Abstract
A comprehensive analytical method based on liquid chromatography–tandem mass spectrometry (LC-MS/MS) was developed for the simultaneous detection of 12 mushroom toxins (ibotenic acid, muscimol, muscarine, β-amanitin, α-amanitin, desoxoviroidin, γ-amanitin, phallisacin, illudin S, phallacidin, phalloidin and illudin M) in mushrooms, serum, urine and simulated gastric fluid. The samples were extracted with water or acetonitrile solution, and the serum sample was further purified with PSA sorbent. Chromatographic separation was performed on an ACQUITY UPLC HSS T3 column with gradient elution using methanol and water containing 1 mM ammonia fluoride as a mobile phase. Mass spectrometric acquisition was performed in electrospray positive ionization mode. Good linearities (R2 > 0.994) were obtained for 12 toxins over the range of 0.05~200 µg/L. Matrix-matched calibration curves were used for quantification. The method limits of quantification were 0.01~0.2 mg/kg for mushrooms and 0.15~2.0 µg/L for three biological liquid samples. The mean recoveries of 12 target toxins (spiked at three concentration levels) ranged from 73.0% to 110.3%, with relative standard deviations not exceeding 19.4%, which meets the requirements for the determination of trace compounds in a biological matrix. This method was applied to the analysis of mushroom samples from Yunnan Province. As a result, 11 toxins, not including illudin M, were detected with a concentration range of 0.61~2143 mg/kg.
Keywords:
mushroom toxins; amanitin; ibotenic acid; muscimol; muscarine; illudin S; serum; urine; LC-MS/MS 1. Introduction
Wild mushrooms are appreciated by many people around the world for their delicious taste and unique flavor. Picking wild mushrooms in some rural areas not only satisfies the taste buds of diners, but also brings economic benefits to local people [1,2,3]. However, some wild mushrooms are poisonous due to the presence of natural toxins [4]. According to The Situation of Food Poisoning Incidents Nationwide published by the China National Health Commission, more than half of all poisoning deaths are attributed to the ingestion of poisonous mushrooms or poisonous animals and plants [5,6,7,8,9]. For example, in Yunnan, a province on the southwestern border of China, where more than half of the world’s edible mushroom production originates, a significant proportion of which is for domestic consumption, there are consistently several or dozens of deaths from mushroom poisoning each year [10,11].
The identification and detection of mushroom toxins is essential for the prevention and control of food poisoning, especially in spring and summer when mushroom poisoning is common. Tests can accurately identify mushrooms containing toxins, thus preventing poisoning incidents caused by accidental ingestion. Toxin analysis in human biosamples can aid clinical decision-making in patient management and pharmacotherapy. Therefore, analytical methods have been developed to determine different types of mushroom toxins in mushrooms, serum or urine samples. Laboratory tests based on immunoassays [12,13] or HPLC [14] as well as LC-MS/MS [15,16] techniques are commonly used. LC-MS/MS, which offers high sensitivity and selectivity in the identification of toxin molecules based on their specific mass-to-charge ratio and retention times, has become the most widely used technology for the detection of mushroom toxins.
In terms of LC-MS/MS techniques, most publications focused on the analysis of amanitin, as it was recognized as one of the most toxic compounds in the mushroom realm, including α-amanitin, β-amanitin, γ-amanitin, phalloidin and phallacidin [17,18]. Muscarine, muscimol and ibotenic acid are other frequently implicated mushroom toxins [19]. In recent decades, Japanese researchers have identified illudin S and illudin M mainly from the genus Omphalotus (such as Omphalotus olearius, Omphalotus nidiformis and Omphalotus guepiniformis), which can cause gastroenteritis poisoning [20,21,22]. At present, most LC-MS/MS analytical methods for the detection of mushroom toxins only follow one class of compounds or no more than six target compounds. However, in real circumstances, there are many types of wild poisonous mushrooms, and edible mushrooms tend to be similar in morphology and difficult to distinguish, resulting in the coexistence of multiple classes of toxins [23]. Therefore, the simultaneous analysis of multiple toxins by rapid LC-MS/MS qualitative and quantitative methods is of great importance. The aim of this study was to develop an efficient sample preparation procedure and an accurate analytical technique for the detection of a total of 12 toxins in mushroom, urine, serum and gastric fluid samples.
2. Materials and Methods
2.1. Reagents and Chemicals
The mushroom toxins α-amanitin, β-amanitin, γ-amanitin, phalloidin, phallacidin, muscarine, muscimol and ibotenic acid (purity ≥ 96.0%) were purchased from Sigma-Aldrich (Alexander, MI, USA). Illudin S and illudin M (purity 98%) were purchased from Cayman Chemicals (Ann Arbor, MI, USA). Phallisacin and desoxoviroidin (purity 98%) were purchased from First Standard (Tianjin, China).
Fisher Scientific (Fair Lawn, NJ, USA) provided LC-MS-grade methanol and acetonitrile. J&K Chemicals (Shanghai, China) provided HPLC-grade formic acid, ammonium formate, ammonium acetate, ammonium carbonate and ammonium fluoride. Simulated gastric fluid (SGF) was purchased from HongFeng Corp. (Dongwan, China). Solid-phase extraction (SPE) sorbents HLB, MCX and PSA used for the purification of serum extracts were purchased from Agela Technologies (Tianjin, China). Ultrapure water (18.2 MΩ/cm) was obtained from a Milli-Q apparatus (Millipore, Bedford, MA, USA).
2.2. Preparation of Standard Solutions
The standard stock solution with a concentration of 100 mg/L was prepared by accurately weighing 1.0 mg toxins and dissolving them individually in 10.0 mL methanol. To prepare calibration standards of different concentrations, aliquots of different stock solutions were combined and then diluted with water.
2.3. LC-MS/MS Analysis
LC-MS/MS analysis was performed using a Waters ACQUITY UPLC system coupled to a Waters Xevo TQ-XS mass spectrometer (Waters Corp., Milford, MA, USA). An ACQUITY (Waters Corp., Milford, MA, USA) UPLC® HSS T3 column (100 mm × 2.1 mm, 1.7 μm) was used for the LC separation. The mobile phase consisted of water containing 1 mM ammonium fluoride and methanol, designated A and B, respectively. The flow rate was kept at 0.3 mL/min. The initial composition of the mobile phase started at 0% B and was held for 0.5 min. This was gradually increased to 65% B over 3 min, held for 2 min and washed with 95% B for 1.5 min. The column was equilibrated with 0% B for 2 min until the next injection. A column oven temperature of 40 °C was set, and a 2 μL injection volume was used. Mass spectrometry was performed using the parameters listed in Table 1. The specific parameters for each compound are given in Section 3.1.

Table 1.
Mass tuning parameters of 12 mushroom toxins.
2.4. Sample Pretreatment
The edible mushroom Lentinus edodes, collected from a local supermarket, was used for the development and validation of the analytical method. Homogenized samples were stained using an Alpha 1-2 LD plus freeze-dyeing machine (Christ/Sigma, Osterode am Harz, Germany). Aliquots of 100 mg dried samples were sonicated with 5 mL of pure water for 30 min and centrifuged at 10,000 rpm for 5 min at 4 °C. Then, 0.5 mL of the supernatant was transferred to a vial for LC-MS/MS analysis.
Serum and urine samples for method development were kindly provided by healthy volunteers in our laboratory. The study was approved by the Medical Ethical Review Committee of the National Institute for Occupational Health and Poison Control, Chinese Center for Disease Control and Prevention [No. NIOHP202323] on 19 June 2023. One-half-milliliter urine samples were mixed with 0.5 mL of water and then centrifuged at 10,000× g rpm for 5 min at 4 °C. For the serum, 0.4 mL samples were placed in 2 mL polypropylene tubes, and then 0.6 mL of acetonitrile was added for protein precipitation. The mixture was sonicated for 15 min and then centrifuged at 10,000× g rpm for 5 min at 4 °C. Then, 0.8 mL of the supernatant was pipetted into a clean Eppendorf tube containing 20 mg of PSA SPE sorbent. The tubes were subjected to vortex-induced vibration at 2000 rpm for 1 min and then centrifuged at 10,000× g rpm for 5 min at 4 °C. The top layer was collected for LC-MS/MS analysis.
SGF samples were diluted with an equal volume of acetonitrile, and the next step was the same as for the urine sample.
2.5. Method Validation
The validation of this method included the assessment of the linear range and linearity of the calibration curves, evaluation of the matrix effect (ME), determination of the method limit of detection (MLOD) and method limit of quantitation (MLOQ), and examination of the accuracy and precision.
3. Results and Discussion
3.1. Optimization of Instrumental Method
3.1.1. MS/MS Parameters
Twelve mushroom toxins, namely ibotenic acid, muscimol, muscarine, β-amanitin, α-amanitin, desoxoviroidin, γ-amanitin, phallisacin, illudin S, phallacidin, phalloidin and illudin M, were selected in this study. The physico-chemical properties and toxicity of these 12 mushroom toxins are given in Table 2.
Table 2.
Physico-chemical properties and toxicity of mushroom toxins in this study.
Optimization of the quadrupole mass spectrometry parameters was performed in ESI positive mode using the single standard (0.1 μg/mL) via a syringe pump at 10 μL/min with direct injection. The calibration of each toxin was initially performed automatically using Waters Intelli Start and then checked manually to ensure accuracy. For the majority of the analytes, [M + H]+ is the most abundant ion, except for illudin S and illudin M. For these two compounds, the dominant ion observed was [M + H − H2O]+, with m/z 247 showing the highest intensity for illudin S, which was produced by in-source collision-induced dissociation with the loss of a water molecule. These ions were selected as precursor ions, and their product ions were subsequently identified by optimization. The underlined transition marked in Table 3 is chosen for quantitation, while the second, less sensitive one is used for confirmation.
Table 3.
LC-MS/MS acquisition parameters for 12 mushroom toxins.
3.1.2. Optimization of LC Columns
The complex variations in chemical composition and physical properties of the 12 toxins present a challenge for their separation and analysis by chromatography. A series of tests were carried out on five different 2.1 mm × 100 mm columns to evaluate the efficacy and specificity of all 12 target compounds. The brand names, particle size, technical characteristics and manufacturers of the columns are summarized in Table 4. The specific details of the mobile phase conditions are given in Tables S1–S5 in Supplementary Materials.
Table 4.
Summary of the chromatographic columns evaluated.
As can be seen from Figure 1, the performance of the HSS T3 column shows favorable sensitivity and efficiency in the analysis of interest, while the retention of ibotenic acid and muscimol on this column is slightly insufficient. The CSH PFP column, with a weak positive charge on the stationary particles, has shown exceptional peak shapes and homogeneous distribution of analyte peaks in various bioanalytical applications and metabolomics [30]. However, in this study, the performance of this column shows deficiencies in either peak shape or response. The Trinity P1 column is designed with a modified inner and outer pore area with an organic layer that provides both reversed-phase and anion-exchange properties and cation-exchange functionality, respectively. Thus, trifunctional retention mechanisms including reversed phase, anion exchange and cation exchange were combined in one column. Although this gave good retention for the highly polar compounds ibotenic acid (RT 2.79 min) and muscimol (RT 1.56 min), the signal intensity of all compounds except muscarine was relatively low, resulting in a decrease in peak intensity, possibly due to the inclusion of 20 mM ammonia formate. A total of twelve toxins were tested on the C18 amide column. This alternative mixed retention phase column showed a comparable elution sequence to that observed on the HSS T3 column, indicating that the predominant retention mechanism is likely to be reversed-phase one. The tailing peak observed for muscarine and the relatively weak response further suggest that the C18 amide column may not be the most appropriate choice (see Figure 1d). In addition, the BEH amide column was also not selected due to suboptimal sensitivity for most compounds and inadequate retention for illudin S and illudin M.
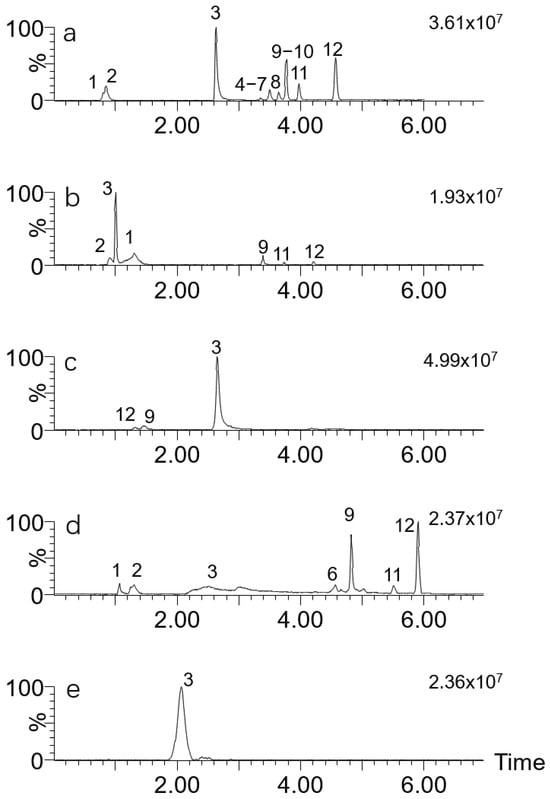
Figure 1.
TIC chromatograms of 12 mushroom toxins on different columns. Toxins: 1. ibotenic acid; 2. muscimol; 3. muscarine; 4. β-amanitin; 5. α-amanitin; 6. desoxoviroidin; 7. γ-amanitin; 8. phallisacin; 9. illudin S; 10. phallacidin; 11. phalloidin; 12. illudin M. Columns: (a) HSS T3; (b) CSH PFP; (c) Trinity P1; (d) C18 amide; (e) BEH amide.
3.1.3. Optimization of Mobile Phase
Based on the above information, the HSS T3 column was selected for this research. The optimization of the mobile phase then took into account the presence of an ammonia buffer along with the organic solvent.
In the literature, ammonia acetate in the concentration range of 1–100 mM is commonly observed as an additive in LC-MS analysis, including in the analysis of mushroom toxins [31]. Other different ammonia buffers have also been reported. Here, we compared ammonium formate, ammonium acetate, ammonium carbonate and ammonium fluoride solution at 1 mM with methanol as a mobile phase component on the effect of LC-MS/MS analysis. Figure 2 shows that ammonium fluoride, ammonium formate and ammonium acetate as modifiers induced similar TIC chromatograms. However, the former gave the best sensitivity for most of the targets. When 1 mM ammonium carbonate was used, strong broadening and tailing of the chromatographic peaks of muscimol and muscarine were observed.
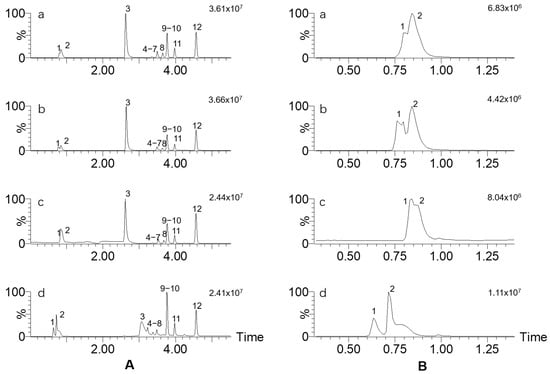
Figure 2.
(A) TIC chromatograms of 12 mushroom toxins using different ammonia buffers and methanol as mobile phase; (B) partial enlargement at 0.25~1.4 min from (A). Ammonia buffer: a, ammonium fluoride; b, ammonium formate; c, ammonium acetate; d, ammonium carbonate. The toxin numbers are the same as in Figure 1.
The composition of the organic solvent and the addition of formic acid were then further optimized. The peak response was investigated using acetonitrile–1 mM ammonium fluoride, acetonitrile–1 mM ammonium fluoride and 0.1% formic acid, methanol–1 mM ammonium fluoride, and methanol–1 mM ammonium fluoride and 0.1% formic acid as mobile phases. The results are shown in Figure 3. The addition of 0.1% formic acid results in a significant decrease in most target compounds in both acetonitrile and methanol systems. The maximum sensitivity was achieved using methanol–1 mM ammonium fluoride.
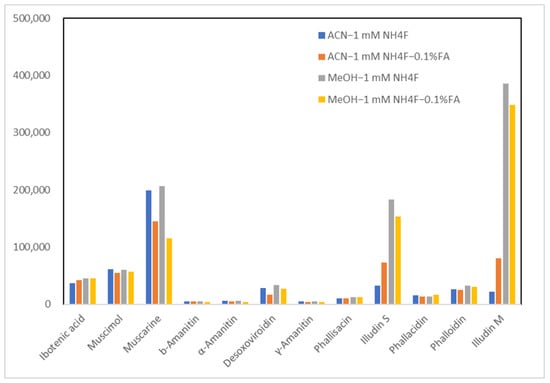
Figure 3.
Peak response of 12 mushroom toxins using different mobile phase components. Abbreviations in the legend: ACN, acetonitrile; MeOH, methanol; NH4F, ammonium fluoride; FA, formic acid.
3.2. Optimization of Sample Pretreatment
As the amount of toxins in poisonous mushrooms is usually high, generally in the mg/kg or even g/kg range, the analysis of mushroom samples is relatively simple compared to other biological matrices. Using an ultrasonic apparatus, 100 mg samples of dried mushrooms were extracted with 5 mL of water, and the resulting extracts were subjected to LC-MS/MS analysis. Urine and SGF samples were diluted with one volume of water and acetonitrile, respectively, followed by centrifugation and transfer of the supernatant for analysis. As for the serum matrix, it contains a high abundance of proteins and phospholipids, which could suppress the spectrometric response during the assay. Acetonitrile extracts of serum were further purified using three types of SPE sorbents: MCX, HLB and PSA. MCX is a mixed-mode exchanger that combines both strong ion exchange and reversed phase in one sorbent, specifically designed for improved cation retention. HLB contains only the reversed-phase component in the sorbent. PSA, the abbreviation for primary secondary silica, has proven to be the most effective sorbent for the removal of various matrices, significantly reducing the matrix effect and playing a vital role in the removal of fatty acids and polar interferences. Purification performance was evaluated in terms of recovery and ME, which is represented by the ratio of the peak area of matrix-matched standards to the pure standard solution at the same concentration level (50 μg/L) and multiplied by 100 for expression in percent. Values of 100 ± 15% indicate that no matrix effect was observed, whereas values above 115% or below 85% indicate the presence of ionization enhancement or suppression, respectively.
The two least retained toxins, ibotenic acid and muscimol, were completely suppressed in serum and urine samples. This result can be explained by the inhibition of ionization caused by the different interferents co-eluting in serum and urine. Therefore, SPE purification of serum samples only gives results for the other 10 toxins as shown in Figure 4. When MCX powder was used, muscarine was tightly bound to the sorbent, and recovery was zero. HLB and PSA showed recoveries between 79 and 112%, while the former achieved ME 41~72%, significantly less than that for PSA (52~84%). It was shown that PSA has a better ability to purify the serum sample compared to MCX sorbent. Therefore, 20 mg of PSA was selected for purifying 0.8 mL of serum extracts.
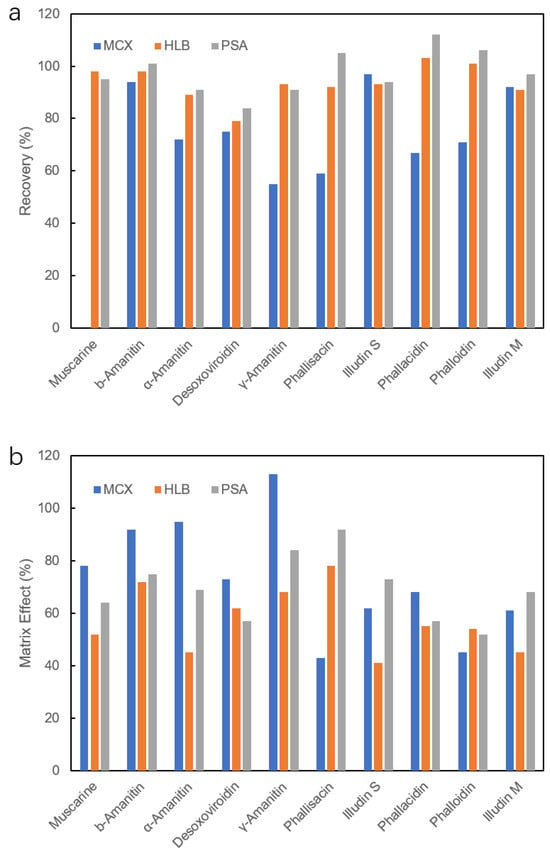
Figure 4.
Recovery (a) and matrix effect (b) of 10 mushroom toxins in serum extracts after different SPE purifications.
3.3. Method Validation
The calibration curve was initially constructed using a clean standard series from 0.02 μg/L to 200 μg/L. Satisfactory linearity was observed over the range of all 12 target toxins from 0.05 to 200 μg/L with correlation coefficients of R2 > 0.994 (Table 5). The isotope dilution method is an ideal way to compensate for the depletion of specific compounds during sample preparation and the interferences of mass spectrometric analysis. However, the lack of isotopic standards for the target toxins in this study poses a significant challenge. Therefore, quantification of real samples was performed using matrix-matched calibration. As shown in Table 5, the ME was obtained by the ratio of the slope of the matrix-matched calibration plot to the slope of the solvent-dissolved standard calibration plot and multiplied by 100 for expression in percent. As a result, 33~91% ME was observed for 12 toxins in mushroom, urine, serum and SGF, indicating varying degrees of response suppression.
Table 5.
Linearity and matrix effects of 12 mushroom toxins.
The MLODs (identified as the concentration giving a signal-to-noise ratio (S/N) of 3) and MLOQs (identified as the concentration giving an S/N of 10) are summarized in Table 6. The MLODs of 12 toxins in mushroom samples were 0.005~0.1 mg/kg, and the MLOQs were 0.01~0.2 mg/kg. Muscarine shows more sensitive results compared to the other toxins in all four matrices. The MLOQs of the 12 toxins in SGF were 0.20~2.0 µg/L. The values for ibotenic acid and muscimol in serum and urine are not available, and the other 10 toxins showed MLOQs of 0.15~2.0 µg/L. These results were in line with or even lower than the majority of previous studies on mushroom toxins [19,32,33,34]. A preliminary summary of the literature on the detection of mushroom toxins by LC-MS/MS is given in Table S6 in Supplementary Materials. Most of these reports focused on mushroom analysis targeting 1~11 toxins, with MDLs ranging from 0.00098 mg/kg to 10 mg/kg. Urine and blood samples were extracted with acidic methanol aqueous solution or other reagents followed by SPE purification, resulting in MDLs of 1~2000 μg/mL. Compared to this literature, this method is very simple, has high sensitivity and has a high coverage of the fungal toxins as well as the sample matrix.
Table 6.
MLODs and MLOQs of 12 mushroom toxins in different matrices.
The accuracy of the method was validated by analyzing the average recovery from spiked blank at three different concentration levels, each performed in six replicates. Four different matrix samples were tested. As shown in Table 7, the accuracy of the method ranged from 73.0% to 110.3%. The precision of this method was represented by the percentage RSD at each spiked level for each toxin, and these values are also summarized in Table 7, with values not exceeding 19.4%.
Table 7.
Recoveries and RSDs of 12 toxins in mushroom, serum, urine and SGF (n = 6).
3.4. Method Application
This novel analytical method was used to analyze 34 samples of wild mushrooms from Yunnan Province. Figure 5 shows LC-MS/MS chromatograms of 12 toxins in standard and real mushroom samples. All toxins except illudin M were detected in these real samples, among which illudin S was found in 15 Omphalotus olearius samples in the concentration range of 4.37~141 mg/kg (Table 8). Ibotenic acid, muscimol and muscarine were detected in five Amanita pseudosychnopyramis samples at 23.6~489 mg/kg. Amanita pallidorosea, Amanita exitialis and Amanita subjunquillea samples contained α-amanitin, β-amanitin, γ-amanitin, phallisacin, phallacidin and phalloidin at 0.61~2143 mg/kg. No toxin was detected in the Chlorophyllum molybdites sample.

Figure 5.
LC-MS/MS chromatograms of 12 mushroom toxins in standard solution (A) and wild mushroom samples (B). Mushroom species: a, Amanita pseudosychnopyramis; b, Amanita pallidorosea; c, Amanita exitialis; d, Amanita subjunquillea; e, Omphalotus olearius.
Table 8.
Occurrence of 12 toxins in wild mushrooms.
4. Conclusions
A comprehensive LC-MS/MS method was developed for the simultaneous detection of 12 mushroom toxins from different species and sources in mushroom and biological samples. The entire method consisted of a simple extraction followed by dilution injection or SPE clean-up using PSA sorbent for serum samples. The method was well validated. The main advantages of the method presented in this paper are its high coverage of mushroom toxins, simplicity, relatively high selectivity and high sensitivity. It can be used in the laboratory for real sample analysis.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/separations11060183/s1, Table S1: Gradient mobile phase conditions of HSS T3 column; Table S2: Gradient mobile phase conditions of CSH PFP column; Table S3: Gradient mobile phase conditions of Trinity P1 column; Table S4: Gradient mobile phase conditions of ACE C18-Amide column; Table S5: Gradient mobile phase conditions of BEH Amide column; Table S6: Literature summary of mushroom toxins detection using LC-MS/MS [14,15,16,19,35,36,37].
Author Contributions
Experiment preparation, writing—original draft preparation, J.L.; validation, H.L.; conceptualization and methodology, J.Z.; writing—review and editing, supervision, C.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Beijing Talent Development Plan for High-level Public Health Technical Personnel Project (Gugan-01-033).
Data Availability Statement
The data supporting reported results can be obtained from the corresponding author upon request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Li, W.W.; Pires, S.M.; Liu, Z.T.; Liang, J.J.; Wang, Y.F.; Chen, W.; Liu, C.W.; Liu, J.K.; Han, H.H.; Fu, P.; et al. Mushroom Poisoning Outbreaks—China, 2010-2020. China CDC Wkly. 2021, 3, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Svanberg, I.; Lindh, H. Mushroom hunting and consumption in twenty-first century post-industrial Sweden. J. Ethnobiol. Ethnomed. 2019, 15, 42. [Google Scholar] [CrossRef] [PubMed]
- Feeney, M.J.; Miler, A.M.; Roupas, P. Mushroom-biologically distinct and nutritionally unique: Exploring a “third food kingdom”. Nutr. Today 2014, 49, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.S.; Lee, Y.Y.; Lee, S.R.; Jang, Y.S.; Ryoo, R.; Park, W.; Kim, S.N.; Lee, S.; Park, C.C.; Kim, K.H. Isolation of Macrocyclic Trichothecene Mycotoxins from the Lethal Toxic Mushroom Podostroma cornu-damae and Their Cytotoxic Activities. Separations 2024, 11, 65. [Google Scholar] [CrossRef]
- Li, H.J.; Zhang, H.S.; Zhang, Y.Z.; Zhang, K.P.; Zhou, J.; Yin, Y.; Jiang, S.F.; Ma, P.B.; He, Q.; Zhang, Y.; et al. Mushroom Poisoning Outbreaks—China, 2019. China CDC Wkly. 2020, 2, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Li, H.J.; Zhang, H.S.; Zhang, Y.Z.; Zhou, J.; Yin, Y.; He, Q.; Jiang, S.F.; Ma, P.B.; Zhang, Y.T.; Wen, K.; et al. Mushroom Poisoning Outbreaks—China, 2020. China CDC Wkly. 2021, 3, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Li, H.J.; Zhang, H.S.; Zhang, Y.Z.; Zhou, J.; Yin, Y.; He, Q.; Jiang, S.F.; Ma, P.B.; Zhang, Y.T.; Yuan, Y.; et al. Mushroom Poisoning Outbreaks—China, 2021. China CDC Wkly. 2022, 4, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Li, H.J.; Zhang, Y.Z.; Zhang, H.S.; Zhou, J.; Liang, J.Q.; Yin, Y.; He, Q.; Jiang, S.F.; Zhang, Y.T.; Yuan, Y.; et al. Mushroom Poisoning Outbreaks—China, 2022. China CDC Wkly. 2023, 5, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Li, H.J.; Zhang, Y.Z.; Zhang, H.S.; Zhou, J.; Chen, Z.H.; Liang, J.Q.; Yin, Y.; He, Q.; Jiang, S.F.; Zhang, Y.T.; et al. Mushroom Poisoning Outbreaks—China, 2023. China CDC Wkly. 2024, 6, 64–68. [Google Scholar] [CrossRef]
- Shen, X.L.; Huang, T.; Jia, Y.; He, J.; Zheng, E.; Peng, X. Epidemiological characteristics and spatial correlation analysis of mushrrom poisoning in Yunnan Province from 2005 to 2019. Chin. J. Food Hyg. 2022, 34, 153–158. [Google Scholar]
- Liu, Z.T.; Su, W.W.; Zhao, J.; Zhang, Q.; Li, J.J.; Dong, H.Y.; Chen, L.P.; Yang, Y.L.; Guo, Y.C.; Min, X.D. The disease burden of wild mushroom poisoning in Yunnan Province from 2017 to 2021. Chin. J. Food Hyg. 2022, 34, 1059–1062. [Google Scholar]
- Zhou, S.Y.; Guo, L.L.; Xu, X.X.; Song, S.S.; Liu, L.Q.; Kuang, H.; Zhu, Y.Y.; Xu, L.G.; Xu, C.L. Ultrasensitive paper sensor for simultaneous detection of alpha-amanitin and beta-amanitin by the production of monoclonal antibodies. Food Chem. 2022, 396, 133660. [Google Scholar] [CrossRef]
- Liang, Y.; Zhou, A.; Bever, C.S.; Cheng, L.W.; Yoon, J.Y. Smartphone-based paper microfluidic competitive immunoassay for the detection of α-amanitin from mushrooms. Mikrochim. Acta 2022, 189, 322. [Google Scholar] [CrossRef] [PubMed]
- Lei, S.; Shi, P.Y.; Wu, w.l.; Xia, B.; Fu, X.; Wan, y.p.; Zhou, Y. Extensive screening of cyclopeptide toxins in mushrooms by ultra-high-performance liquid chromatography coupled with quadrupole-Orbitrap mass spectrometry. Food Chem. 2020, 329, 127–146. [Google Scholar] [CrossRef]
- Bambauer, T.P.; Wagmann, L.; Maurer, H.H.; Weber, A.A.; Meyer, M.R. Development and application of a strategy for analyzing eight biomarkers in human urine to verify toxic mushroom or ricinus communis ingestions by means of hydrophilic interaction LC coupled to HRMS/MS. Talanta 2020, 213, 120847. [Google Scholar] [CrossRef]
- Zhang, Y.Z.; Yan, Y.Y.; Li, H.J.; Fan, Y.G.; Xu, F. Toxin screening of Pseudosperma umbrinellum (Agaricals, Basidiomycota): First report of phalloidin in Inocybaceae mushroom. Toxicon 2022, 217, 155–161. [Google Scholar] [CrossRef]
- Honeychurch, K. Review: The Application of Liquid Chromatography Electrochemical Detection for the Determination of Drugs of Abuse. Separations 2016, 3, 28. [Google Scholar] [CrossRef]
- Abbott, N.L.; Hill, K.L.; Garrett, A.; Carter, M.D.; Hamelin, E.I.; Johnson, R.C. γ-amanitin in urine by LC-MS/MS using 15N10-αamanitin as the internal standard. Toxicon 2018, 152, 71–77. [Google Scholar] [CrossRef]
- Gonmori, K.; Hasegawa, K.; Fujita, H.; Kamijo, Y.; Nozawa, H.; Yamagishi, I.; Minakata, K.; Watanabe, K.; Suzuki, O. Analysis of ibotenic acid and muscimol in Amanita mushrooms by hydrophilic interaction liquid chromatography-tandem mass spectrometry. Forensic Toxicol. 2012, 30, 168–172. [Google Scholar] [CrossRef]
- Engler, M.; Anke, T.; Sterner, O. Production of antibiotics by Collybia nivalis, Omphalotus olearis, a Favolaschia and a Pterula species on natural substrates. Z. Naturforschung C J. Biosci. 1998, 53, 318–324. [Google Scholar] [CrossRef]
- Burgess, M.L.; Zhang, Y.L.; Barrow, K.D. Characterization of new illudanes, illudins F, G, and H from the basidiomycete omphalotus nidiformis. J. Nat. Prod. 1999, 62, 1542–1544. [Google Scholar] [CrossRef] [PubMed]
- Aoki, S.; Aboshi, T.; Shiono, Y.; Kimura, K.I.; Murata, T.; Arai, D.; Iizuka, Y.; Murayama, T. Constituents of the Fruiting Body of Poisonous Mushroom Omphalotus japonicus. Chem. Pharm. Bull. 2020, 68, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Flament, E.; Guitton, J.; Gaulier, J.M.; Gaillard, Y. Human Poisoning from Poisonous Higher Fungi: Focus on Analytical Toxicology and Case Reports in Forensic Toxicology. Pharmaceuticals 2020, 13, 454. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Tian, E.J.; Bao, H.Y. The classification and structures of mushroom toxins. J. Mushroom Res. 2022, 20, 128–140. [Google Scholar] [CrossRef]
- Marciniak, B.; Lopaczyńska, D.; Kowalczyk, E.; Skośkiewicz, J.; Witczak, M.; Majczyk, M.; Grabowicz, W.; Ferenc, T. Evaluation of micronuclei in mice bone marrow and antioxidant systems in erythrocytes exposed to α-amanitin. Toxicon 2013, 63, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, B.; Łopaczyńska, D.; Ferenc, T. Evaluation of the genotoxicity of alpha-amanitin in mice bone marrow cells. Toxicon 2017, 137, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Schobert, R.; Biersack, B.; Knauer, S.; Ocker, M. Conjugates of the mushroom cytotoxin illudin M with improved tumour specificity. Bioorg. Med. Chem. 2008, 16, 8592–8597. [Google Scholar] [CrossRef] [PubMed]
- Anchel, M.; Hervey, A.; Robbins, W.J. Production of Illudin M and of a Fourth Crystalline Compound by Clitocybe Illudens. Proc. Natl. Acad. Sci. USA 1952, 38, 927–928. [Google Scholar] [CrossRef] [PubMed]
- Kirchmair, M.; Pöder, R.; Huber, C.G. Identification of illudins in Omphalotus nidiformis and Omphalotus olivascens var. indigo by column liquid chromatography-atmospheric pressure chemical ionization tandem mass spectrometry. J. Chromatogr. A 1999, 832, 247–252. [Google Scholar] [CrossRef]
- Girel, S.; Guillarme, D.; Fekete, S.; Rudaz, S.; González-Ruiz, V. Investigation of several chromatographic approaches for untargeted profiling of central carbon metabolism. J. Chromatogr. A 2023, 1697, 463994. [Google Scholar] [CrossRef]
- Zhang, S.; Zhao, Y.; Li, H.; Zhou, S.; Chen, D.; Zhang, Y.; Yao, Q.; Sun, C. A Simple and High-Throughput Analysis of Amatoxins and Phallotoxins in Human Plasma, Serum and Urine Using UPLC-MS/MS Combined with PRiME HLB μElution Platform. Toxins 2016, 8, 128. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, I.; Domingues, C.; Barbosa, R.M.; Ramos, F. Amanitins in Wild Mushrooms: The Development of HPLC-UV-EC and HPLC-DAD-MS Methods for Food Safety Purposes. Foods 2022, 11, 3929. [Google Scholar] [CrossRef] [PubMed]
- Uto, Y.; Sasaki, K.; Takahashi, M.; Morimoto, K.; Inoue, K. Application of High-speed Countercurrent Chromatography for the Purification of High-purity Illudin S from Omphalotus japonicus. Anal. Sci. 2019, 35, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, Y.; Itou, T. Determination of illudin S in Omphalotus guepiniformis and foods that caused food poisoning by liquid chromatography with tandem mass spectrometry. Shokuhin Eiseigaku Zasshi 2009, 50, 167–172. (In Japanese) [Google Scholar] [CrossRef]
- Bambauer, T.P.; Wagmann, L.; Weber, A.A.; Meyer, M.R. Further development of a liquid chromatography-high-resolution mass spectrometry/mass spectrometry-based strategy for analyzing eight biomarkers in human urine indicating toxic mushroom or Ricinus communis ingestions. Drug Test. Anal. 2021, 13, 1603–1613. [Google Scholar] [CrossRef] [PubMed]
- Bambauer, T.P.; Wagmann, L.; Weber, A.A.; Meyer, M.R. Analysis of α- and β-amanitin in human plasma at subnanogram per milliliter levels by reversed phase ultra-high performance liquid chromatography coupled to orbitrap mass spectrometry. Toxins 2020, 12, 671. [Google Scholar] [CrossRef]
- Yoshioka, N.; Akamatsu, S.; Mitsuhashi, T.; Todo, C.; Asano, M.; Ueno, Y. A simple method for the simultaneous determination of mushroom toxins by liquid chromatography–time-of-flight mass spectrometry. Forensic Toxicol. 2014, 32, 89–96. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).