
Aggregation-Dispersion Chromatography: Application of Elastin-like Polypeptides


Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmid Construction
2.2. Protein Production
2.3. Protein Purification by Aggregation-Dispersion Chromatography
3. Results and Discussion
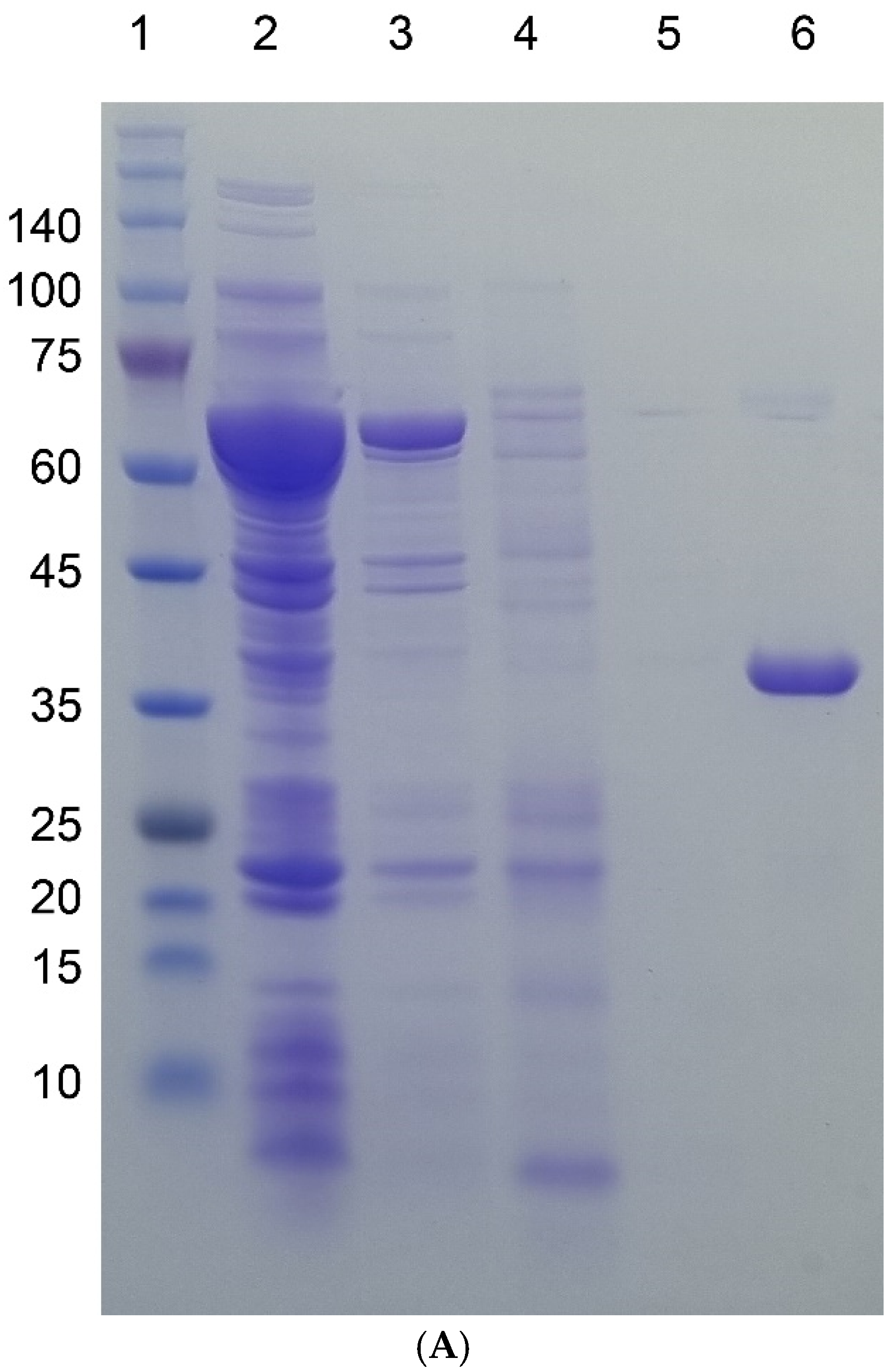
3.1. Protein Production in E. coli
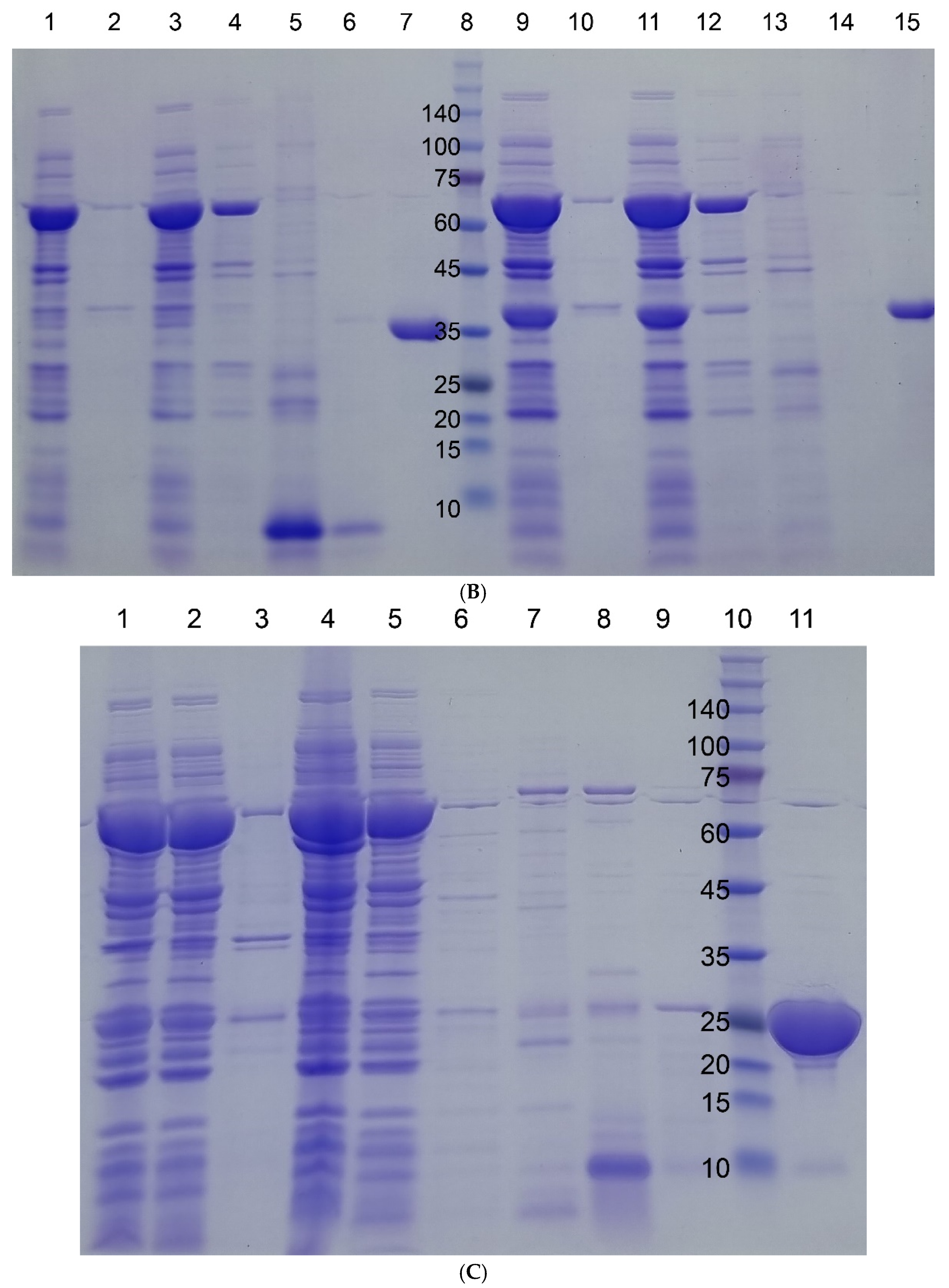
3.2. Protein Purification by Aggregation-Dispersion Chromatography
3.3. Comparison with Other Aggregation Tags
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Labrou, N.E. (Ed.) Protein Purification: An Overview. In Protein Downstream Processing: Design, Development and Application of High and Low-Resolution Methods; Humana Press: Totowa, NJ, USA, 2014; pp. 3–10. [Google Scholar] [CrossRef]
- Batool, M.; Ahmad, B.; Choi, S. A Structure-Based Drug Discovery Paradigm. Int. J. Mol. Sci. 2019, 20, 2783. [Google Scholar] [CrossRef] [PubMed]
- Wingfield, P.T. Overview of the Purification of Recombinant Proteins. Curr. Protoc. Protein Sci. 2015, 80, 6.1.1-6.1.35. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, N.K.; Shrivastava, A. Recent Developments in Bioprocessing of Recombinant Proteins: Expression Hosts and Process Development. Front. Bioeng. Biotechnol. 2019, 7, 420. [Google Scholar] [CrossRef] [PubMed]
- Rosano, G.L.; Ceccarelli, E.A. Recombinant protein expression in Escherichia coli: Advances and challenges. Front. Microbiol. 2014, 5, 172. [Google Scholar] [CrossRef]
- Baneyx, F. Recombinant protein expression in Escherichia coli. Curr. Opin. Biotechnol. 1999, 10, 411–421. [Google Scholar] [CrossRef]
- Rudge, S.R.; Ladisch, M.R. Industrial Challenges of Recombinant Proteins. In Current Applications of Pharmaceutical Biotechnology; Silva, A.C., Moreira, J.N., Lobo, J.M.S., Almeida, H., Eds.; Springer: Cham, Switzerland, 2020; pp. 1–22. [Google Scholar] [CrossRef]
- Jayakrishnan, A.; Rosli, W.R.W.; Tahir, A.R.M.; Razak, F.S.A.; Kee, P.E.; Ng, H.S.; Chew, Y.-L.; Lee, S.-K.; Ramasamy, M.; Tan, C.S.; et al. Evolving Paradigms of Recombinant Protein Production in Pharmaceutical Industry: A Rigorous Review. Sci 2024, 6, 9. [Google Scholar] [CrossRef]
- Zhang, Z.-X.; Nong, F.-T.; Wang, Y.-Z.; Yan, C.-X.; Gu, Y.; Song, P.; Sun, X.-M. Strategies for efficient production of recombinant proteins in Escherichia coli: Alleviating the host burden and enhancing protein activity. Microb. Cell Factories 2022, 21, 191. [Google Scholar] [CrossRef]
- Pina, A.S.; Lowe, C.R.; Roque, A.C.A. Challenges and opportunities in the purification of recombinant tagged proteins. Biotechnol. Adv. 2014, 32, 366–381. [Google Scholar] [CrossRef]
- Lebendiker, M.; Danieli, T. Purification of proteins fused to maltose-binding protein. Methods Mol. Biol. 2011, 681, 281–293. [Google Scholar] [CrossRef]
- Harper, S.; Speicher, D.W. Purification of proteins fused to glutathione S-transferase. Methods Mol. Biol. 2011, 681, 259–280. [Google Scholar] [CrossRef]
- Loughran, S.T.; Walls, D. Purification of poly-histidine-tagged proteins. Methods Mol. Biol. 2011, 681, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Karyolaimos, A.; de Gier, J.-W. Strategies to Enhance Periplasmic Recombinant Protein Production Yields in Escherichia coli. Front. Bioeng. Biotechnol. 2021, 9, 797334. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Zhao, Q.; Xing, L.; Zhou, B.; Wang, X. Aggregating tags for column-free protein purification. Biotechnol. J. 2015, 10, 1877–1886. [Google Scholar] [CrossRef]
- Hassouneh, W.; Christensen, T.; Chilkoti, A. Elastin-Like Polypeptides as a Purification Tag for Recombinant Proteins. Curr. Protoc. Protein Sci. 2010, 61, 6.11.1-6.11.16. [Google Scholar] [CrossRef]
- Chae, Y.K.; Um, Y.; Kim, H. A simple and sensitive detection of the binding ligands by using the receptor aggregation and NMR spectroscopy: A test case of the maltose binding protein. J. Biomol. NMR 2021, 75, 371–381. [Google Scholar] [CrossRef]
- Chae, Y.K.; Kim, H. Development of an Autoinducible Plasmid for Recombinant Protein Production. Protein Pept. Lett. 2021, 28, 1398–1407. [Google Scholar] [CrossRef] [PubMed]
- Shi, P.; Aluri, S.; Lin, Y.-A.; Shah, M.; Edman, M.; Dhandhukia, J.; Cui, H.; MacKay, J.A. Elastin-based protein polymer nanoparticles carrying drug at both corona and core suppress tumor growth in vivo. J. Control. Release 2013, 171, 330–338. [Google Scholar] [CrossRef]
- Bartlow, P.; Uechi, G.T.; Cardamone, J.J.; Sultana, T.; Fruchtl, M.; Beitle, R.R.; Ataai, M.M. Identification of native Escherichia coli BL21 (DE3) proteins that bind to immobilized metal affinity chromatography under high imidazole conditions and use of 2D-DIGE to evaluate contamination pools with respect to recombinant protein expression level. Protein Expr. Purif. 2011, 78, 216–224. [Google Scholar] [CrossRef]
- Sun, P.; Tropea, J.E.; Waugh, D.S. Enhancing the solubility of recombinant proteins in Escherichia coli by using hexahistidine-tagged maltose-binding protein as a fusion partner. Methods Mol. Biol. 2011, 705, 259–274. [Google Scholar] [CrossRef]
- Fan, Y.; Miozzi, J.M.; Stimple, S.D.; Han, T.-C.; Wood, D.W. Column-Free Purification Methods for Recombinant Proteins Using Self-Cleaving Aggregating Tags. Polymers 2018, 10, 468. [Google Scholar] [CrossRef]
- Singh, A.; Upadhyay, V.; Upadhyay, A.K.; Singh, S.M.; Panda, A.K. Protein recovery from inclusion bodies of Escherichia coli using mild solubilization process. Microb. Cell Factories 2015, 14, 41. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Number of Amino Acids | MW | pI | Instability Index | Aliphatic Index | Grand Average of Hydropathicity (GRAVY) |
|---|---|---|---|---|---|---|
| CcdB | 101 | 11,706 | 6.08 | 28.88 | 94.46 | −0.162 |
| MazE | 82 | 9355 | 4.72 | 52.26 | 109.27 | −0.299 |
| MazF | 111 | 12,098 | 8.49 | 39.97 | 85.14 | −0.093 |
| S-peptide | 20 | 2166 | 6.76 | 38.39 | 25.00 | −0.980 |
| GLRX3 | 83 | 9137 | 6.71 | 55.92 | 85.90 | −0.314 |
| GLRX4 | 115 | 12,878 | 4.69 | 59.40 | 87.48 | −0.272 |
| SH3BGRL1 | 114 | 12,774 | 5.22 | 49.88 | 65.18 | −0.691 |
| MBP | 366 | 40,209 | 5.07 | 19.39 | 83.28 | −0.331 |
| I48 | 243 | 20,698 | 5.27 | 30.88 | 134.32 | 1.245 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, H.B.; Chae, Y.K. Aggregation-Dispersion Chromatography: Application of Elastin-like Polypeptides. Separations 2024, 11, 335. https://doi.org/10.3390/separations11120335
Shin HB, Chae YK. Aggregation-Dispersion Chromatography: Application of Elastin-like Polypeptides. Separations. 2024; 11(12):335. https://doi.org/10.3390/separations11120335
Chicago/Turabian StyleShin, Han Bin, and Young Kee Chae. 2024. "Aggregation-Dispersion Chromatography: Application of Elastin-like Polypeptides" Separations 11, no. 12: 335. https://doi.org/10.3390/separations11120335
APA StyleShin, H. B., & Chae, Y. K. (2024). Aggregation-Dispersion Chromatography: Application of Elastin-like Polypeptides. Separations, 11(12), 335. https://doi.org/10.3390/separations11120335