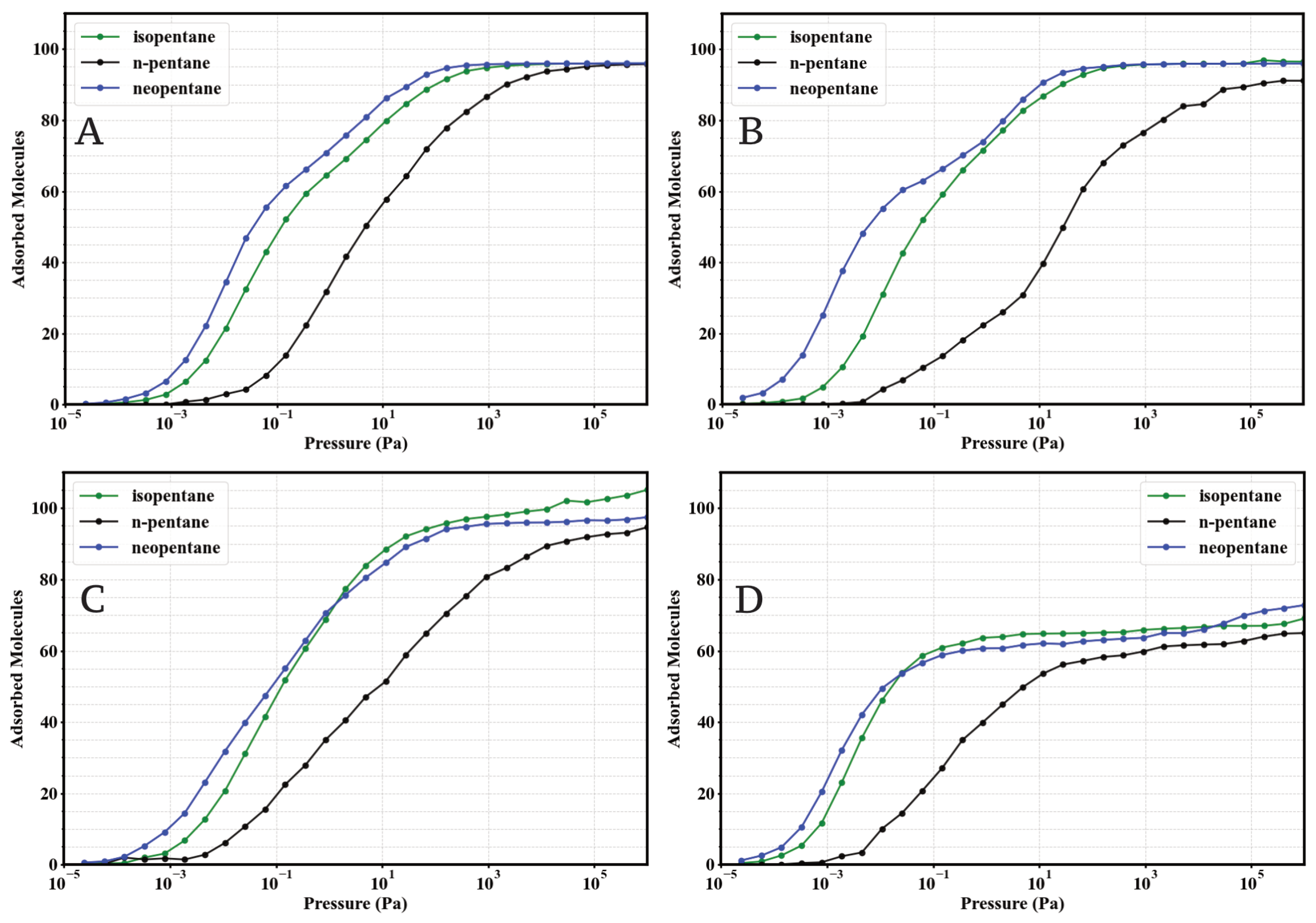
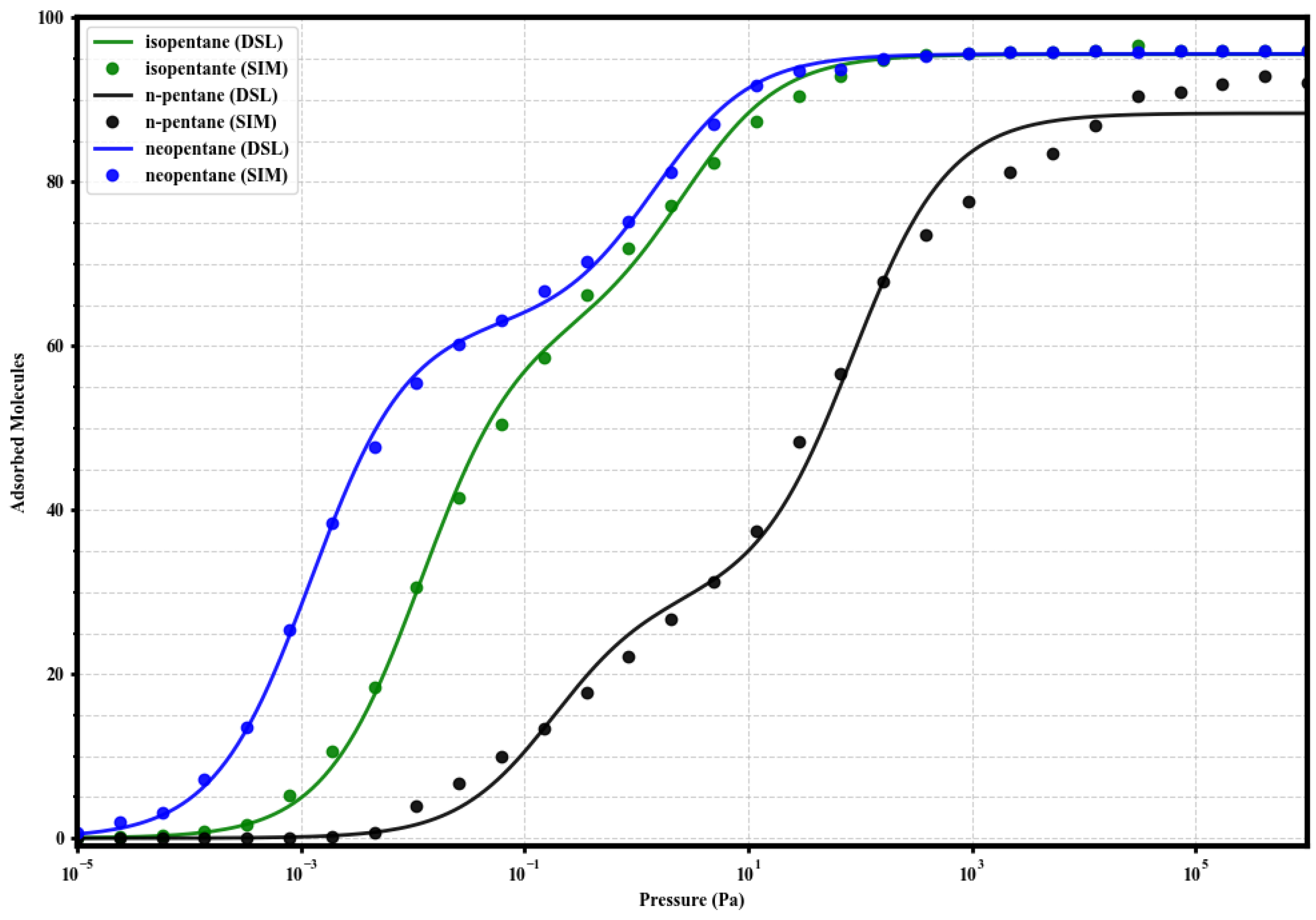
To evaluate the impact of linker functionalization on pentane isomer capture, GCMC simulations were performed to obtain the single-component adsorption isotherms of n-pentane (nP), isopentane (iP), and neopentane (neoP) in the four UiO-66 variants (UiO-66-H, UiO-66-NH2, UiO-66-CH3, and UiO-66-COOH) at 298 . The results are presented in Figure 2.
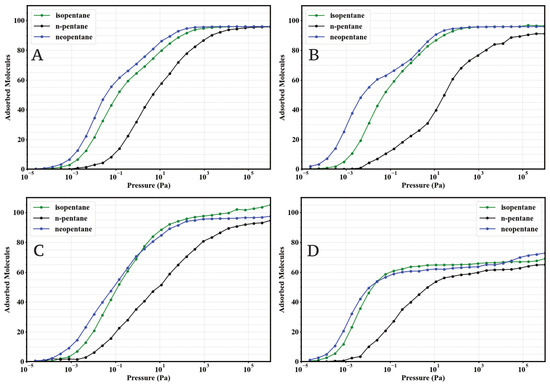
Figure 2.
Simulated single-component adsorption isotherms (GCMC) for isopentane (green), n-pentane (black), and neopentane (blue) at 298 K in: (A) UiO-66-H, (B) UiO-66-NH2, (C) UiO-66-CH3, and (D) UiO-66-COOH. Adsorbed loading (molecules per simulated unit cell) is plotted against pressure (Pa) on a logarithmic scale.
A general and consistent observation across the four materials is the marked preference for the adsorption of branched isomers (iP and neoP) over the linear isomer (nP), especially in the low-pressure regime (). This indicates an intrinsically higher affinity of the UiO-66 porous environment towards the more compact or branched C5 structures. At high pressures (), the saturation capacities for the three isomers tend to converge to similar values within UiO-66-H, -NH2, and -CH3, reaching approximately 100 molecules per simulation cell. However, n-pentane requires significantly higher pressures to reach saturation compared to its branched counterparts.
Examining the specific effect of functionalization reveals notable differences. The parent MOF UiO-66-H (Figure 2A) and the methylated variant UiO-66-CH3 (Figure 2C) exhibit very similar behaviors. In both, isopentane and neopentane show comparable affinities, substantially higher than n-pentane at low pressures, suggesting an affinity order of neoP ≈ iP nP.
The introduction of the amino group (UiO-66-NH2, Figure 2B) induces a drastic change in low-pressure preference. This material demonstrates exceptionally enhanced and selective affinity towards neopentane. The adsorption curve for neoP rises much more steeply than those for isopentane and n-pentane, establishing a clear affinity hierarchy: neoP iP > nP. Additionally, neopentane also reaches a slightly higher saturation capacity in UiO-66-NH2 compared to the other two isomers in the same material.
On the other hand, the carboxylated variant UiO-66-COOH (Figure 2D) displays a low-pressure affinity profile similar to UiO-66-H and -CH3 (neoP ≈ iP nP). However, it is distinguished by a notable reduction in the total saturation capacity for all three isomers, settling around 70–75 molecules. This suggests that the carboxyl group might impose significant steric or volumetric constraints within the pores, or unfavorably alter host-guest interactions.
These single-component adsorption results establish a comparative baseline for the intrinsic behavior of each MOF towards pentane isomers. The observed differences, particularly the strong preference for neopentane in UiO-66-NH2 and the reduced capacity in UiO-66-COOH, suggest that linker functionalization effectively modulates both affinity and adsorption capacity, which will have direct implications for mixture separation properties discussed later.
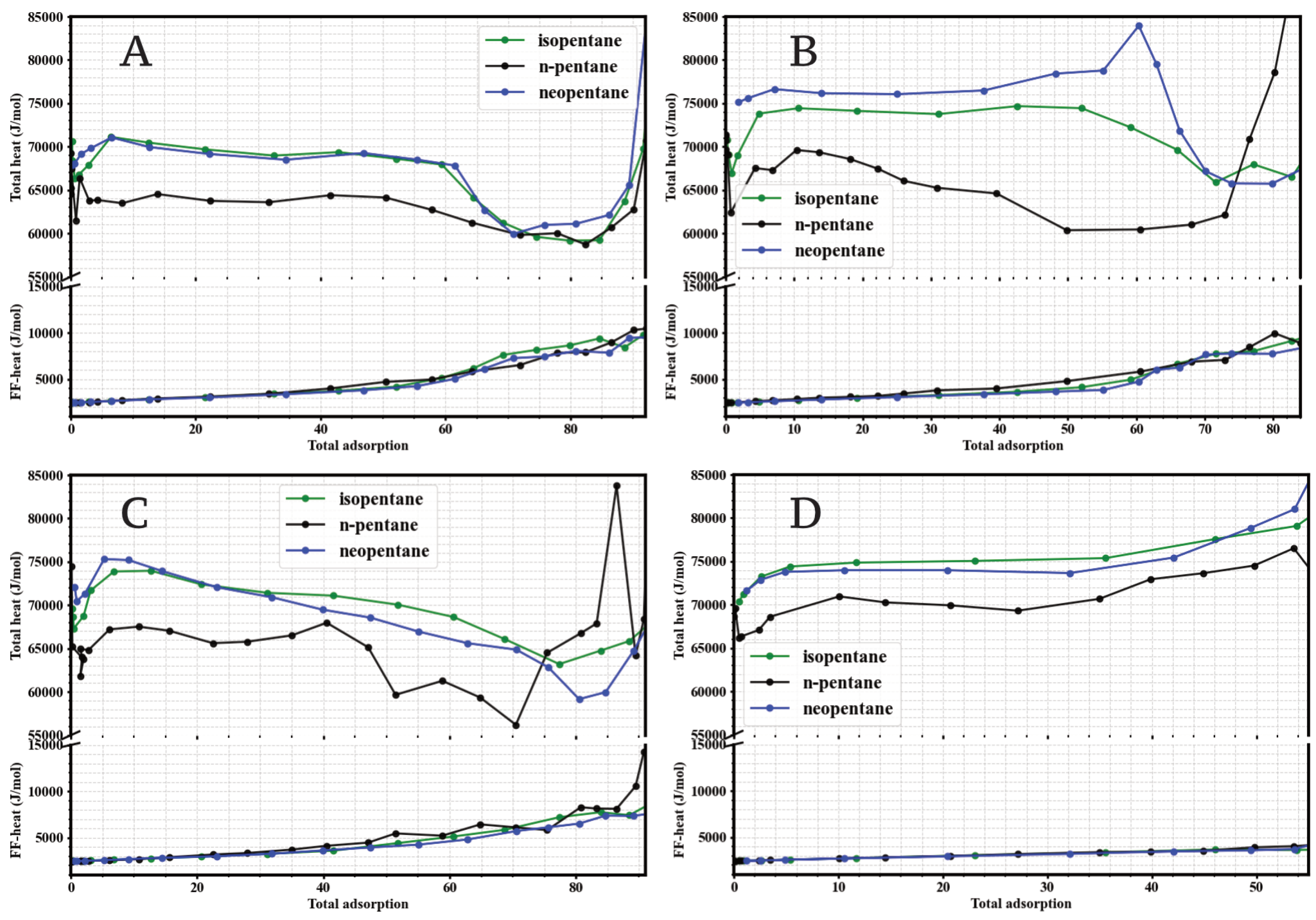
3.2. Isosteric Heats of Adsorption
To quantify the strength of host-guest interactions and better understand the affinities observed in the single-component isotherms, the isosteric heats of adsorption () were calculated for n-pentane, isopentane, and neopentane in the four UiO-66 variants as a function of loading (adsorbed molecules). The results are shown in Figure 3.
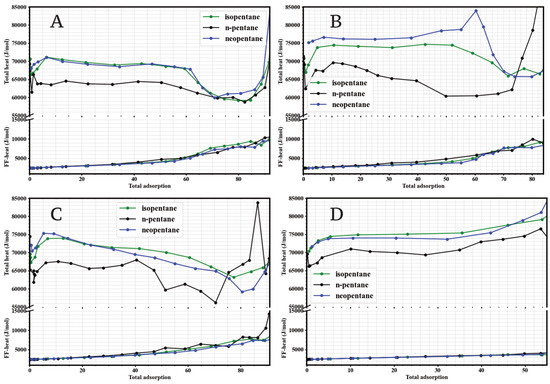
Figure 3.
Simulated isosteric heats of adsorption (, top curves) and average fluid-fluid interaction energy per molecule (bottom curves) for isopentane (green), n-pentane (black), and neopentane (blue) at 298 K as a function of adsorbed loading (molecules per simulated unit cell) in: (A) UiO-66-H, (B) UiO-66-NH2, (C) UiO-66-CH3, and (D) UiO-66-COOH.
A consistent finding across the four materials is that, at low loading (approaching zero coverage), the isosteric heats for the branched isomers (isopentane and neopentane) are significantly higher than for the linear isomer (n-pentane). Specifically, for UiO-66-H (Figure 3A), UiO-66-CH3 (Figure 3C), and UiO-66-COOH (Figure 3D), the values at low loading () for isopentane and neopentane are in the range of 71 mol−1 to 75 mol−1, while for n-pentane they are in the range of 62 mol−1 to 68 mol−1. This higher exothermicity for the branched isomers quantitatively corroborates the greater affinity inferred from the initial slopes of the isotherms (Figure 2), indicating an energetically more favorable interaction of the branched/compact molecules with the UiO-66 framework. The trend of the isosteric heat of adsorption (), which is generally higher at low loadings and then decreases or stabilizes as the number of adsorbed molecules increases, is compatible with an initial filling of the most energetically favorable sites. Consistent with the porous structure of UiO-66, which features tetrahedral (smaller, ≈ Å) and octahedral (larger, ≈12 Å) cavities[cite: 2129, 2130], and supported by the analysis of molecular configurations at different pressures (Figures 12–15), we identify these initial high-energy sites with the more constrictive tetrahedral cavities. Subsequent adsorption into the larger octahedral cavities, and the increase in fluid-fluid interactions (shown in Figure 3), would contribute to the evolution of at higher loadings.
The effect of functionalization is particularly pronounced in UiO-66-NH2 (Figure 3B). In this material, the for neopentane reaches approximately 80 mol−1, a value notably higher than that for isopentane (∼70 mol−1) and n-pentane (∼65 mol−1) in the same MOF, and also higher than the of any isomer in the other three UiO-66 variants. This exceptionally strong interaction of neopentane with the UiO-66-NH2 framework directly explains the steep slope of its isotherm at low pressures and its clear preference over the other isomers in this material.
The dependence of on loading also reveals information about the energetic heterogeneity of adsorption sites and adsorbate-adsorbate interactions. Generally, for isopentane and neopentane, tends to decrease slightly or remain relatively constant as loading increases, suggesting initial adsorption on the most favorable sites followed by occupation of slightly less energetic sites or the onset of repulsive interactions between adsorbed molecules. In contrast, for n-pentane, often shows a slight initial increase before decreasing or stabilizing, which could indicate reorganization of adsorbed molecules or the influence of attractive intermolecular interactions as the concentration in the pores increases. The curves labeled “Fluid-Fluid Energy” in Figure 3 represent the contribution of adsorbate-adsorbate interactions to the total energy, showing their increasing importance at higher loadings.
The isosteric heats confirm that energetic interactions are the main driver of the preferential affinity for branched isomers in the UiO-66 family at low pressures. More importantly, they quantify the outstanding effect of the -NH2 group in selectively strengthening the interaction with neopentane, providing an energetic basis for the observed differences in isotherms and anticipating a potential impact on mixture separation selectivity.
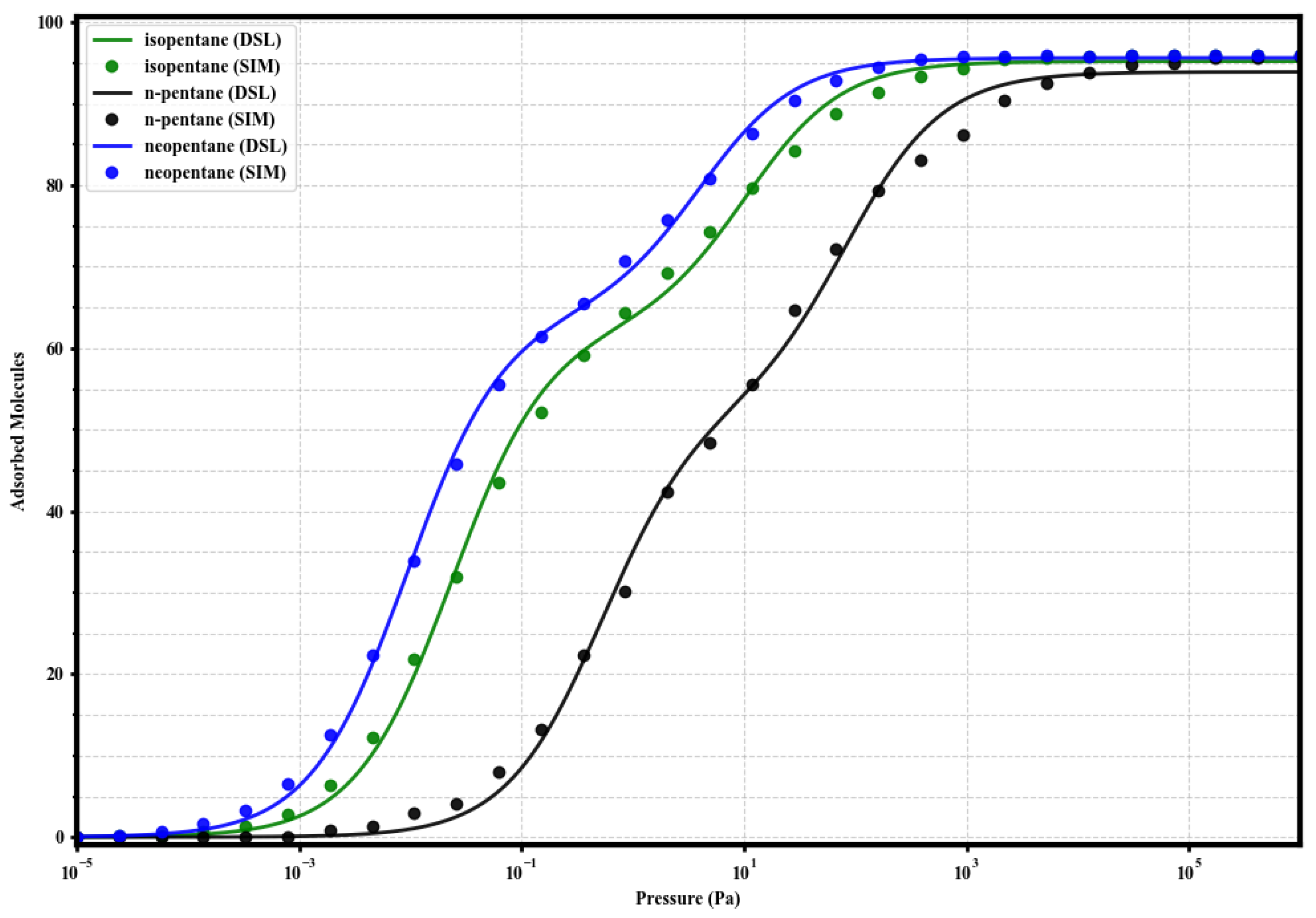
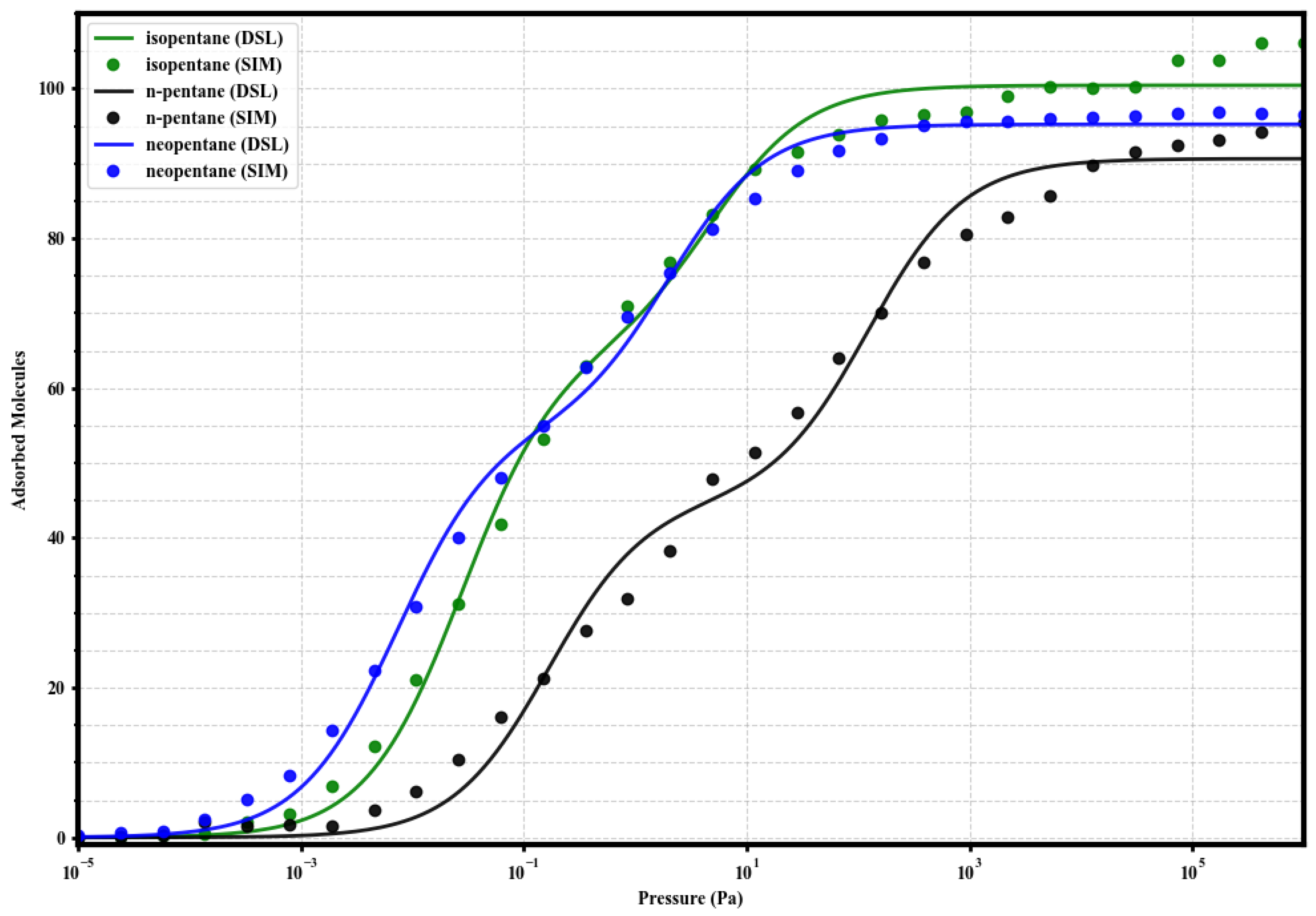
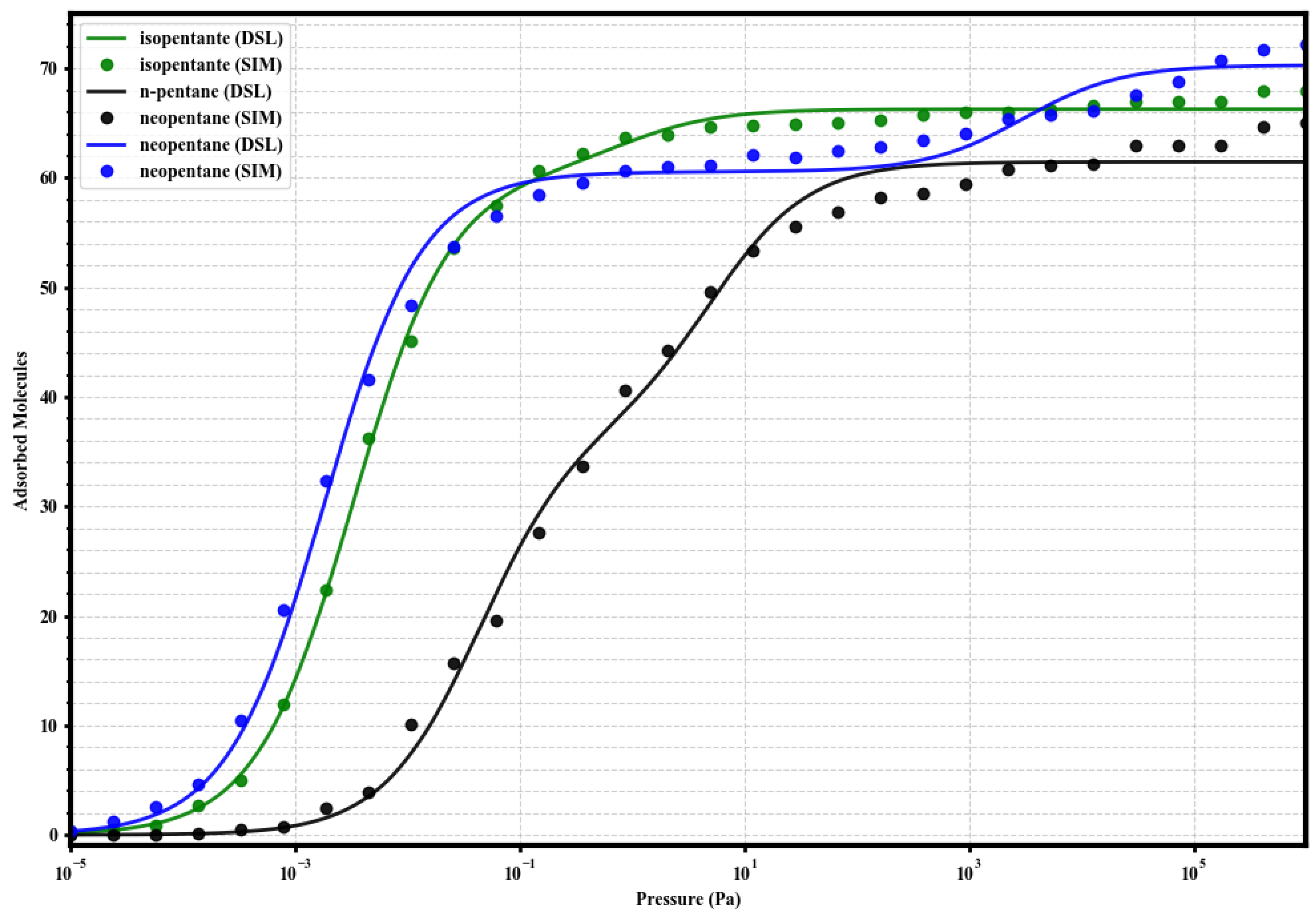
3.3. Modeling of Single-Component Isotherms with Dual-Site Langmuir
In order to quantify the energetic heterogeneity of the adsorption sites and correlate the shape of the isotherms with affinities, the simulated single-component adsorption data were fitted using the Dual-Site Langmuir (DSL) model:
where q is the adsorbed loading, P is the pressure, and are the saturation capacities of sites 1 (strong) and 2 (weak), and and are the respective affinity constants. The resulting fits, shown along with the simulated data in Figure 4, Figure 5, Figure 6 and Figure 7, adequately describe the adsorption behavior over the entire pressure range. The fitting parameters are summarized in Table 2.
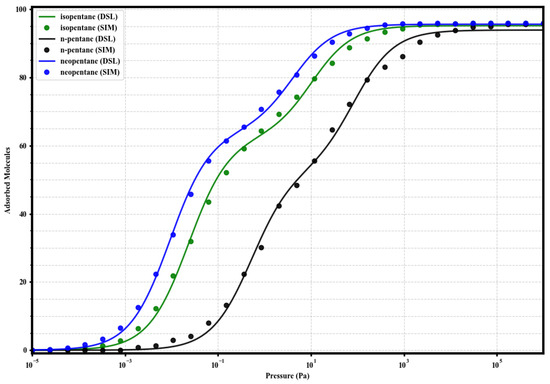
Figure 4.
Dual-Site Langmuir model fits (solid lines) to the simulated single-component adsorption data (circles) for isopentane (green), n-pentane (black), and neopentane (blue) in UiO-66-H at 298 K. Fitted parameters are given in Table 2.
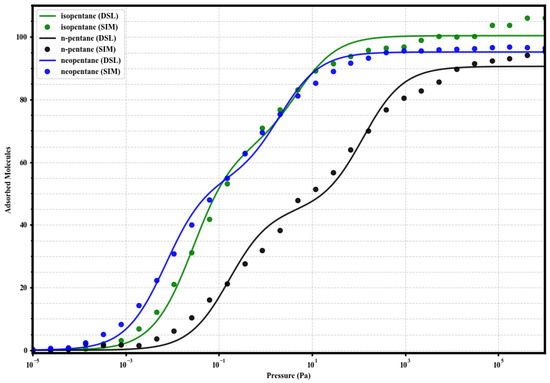
Figure 5.
Dual-Site Langmuir model fits (solid lines) to the simulated single-component adsorption data (circles) for isopentane (green), n-pentane (black), and neopentane (blue) in UiO-66-CH3 at 298 K. Fitted parameters are given in Table 2.
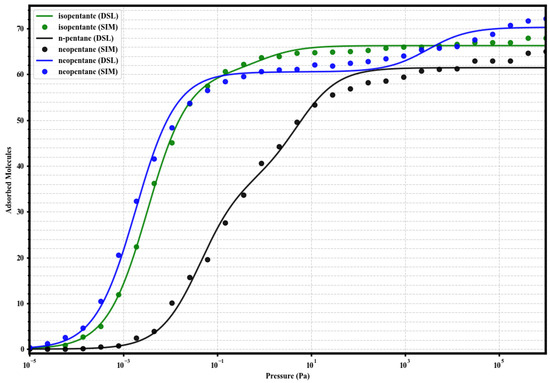
Figure 6.
Dual-Site Langmuir model fits (solid lines) to the simulated single-component adsorption data (circles) for isopentane (green), n-pentane (black), and neopentane (blue) in UiO-66-COOH at 298 K. Fitted parameters are given in Table 2.
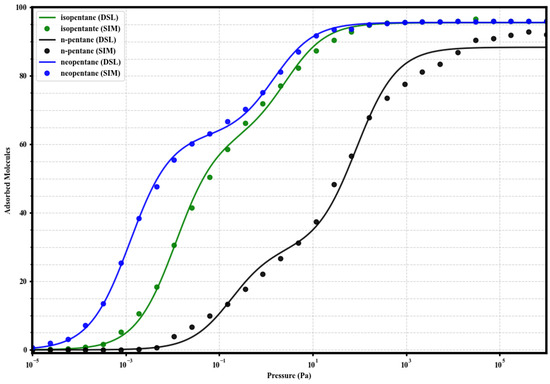
Figure 7.
Dual-Site Langmuir model fits (solid lines) to the simulated single-component adsorption data (circles) for isopentane (green), n-pentane (black), and neopentane (blue) in UiO-66-NH2 at 298 K. Fitted parameters are given in Table 2.
Table 2.
Parameters of the Dual-Site Langmuir (DSL) model obtained from fitting the simulated single-component isotherms at 298 K. Units of are molecules per simulated unit cell, and units of are Pa−1.
Upon analyzing the Dual-Site Langmuir (DSL) model parameters (Table 2), the two-site nature observed in the isotherms (Figure 4, Figure 5, Figure 6 and Figure 7) is consistent with the sequential occupation of the two types of cavities present in the fcu topology of UiO-66: the tetrahedral and octahedral ones. Our analyses, which include the visualization of molecular configurations at increasing pressures (Figures 12–15), suggest that the higher affinity sites (site 1, reflected in the parameter) predominantly correspond to the filling of the tetrahedral cages. These, being smaller (approximately Å) and offering greater confinement, could generate more energetically favorable interactions with the pentane isomers. Consequently, the lower affinity sites (site 2, parameter) are associated with the filling of the larger octahedral cages (approximately 12 Å), and/or with less specific interactions occurring at higher loadings once the primary sites begin to saturate. This interpretation allows for a more concrete physical meaning to be assigned to the , , , and parameters, relating them to the structural characteristics of the adsorbent.
The analysis of the DSL parameters (Table 2) quantitatively confirms the trends observed in the isotherms and isosteric heats. In UiO-66-H, the high values for neoP (108.5) and isoP (43.0) contrast with the low value for nP (1.89), reflecting the preferential affinity for the branched isomers. The difference quantifies the slight energetic advantage of neopentane. The total saturation capacity () is similar for the three (≈95 molecules).
In UiO-66-CH3, the affinity hierarchy is maintained (neoP > isoP nP), but for nP (5.86) increases notably compared to UiO-66-H, suggesting an improved initial interaction. The capacity of the strong site for isopentane (65.0) is the largest, indicating possible specific favorable packing for this isomer.
In UiO-66-COOH, the total capacity is significantly reduced (≈61–70 molecules). The values for neoP (551.7) and isoP (306.9) are exceptionally high, indicating very energetic primary sites created by the carboxyl group, while for nP (2.24) remains low. The behavior of the secondary site () is complex, with an unexpectedly high value for isopentane (1.16).
Finally, in UiO-66-NH2, the selective effect of the amino group is evident. for neoP reaches an extreme value (819.9), far exceeding that of isoP (85.5), and both are much larger than for nP (4.51). This quantifies the strong intrinsic selectivity neoP >> isoP > nP. Furthermore, the capacity of the primary site for nP (≈ 30) is drastically reduced, forcing its majority adsorption onto weak secondary sites.
In conclusion, the DSL model effectively captures the adsorption heterogeneity and quantifies how functionalization differentially modulates the affinities () and capacity distribution () for each isomer, laying the groundwork for analyzing mixture separation.
3.4. Simulation of Competitive Adsorption and Comparison with Predictive Models
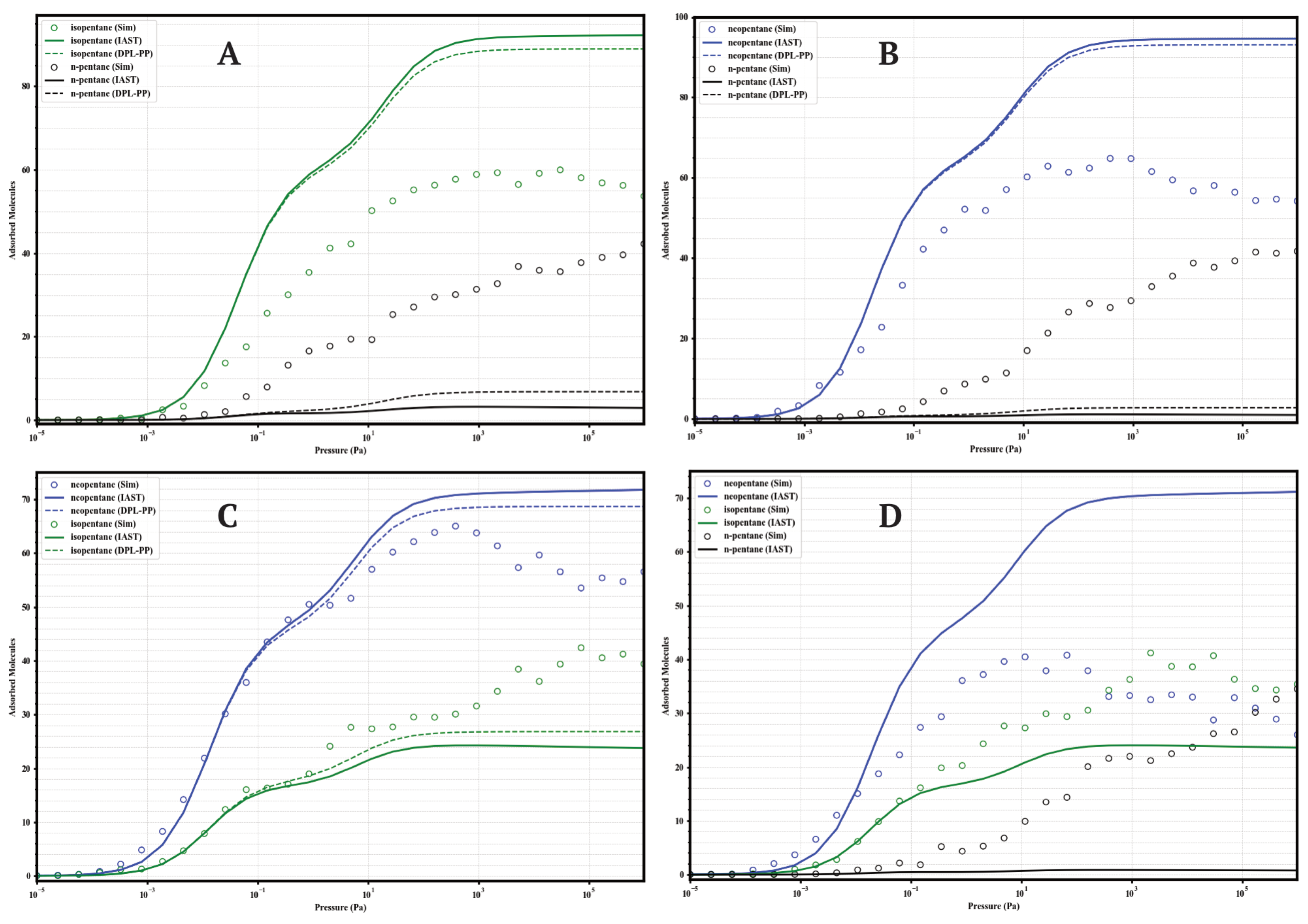
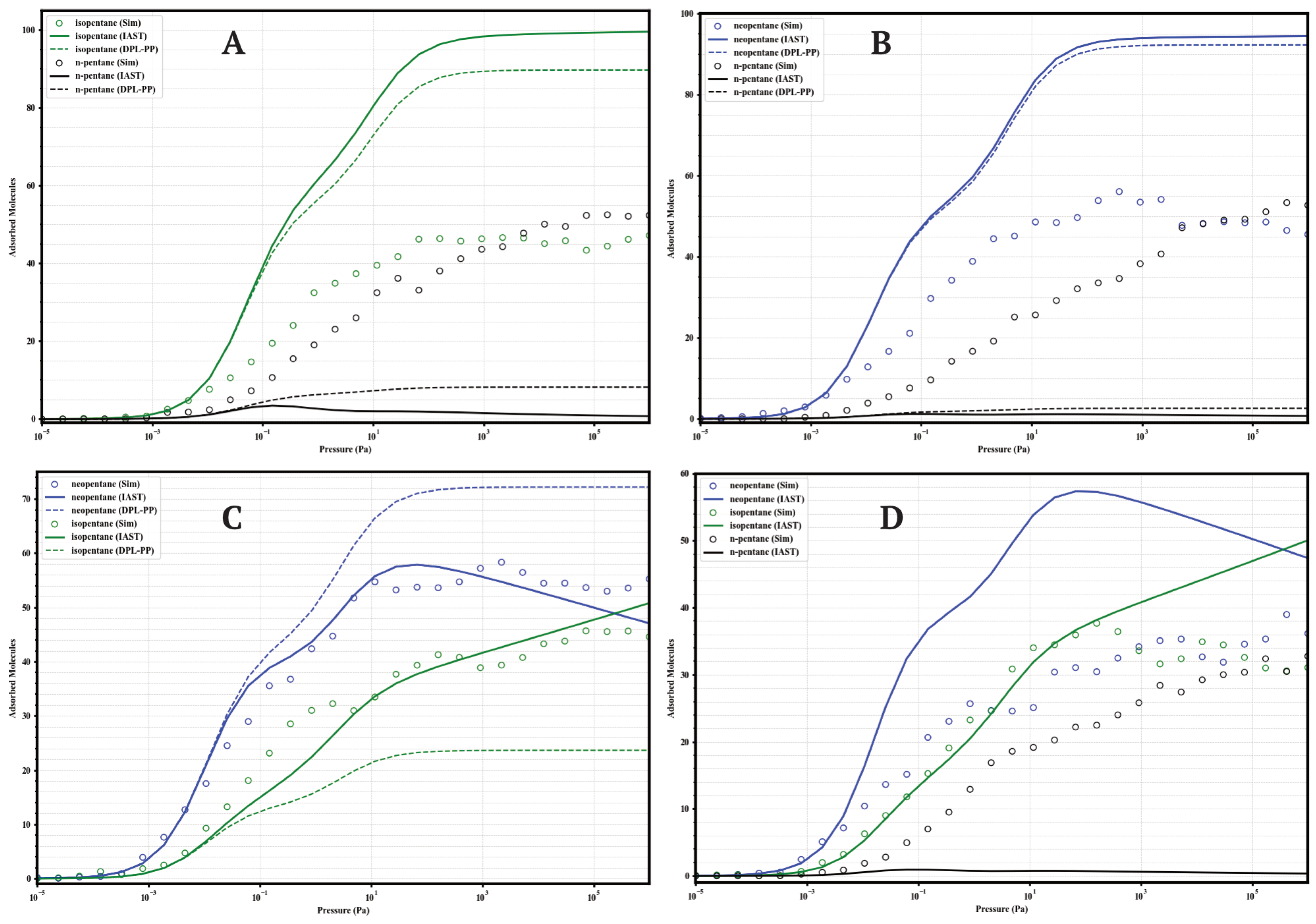
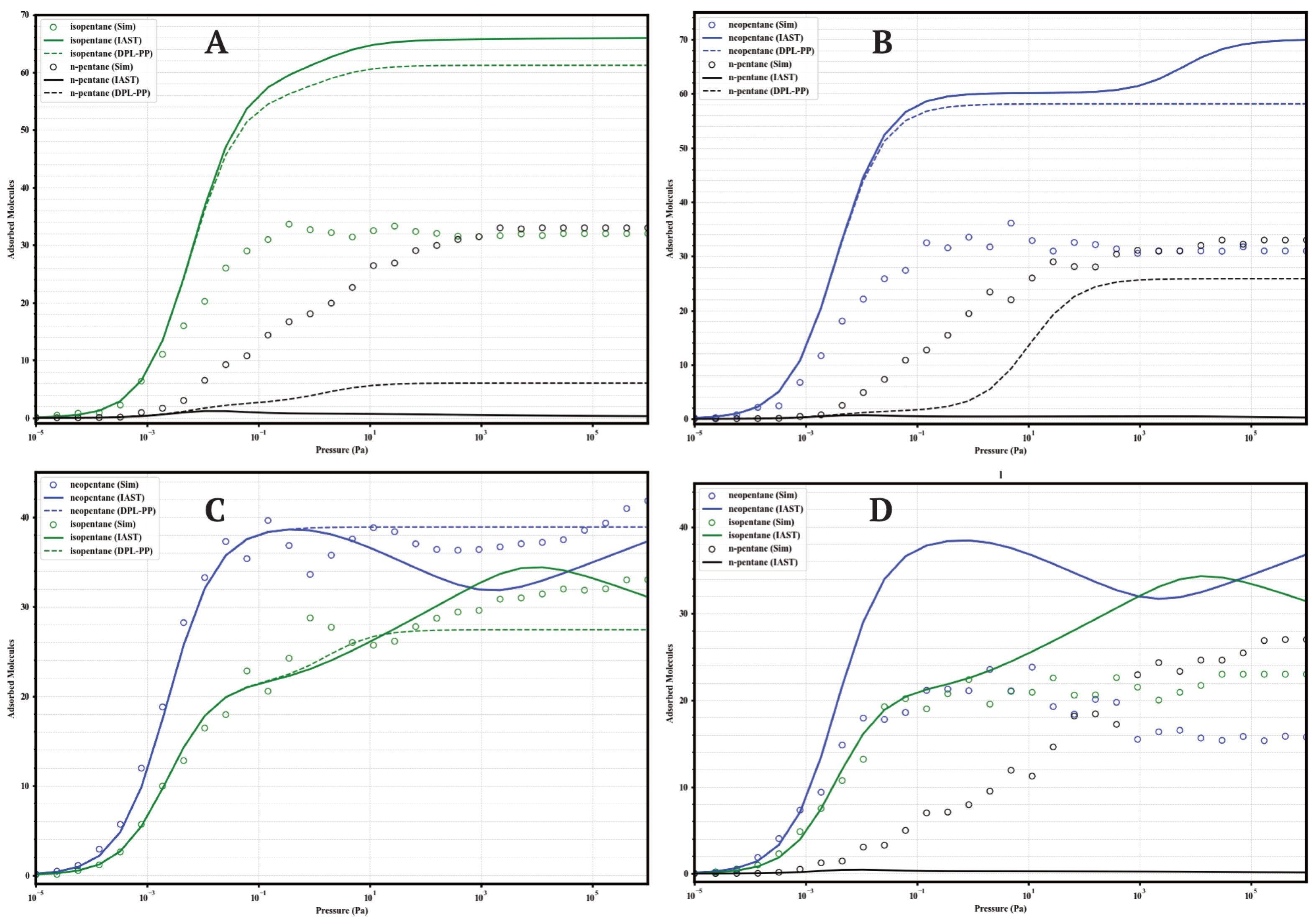
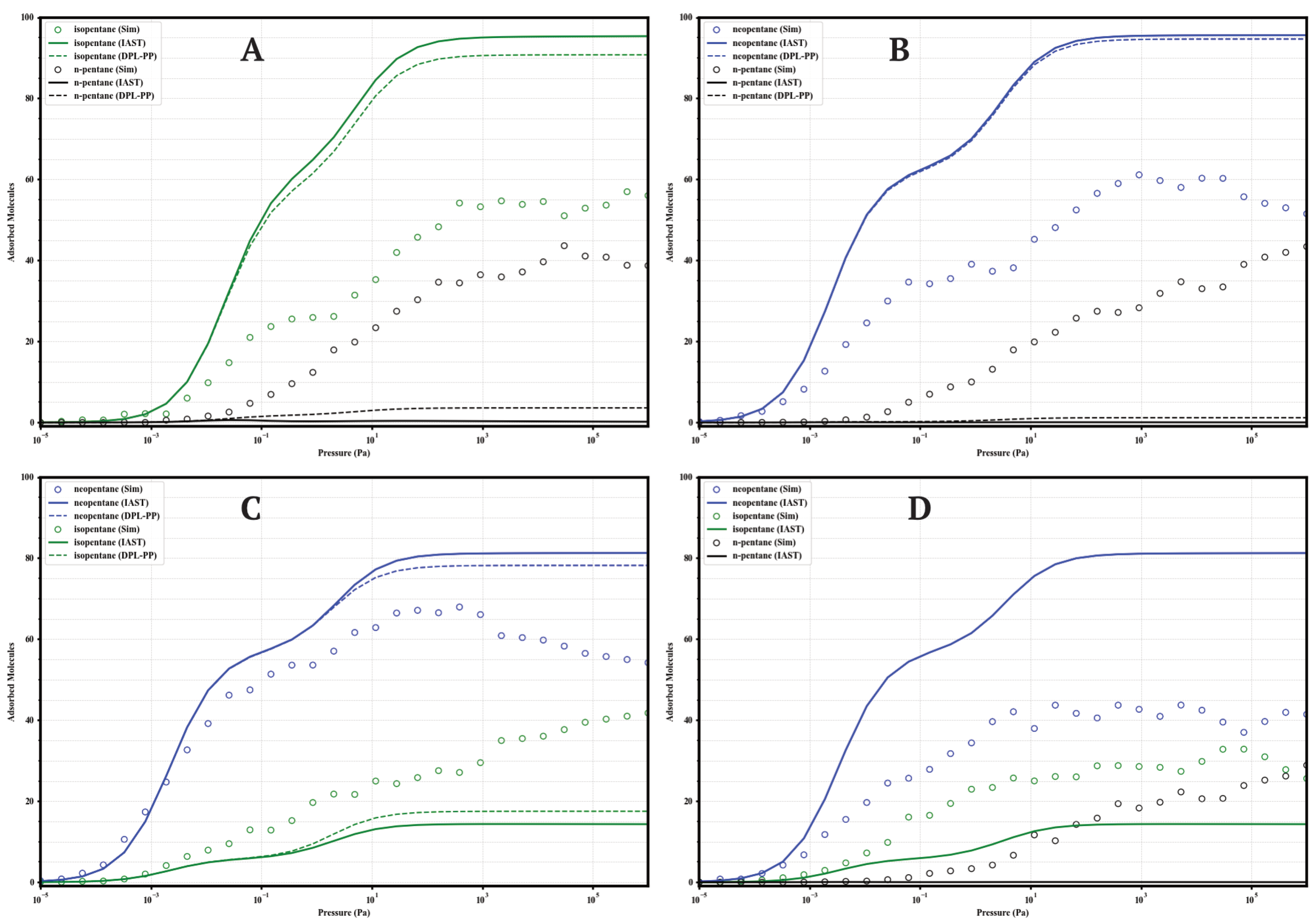
The separation performance of the UiO-66 variants under competitive conditions was evaluated through GCMC simulations of equimolar binary mixtures (isopentane/n-pentane, neopentane/n-pentane, neopentane/isopentane; 50/50) and a ternary mixture (neopentane/isopentane/n-pentane; 33/33/33) at 298 . These results provide direct insight into the real selectivity in the presence of multiple components and serve to validate the accuracy of predictive models like the Ideal Adsorbed Solution Theory (IAST) and the Dual Process Langmuir Formulation (DPL), calculated from the Dual-Site Langmuir (DSL) fits of the single-component isotherms. The comparison between GCMC data and IAST predictions is presented in Figure 8, Figure 9, Figure 10 and Figure 11.
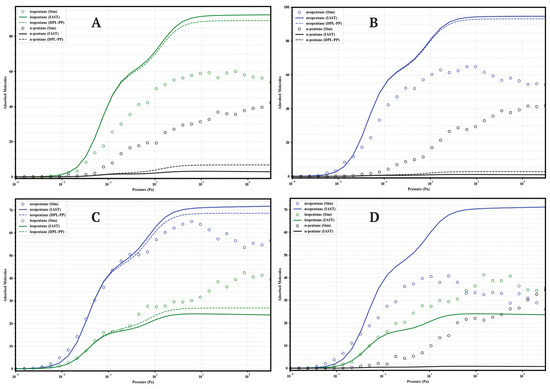
Figure 8.
Comparison of adsorption in equimolar mixtures simulated by GCMC (circles) and predicted by IAST (solid lines) and DPL (dashed lines) for UiO-66-H at 298 K. Panels: (A) Iso/nP, (B) Neo/nP, (C) Neo/Iso, (D) Neo/Iso/nP. Colors: isopentane (green), n-pentane (black), neopentane (blue).
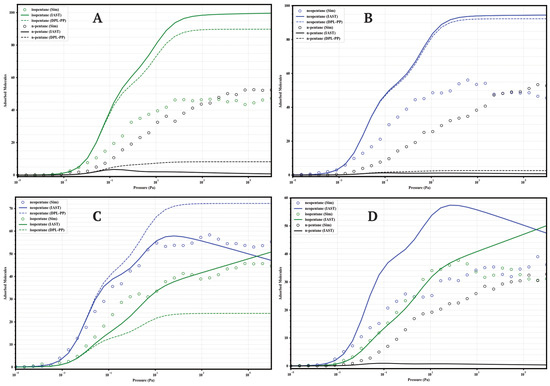
Figure 9.
Comparison of adsorption in equimolar mixtures simulated by GCMC (circles) and predicted by IAST (solid lines) and DPL (dashed lines) for UiO-66-CH3 at 298 K. Panels: (A) Iso/nP, (B) Neo/nP, (C) Neo/Iso, (D) Neo/Iso/nP. Colors: isopentane (green), n-pentane (black), neopentane (blue).
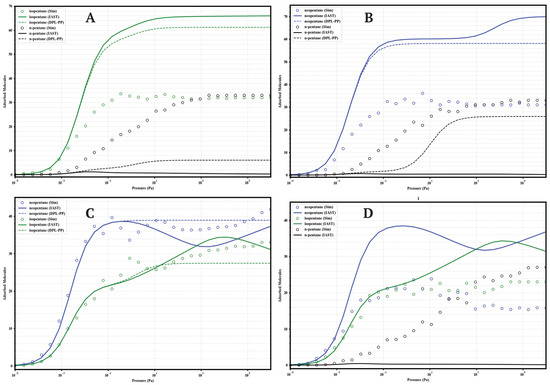
Figure 10.
Comparison of adsorption in equimolar mixtures simulated by GCMC (circles) and predicted by IAST (solid lines) and DPL (dashed lines) for UiO-66-COOH at 298 K. Panels: (A) Iso/nP, (B) Neo/nP, (C) Neo/Iso, (D) Neo/Iso/nP. Colors: isopentane (green), n-pentane (black), neopentane (blue).
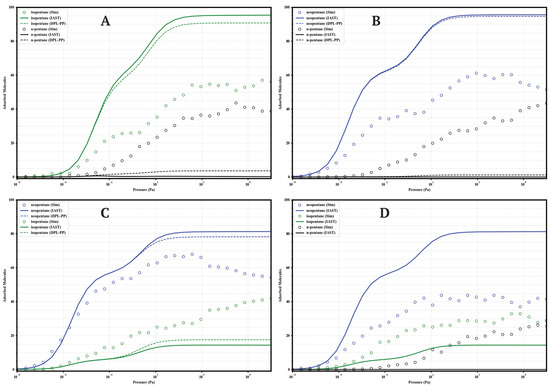
Figure 11.
Comparison of adsorption in equimolar mixtures simulated by GCMC (circles) and predicted by IAST (solid lines) and DPL (dashed lines) for UiO-66-NH2 at 298 K. Panels: (A) Iso/nP, (B) Neo/nP, (C) Neo/Iso, (D) Neo/Iso/nP. Colors: isopentane (green), n-pentane (black), neopentane (blue).
A general finding across the four materials is that direct GCMC simulations of the mixtures consistently confirm the intrinsic preference for branched isomers over the linear isomer, in line with the single-component analyses. Likewise, IAST and DPL qualitatively predict this preference order correctly in practically all cases. However, a recurring quantitative discrepancy is that IAST systematically tends to overestimate selectivity: it predicts higher loadings for strongly adsorbed components and lower loadings (often near zero) for weakly adsorbed components, compared to the GCMC results, where co-adsorption is generally more significant. DPL predictions are even worse.
4. Discussion
4.1. Performance of Individual MOFs in Mixtures
4.1.1. UiO-66-H (Parent)
The parent MOF (Figure 8) establishes the baseline behavior. In the Iso/nP and Neo/nP binary mixtures, GCMC confirms the preference for branched isomers but with substantial co-adsorption of nP (≈40–43 molecules at high P). Selectivity is moderate. In the Neo/Iso mixture, GCMC shows very low selectivity, with neoP slightly preferred. The ternary mixture confirms this: Neo ≈ Iso > nP, with significant loadings for all three. IAST and DPL overestimate the separation between linear and branched isomers in all cases.
4.1.2. UiO-66-CH3 (Methylated)
The methylated variant (Figure 9) behaves very similarly to UiO-66-H. It shows moderate selectivity in binary mixtures with nP, high co-adsorption of nP (≈45 molecules), and slight preference for neoP over isoP. In the ternary mixture, selectivity is the lowest of the series (Neo ≈ 35, Iso ≈ 32, nP ≈ 30 molecules), indicating that the methyl group does not enhance competitive separation. IAST continues to overestimate selectivity.
4.1.3. UiO-66-NH2 (Aminated)
Amino functionalization (Figure 11) induces the highest selectivity. GCMC confirms the almost complete exclusion of nP (loading < 5 molecules) in binary and ternary mixtures. The Neo/Iso competition is highly selective in favor of neoP, especially at P < (loading Neo ≈ 65–70 vs. Iso ≈ 40–45). In the ternary mixture, high selectivity is maintained (Neo ≈ 45 >> Iso ≈ 28 >> nP < 5). IAST correctly predicts the qualitative order but overestimates the loadings of Neo/Iso, failing quantitatively. Maximum selectivity is achieved at intermediate pressures (10−1 to 103 ).
4.1.4. UiO-66-COOH (Carboxylated)
This material (Figure 10) shows the most complex behavior. In binary mixtures with nP, GCMC indicates moderate selectivity with high co-adsorption of nP (≈32–36 molecules). In the Neo/Iso mixture, GCMC reveals a preference for neoP at P < 10 , but at higher pressures, the loadings equalize or isopentane becomes slightly preferred, indicating low or inverted selectivity. The ternary mixture confirms the complexity: high co-adsorption of all three (≈22–27 molecules), with a pressure-dependent preference order (Neo > Iso > nP at low P, but Iso ≈ nP ≈ Neo at high P). IAST is unable to predict this complexity.
4.2. Limitations of Predictive Models
The systematic GCMC vs IAST vs DPL comparison reveals consistent limitations of the analytic models. While IAST and DPL generally predict the qualitative order correctly, they recurrently fails in the quantitative prediction of partial loadings in mixtures. The tendency is to overestimate the loading of the most affine component and underestimate the co-adsorption of the less affine ones. These deviations, pronounced in -NH2 and -COOH but also significant in -H and -CH3, underscore the importance of competitive, packing, and possibly entropic effects not captured by the ideality assumed in IAST. Consequently, direct GCMC simulations are indispensable for an accurate quantitative assessment of the separation performance of these functionalized materials.
4.3. Mechanisms of Pentane Isomer Separation in Functionalized UiO-66
The results obtained from GCMC simulations and the analysis of isosteric heats () indicate that the separation of pentane isomers in the studied UiO-66 variants is predominantly governed by differences in adsorption affinity (thermodynamic mechanism). These affinities arise exclusively from the non-bonded interactions described by the Lennard-Jones (LJ) potential between the isomer molecules and the MOF framework atoms, as explicit electrostatic interactions were not included in the force field model used. Since all C5 isomers are adsorbed in the four variants (albeit with different affinities and capacities), strict size exclusion is not the primary mechanism, and kinetic effects were not evaluated in this study.
4.3.1. General Preference for Branched Isomers (Shape Complementarity)
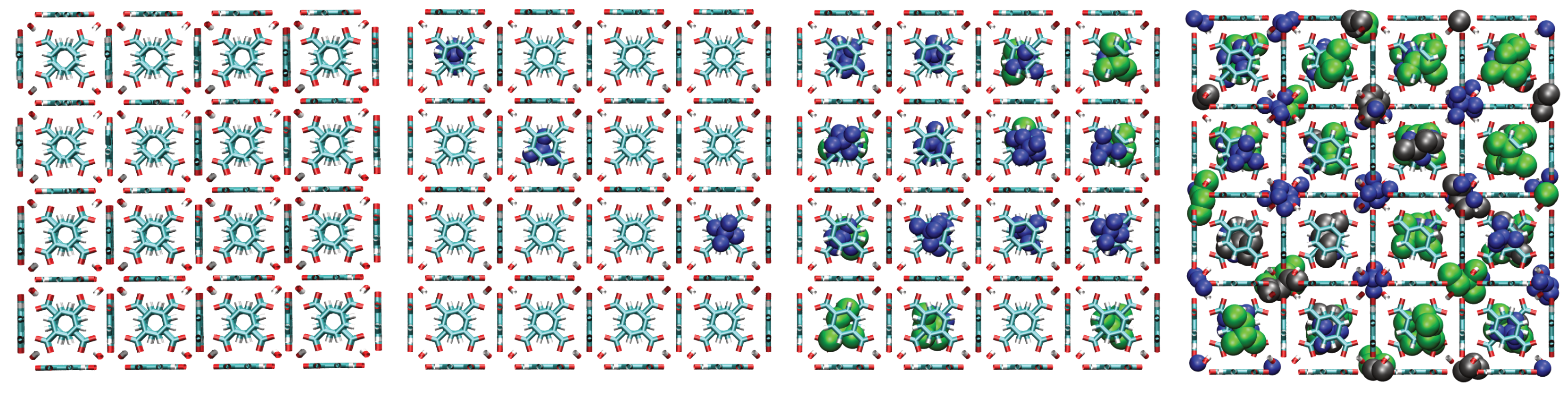
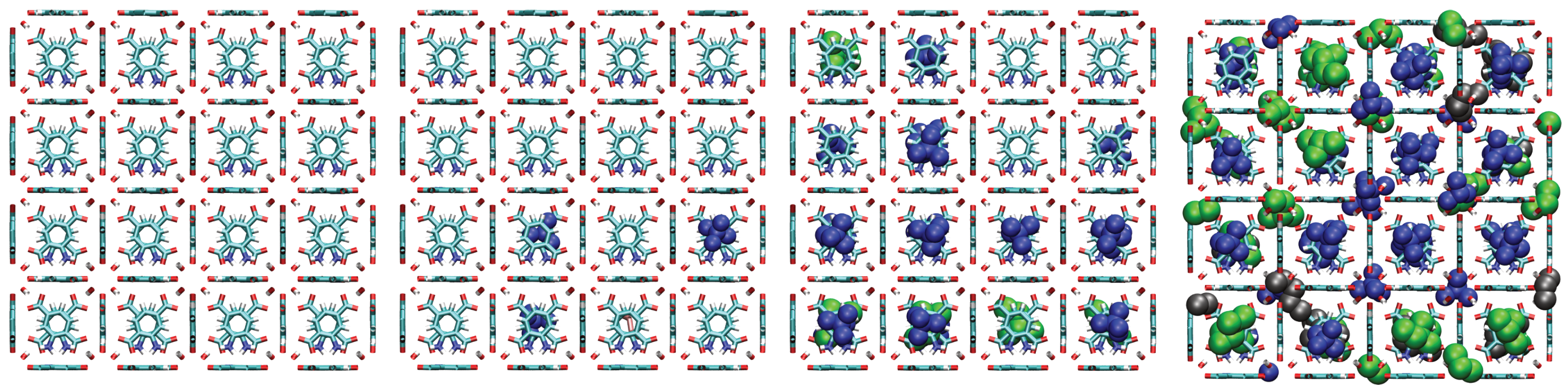
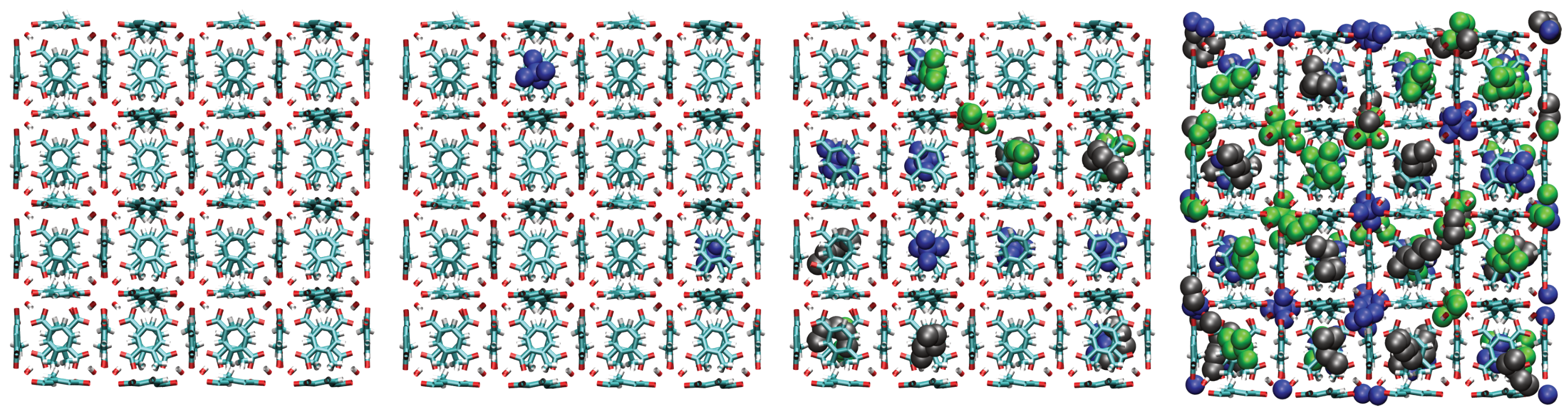
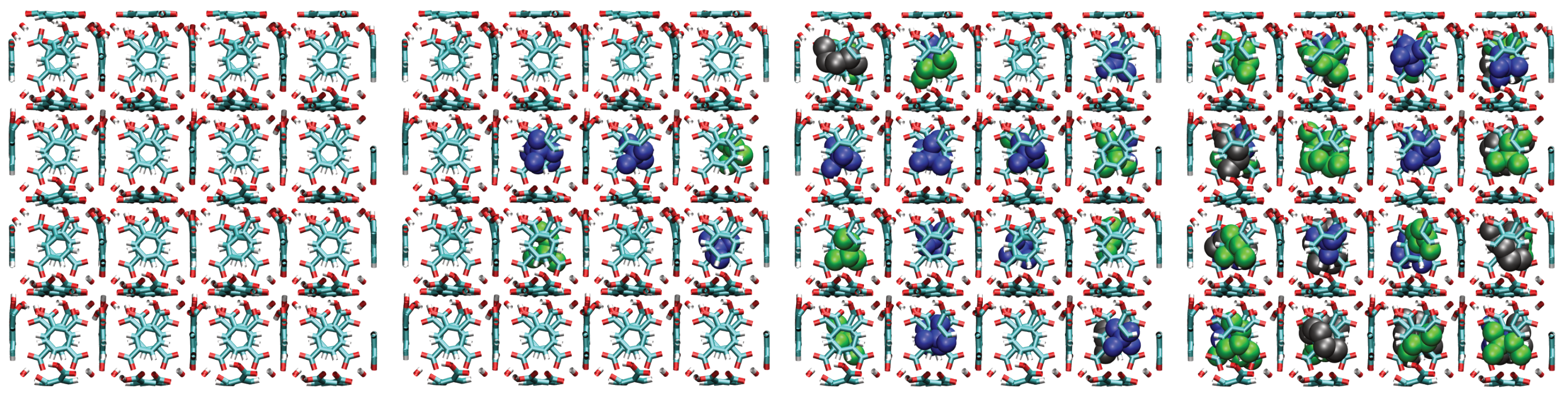
A consistent observation in UiO-66-H, -CH3, and -COOH is the intrinsically higher affinity (higher values) for the branched isomers (isopentane and neopentane) over n-pentane, especially at low pressures. Unlike flat surfaces or one-dimensional channels where linear chains can maximize van der Waals contacts, the three-dimensional cavities (tetrahedral and octahedral) characteristic of the UiO-66 fcu topology appear to offer better shape complementarity and more efficient volumetric packing for the more compact and globular molecular structures of isopentane and, particularly, neopentane. Indeed, combined experimental (2H NMR) and computational (MD) studies have confirmed that these tetrahedral cages are the preferential adsorption sites for pentane isomers [33]. As evidenced in the molecular configurations (Figure 12, Figure 13, Figure 14 and Figure 15), this process appears to occur sequentially; the observed preference for branched isomers, especially neopentane, could be explained by a particularly efficient and energetically favorable packing within the tetrahedral cages, which would preferentially fill at low pressures due to optimized LJ interactions. These molecules can fit better within the “cages”, optimizing the sum of attractive LJ (dispersion) interactions with a larger number of framework atoms simultaneously, compared to the flexible and more extended n-pentane chain. As pressure increases and these primary (tetrahedral) sites become saturated, adsorption would begin to occur in the octahedral cages, where packing and affinity differences might vary, but the initial advantage gained in the smaller cages has already established a preference. This geometric complementarity is visually illustrated in Figure 12, which shows how the different isomers occupy the MOF cavities, highlighting the differential packing. The spatial arrangement of adsorbed molecules within the octahedral and tetrahedral pores of the MOF underscores the importance of shape complementarity and steric effects in the observed differential adsorption affinity, particularly the favorable fit and distinct packing of branched isomers compared to the linear one. Figure 12, Figure 13, Figure 14 and Figure 15 display unit cells at system pressures of 1 × 10−5 Pa, 2 × 10−4 Pa, 1 × 10−2 Pa, and 160 Pa, respectively. Each figure represents the corresponding pressure state. A detailed analysis of the representative molecular configurations obtained from GCMC at increasing pressures (Figure 12, Figure 13, Figure 14 and Figure 15) supports the hypothesis of a sequential filling of the MOF cavities. It is observed that at low pressures (e.g., 1 × 10−5 Pa and 2 × 10−4 Pa), the isomers tend to preferentially occupy the tetrahedral cavities. As the pressure increases (e.g., 1 × 10−2 Pa and 160 Pa), the progressive occupation of the octahedral cavities also becomes evident, once the tetrahedral sites are more populated. This visual behavior supports the interpretation of isotherm and isosteric heat data in terms of primary (tetrahedral) and secondary (octahedral) adsorption sites.
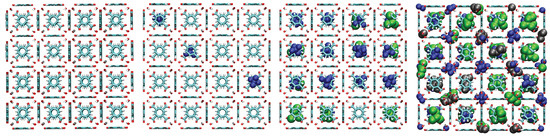
Figure 12.
Representative snapshots from GCMC simulations illustrating the molecular packing of pentane isomers within the cavities of UiO-66-H at 298 K for four different pressures. Molecules shown are neopentane (blue, compact space-filling), isopentane (green), and n-pentane (black).
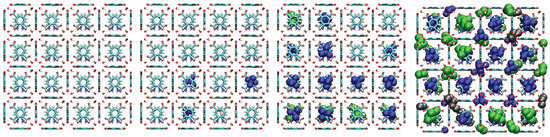
Figure 13.
Representative snapshots from GCMC simulations illustrating the molecular packing of pentane isomers within the cavities of UiO-66- at 298 K for four different pressures. Molecules shown are neopentane (blue, compact space-filling), isopentane (green), and n-pentane (black).
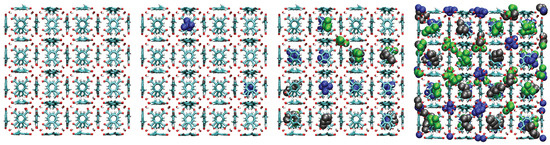
Figure 14.
Representative snapshots from GCMC simulations illustrating the molecular packing of pentane isomers within the cavities of UiO-66- at 298 K for four different pressures. Molecules shown are neopentane (blue, compact space-filling), isopentane (green), and n-pentane (black).

Figure 15.
Representative snapshots from GCMC simulations illustrating the molecular packing of pentane isomers within the cavities of UiO-66-COOH at 298 K for four different pressures. Molecules shown are neopentane (blue, compact space-filling), isopentane (green), and n-pentane (black).
4.3.2. Impact of Linker Functionalization on LJ Interactions and Sterics
Modification of the BDC linker modulates these geometry-based interactions:
- -H (Parent) and -CH3 (Methyl) Groups: The parent MOF (UiO-66-H) establishes the baseline preference for branched isomers. Introducing the methyl group (-CH3), which has a moderate steric effect and its own LJ parameters, does not significantly alter this relative preference. Both materials show very similar affinities () and adsorption behaviors, suggesting that the impact of -CH3 on the overall LJ interactions and accessible pore geometry is minimal for discriminating between C5 isomers compared to the parent material.
- -NH2 (Amino) Group: This functionalization induces the most selective effect, strongly favoring neopentane. Within the LJ model, this is explained by the creation of a geometrically specific and energetically favorable adsorption environment in terms of LJ interactions. The shape, size, and LJ parameters of the N and H atoms of the amino group, situated at a specific position within the cavity, optimize the dispersion (vdW) interactions with the quasi-spherical neopentane molecule. Figure 13 helps visualize how this molecule might fit uniquely and efficiently into this modified “niche”, maximizing its attractive contacts with the framework in a way not accessible to isopentane or n-pentane. This is a clear example of molecular recognition based on shape and optimization of site-specific LJ interactions.
- -COOH (Carboxyl) Group: The main impact of this group is steric hindrance, reflected in the drastic reduction of saturation capacity. Although the initial values for branched isomers are high (suggesting strong initial LJ interactions, possibly with the additional oxygen atoms), the excluded volume by the -COOH group limits the number of molecules that can be adsorbed and hinders efficient packing. The complex selectivity in mixtures suggests a delicate balance between these strong initial interactions and severe steric constraints at higher loadings.
4.3.3. Conclusion on Mechanisms
Within the framework of the computational model employed (united-atom, LJ potential, rigid framework), the separation of C5 isomers in the UiO-66 family is governed by adsorption thermodynamics, dictated by Lennard-Jones interactions and modulated by geometric complementarity and steric effects. The general preference for branched isomers in the fcu topology is due to better shape fitting in the 3D cavities, as visualized in the molecular configurations in Figure 12. Functionalization acts by tuning these interactions: the -CH3 group has minimal effect; the -COOH group introduces strong steric hindrance limiting capacity; and the -NH2 group creates sites with specific geometric recognition that maximize LJ interactions with neopentane, leading to high intrinsic selectivity.
5. Conclusions
In this work, we employed GCMC and MD molecular simulations to systematically investigate the adsorption and separation of pentane isomers (n-pentane, isopentane, and neopentane) in the UiO-66 MOF and three of its directly functionalized variants (-NH2, -CH3, -COOH) at 298 . The main objective was to elucidate the impact of linker functionalization on the affinity and selectivity towards these C5 isomers within a robust and well-defined topology, using a force field model based on Lennard-Jones interactions and treating the framework as rigid.
Single-component adsorption results and isosteric heats of adsorption () consistently reveal an intrinsic preference of the UiO-66 structure for branched isomers over the linear isomer at low pressures. In the parent material UiO-66-H and the methylated variant UiO-66-CH3, the affinity order is neopentane ≈ isopentane n-pentane. This behavior is attributed to better geometric complementarity between the compact molecular shapes of the branched isomers and the three-dimensional cavities of the fcu topology, allowing for optimization of van der Waals (LJ) interactions, as visualized in Figure 12.
Linker functionalization significantly modulates this intrinsic behavior. The introduction of the amino group in UiO-66-NH2 induces a notable increase in affinity, highly selectively towards neopentane ( 80 mol−1), establishing strong intrinsic selectivity: neopentane isopentane > n-pentane. This pronounced effect is explained by the creation of a geometrically specific adsorption environment where LJ interactions between neopentane and the framework (including the -NH2 group) are maximized. Conversely, the carboxylated variant UiO-66-COOH, while maintaining high initial affinity for branched isomers, exhibits significantly reduced saturation capacity, indicating strong steric hindrance caused by the -COOH group that limits access and/or efficient packing in the pores.
Equimolar mixture simulations (binary and ternary) confirm the trends observed in single-component studies and the effectiveness of functionalization in modulating competitive selectivity. UiO-66-NH2 demonstrates the highest selectivity under competitive conditions, particularly for neopentane/isopentane separation and n-pentane exclusion, especially at intermediate pressures. UiO-66-H and UiO-66-CH3 show moderate and similar selectivities, while UiO-66-COOH presents complex behavior with low overall selectivity at high pressures due to significant co-adsorption and steric effects. The systematic comparison with Ideal Adsorbed Solution Theory (IAST) predictions reiterates its limitations in quantitatively predicting mixture behavior in these heterogeneous systems, although it correctly captures the qualitative order of preference in most cases.
In summary, this computational study demonstrates that linker functionalization within the UiO-66 platform is an effective strategy for tuning affinity and selectivity towards pentane isomers based on Lennard-Jones interactions and geometric/steric effects. Particularly, UiO-66-NH2 emerges as a promising candidate for affinity-based selective separation, with a notable preference for neopentane. These findings provide fundamental molecular understanding and comparative insights into the recognition mechanisms in functionalized MOFs and offer guidelines for the rational design of future porous materials for light alkane separations.
Author Contributions
Conceptualization, N.A.P.-C. and A.G.A.; methodology, N.A.P.-C. and A.G.A.; software, A.G.A.; formal analysis, N.A.P.-C. and A.G.A.; investigation, N.A.P.-C., M.R. and A.G.A.; writing—original draft preparation, N.A.P.-C., M.R. and A.G.A.; writing—review and editing, N.A.P.-C., M.R., and A.G.A.; visualization, N.A.P.-C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable. This study involves only computational modeling.
Informed Consent Statement
Not applicable. This study did not involve humans.
Data Availability Statement
The simulation input files and raw data supporting reported results can be made available upon reasonable request to the corresponding author.
Acknowledgments
The author N.A.P.-C. thank CONICET for a postdoctoral scholarship. M.R. and A.G.A. are staff members of CONICET.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
The following abbreviations are used in this manuscript:
| BDC | 1,4-Benzenedicarboxylic acid |
| DSL | Dual-Site Langmuir |
| GCMC | Grand Canonical Monte Carlo |
| IAST | Ideal Adsorbed Solution Theory |
| iP | Isopentane |
| LJ | Lennard-Jones |
| MD | Molecular Dynamics |
| MOF | Metal-Organic Framework |
| neoP | Neopentane |
| nP | n-Pentane |
| NVT | Canonical Ensemble (Constant Number of particles, Volume, Temperature) |
| Qst | Isosteric Heat of Adsorption |
| RON | Research Octane Number |
| TraPPE-UA | Transferable Potentials for Phase Equilibria United-Atom |
| UA | United-Atom |
| UFF | Universal Force Field |
| vdW | van der Waals |
References
- Wang, H.; Li, J. Microporous metal–organic frameworks for adsorptive separation of C5–C6 alkane isomers. Acc. Chem. Res. 2019, 52, 1968–1978. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zhou, F.; Sheng, B.; Zhang, Z.; Yang, Q.; Yang, Y.; Ren, Q.; Bao, Z. Robust Nickel Aspartate Framework for Shape Recognition of Hexane Isomers. ACS Sustain. Chem. Eng. 2022, 10, 11330–11337. [Google Scholar] [CrossRef]
- Xie, F.; Yu, L.; Wang, H.; Li, J. Metal-Organic Frameworks for C6 Alkane Separation. Angew. Chem. Int. Ed. 2023, 62, e202300722. [Google Scholar] [CrossRef]
- Zhang, Z.; Peh, S.B.; Kang, C.; Yu, K.; Zhao, D. Efficient Splitting of Alkane Isomers by a Bismuth-Based Metal-Organic Framework with Auxetic Reentrant Pore Structures. Angew. Chem. Int. Ed. 2022, 61, e202211808. [Google Scholar] [CrossRef] [PubMed]
- Luna-Triguero, A.; Gómez-Álvarez, P.; Calero, S. Adsorptive Process Design for the Separation of Hexane Isomers Using Zeolites. Phys. Chem. Chem. Phys. 2017, 19, 5037–5042. [Google Scholar] [CrossRef]
- Wang, H.; Dong, X.; Lin, J.; Teat, S.J.; Jensen, S.; Cure, J.; Alexandrov, E.V.; Xia, Q.; Tan, K.; Wang, Q.; et al. Topologically Guided Tuning of Zr-Mof Pore Structures for Highly Selective Separation of C6 Alkane Isomers. Nat. Commun. 2018, 9, 1745. [Google Scholar] [CrossRef]
- Schenk, M.; Vidal, S.; Vlugt, T.J.H.; Smit, B.; Krishna, R. Separation of Alkane Isomers by Exploiting Entropy Effects During Adsorption on Silicalite-1: A Configurational-Bias Monte Carlo Simulation Study. Langmuir 2001, 17, 7110–7117. [Google Scholar] [CrossRef]
- Cui, W.G.; Hu, T.L.; Bu, X.H. Metal–organic framework materials for the separation and purification of light hydrocarbons. Adv. Mater. 2020, 32, 1806445. [Google Scholar] [CrossRef]
- Arcidiácono, M.; Mártire, A.P.; Allegretto, J.A.; Rafti, M.; Marmisollé, W.A.; Azzaroni, O. Nanoarchitectonics of Metal–Organic Frameworks (MOFs) for energy and sensing applications. Mater. Nanoarchitectonics 2024, 387–428. [Google Scholar] [CrossRef]
- Bobbitt, N.S.; Rosen, A.S.; Snurr, R.Q. Topological effects on separation of alkane isomers in metal- organic frameworks. Fluid Phase Equilib. 2020, 519, 112642. [Google Scholar] [CrossRef]
- Yu, L.; Dong, X.; Gong, Q.; Acharya, S.R.; Lin, Y.; Wang, H.; Han, Y.; Thonhauser, T.; Li, J. Splitting Mono- and Dibranched Alkane Isomers by a Robust Aluminum-Based Metal–Organic Framework Material with Optimal Pore Dimensions. J. Am. Chem. Soc. 2020, 142, 6925–6929. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Ullah, S.; Wang, H.; Xia, Q.; Thonhauser, T.; Li, J. High-Capacity Splitting of Mono- and Dibranched Hexane Isomers by a Robust Zinc-Based Metal–Organic Framework. Angew. Chem. Int. Ed. 2022, 61, e202211359. [Google Scholar] [CrossRef] [PubMed]
- Dubbeldam, D.; Krishna, R.; Calero, S.; Yazaydın, A.O. Computer-Assisted Screening of Ordered Crystalline Nanoporous Adsorbents for Separation of Alkane Isomers. Angew. Chem. Int. Ed. 2012, 51, 11867–11871. [Google Scholar] [CrossRef] [PubMed]
- Cavka, J.H.; Jakobsen, S.; Olsbye, U.; Guillou, N.; Lamberti, C.; Bordiga, S.; Lillerud, K.P. A New Zirconium Inorganic Building Brick Forming Metal Organic Frameworks with Exceptional Stability. J. Am. Chem. Soc. 2008, 130, 13850–13851. [Google Scholar] [CrossRef]
- Kandiah, M.; Nilsen, M.H.; Usseglio, S.; Jakobsen, S.; Olsbye, U.; Tilset, M.; Larabi, C.; Quadrelli, E.A.; Bonino, F.; Lillerud, K.P. Synthesis and Stability of Tagged UiO-66 Zr-MOFs. Chem. Mater. 2010, 22, 6632–6640. [Google Scholar] [CrossRef]
- DeCoste, J.B.; Peterson, G.W.; Jasuja, H.; Glover, T.G.; Huang, Y.G.; Walton, K.S. Stability and Degradation Mechanisms of Metal–organic Frameworks Containing the Zr6O4(OH)4 Secondary Building Unit. J. Mater. Chem. A 2013, 1, 5642–5652. [Google Scholar] [CrossRef]
- Lü, G.; Cui, C.; Zhang, W.; Liu, Y.; Huo, F. Synthesis and Self-Assembly of Monodispersed Metal-Organic Framework Microcrystals. Chem. Asian J. 2012, 7, 2298–2301. [Google Scholar] [CrossRef]
- Rada, Z.H.; Abid, H.R.; Shang, J.; Sun, H.; He, Y.; Webley, P.A.; Liu, S.; Wang, S. Functionalized UiO-66 by Single and Binary (OH)2 and NO2 Groups for Uptake of CO2 and CH4. Ind. Eng. Chem. Res. 2016, 55, 7924–7934. [Google Scholar] [CrossRef]
- Cmarik, G.E.; Kim, M.; Cohen, S.M.; Walton, K.S. Tuning the Adsorption Properties of UiO-66 via Ligand Functionalization. Langmuir 2012, 28, 15606–15613. [Google Scholar] [CrossRef]
- Marshall, R.J.; Forgan, R.S. Postsynthetic Modification of Zirconium Metal-Organic Frameworks. Eur. J. Inorg. Chem. 2016, 2016, 4310–4331. [Google Scholar] [CrossRef]
- Smit, B.; Maesen, T.L.M. Molecular simulations of zeolites: Absorption, diffusion, and reaction. Chem. Rev. 2008, 108, 4125–4184. [Google Scholar] [CrossRef] [PubMed]
- Vaddanam, V.S.; Bootharajan, M.; Sengupta, S.; Sreenivasulu, B.; Rao, C.V.S.B.; Ammath, S. Hydrogen Adsorption Behavior of Highly Stable Assorted Functionalized UiO-66-MOFs at Low Pressures: A Combined Experimental and DFT Study. J. Phys. Chem. C 2023, 127, 14757–14768. [Google Scholar] [CrossRef]
- Hermosilla, M.E.F.; Chávez, N.A.P.; Albesa, A.G. Monte Carlo simulations of n-butane and n-octane adsorbed onto graphite and a molecular model of activated carbon. Adsorption 2019, 25, 1419–1424. [Google Scholar] [CrossRef]
- Gallaba, D.H.; Albesa, A.G.; Migone, A.D. Evidence of gate-opening on xenon adsorption on ZIF-8: An adsorption and computer simulation study. J. Phys. Chem. C 2016, 120, 16649–16657. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Martin, M.G.; Siepmann, J.I. Transferable potentials for phase equilibria. 1. United-atom description of n-alkanes. J. Phys. Chem. B 1998, 102, 2569–2577. [Google Scholar] [CrossRef]
- Eggimann, B.L.; Sunnarborg, A.J.; Stern, H.D.; Bliss, A.P.; Siepmann, J.I. An Online Parameter and Property Database for the TraPPE Force Field. Mol. Simul. 2014, 40, 101–105. [Google Scholar] [CrossRef]
- Salam, A.; Deleuze, M.S. High-level theoretical study of the conformational equilibrium of n-pentane. J. Chem. Phys. 2002, 116, 1296–1302. [Google Scholar] [CrossRef]
- Yang, Q.; Guillerm, V.; Ragon, F.; Wiersum, A.D.; Llewellyn, P.L.; Zhong, C.; Devic, T.; Serre, C.; Maurin, G. CH4 Storage and CO2 Capture in Highly Porous Zirconium Oxide Based Metal–Organic Frameworks. Chem. Commun. 2012, 48, 9831–9833. [Google Scholar] [CrossRef]
- Mayo, S.L.; Olafson, B.D.; Goddard, W.A. DREIDING: A generic force field for molecular simulations. J. Phys. Chem. 1990, 94, 8897–8909. [Google Scholar] [CrossRef]
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A., III; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Nicholson, D.; Parsonage, N.G. Computer Simulation and the Statistical Mechanics of Adsorption; Academic Press: London, UK, 1982. [Google Scholar]
- Khudozhitkov, A.E.; Plekhanov, M.S.; Arzumanov, S.S.; Kolokolov, D.I.; Stepanov, A.G. Mobility and separation of linear and branched C5 alkanes in UiO-66 (Zr) probed by 2H NMR and MD simulations. Phys. Chem. Chem. Phys. 2023, 25, 27516–27523. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).