Abstract
Tepotinib (MSC2156119) is a potent mesenchymal–epithelial transition (MET) factor inhibitor, a receptor tyrosine kinase that plays a crucial role in promoting cancer cell malignant progression. Adverse effects of tepotinib (TEP), such as peripheral edema, interstitial lung disease, nausea and diarrhea, occur due to drug accumulation and lead to termination of therapy. Therefore, the in silico and experimental metabolic susceptibility of TEP was investigated. In the current work, an LC-MS/MS analytical method was developed for TEP estimation with metabolic stability assessment. TEP and lapatinib (LTP) used as internal standards (ISs) were separated on a reversed-phase C18 column using the isocratic mobile phase. Protein precipitation steps were used to extract TEP from the human liver microsome (HLM) matrix. An electrospray ionization multi-reaction monitoring (MRM) acquisition was conducted at m/z 493→112 for TEP, at m/z 581→350, and 581→365 for the IS. Calibration was in the range of 5 to 500 ng/mL (R2 = 0.999). The limit of detection (LOD) was 0.4759 ng/mL, whereas the limit of quantification (LOQ) was 1.4421 ng/mL. The reproducibility of the developed analytical method (inter- and intra-day precision and accuracy) was within 4.39%. The metabolic stability of TEP in HLM was successfully assessed using the LC-MS/MS method. The metabolic stability assessment of TEP showed intermediate Clint (35.79 mL/min/kg) and a moderate in vitro t1/2 (22.65 min), proposing the good bioavailability and moderate extraction ratio of TEP. The in silico results revealed that the N-methyl piperidine group is the main reason of TEP metabolic lability. The in silico Star Drop software program could be used in an effective protocol to confirm and propose the practical in vitro metabolic experiments to spare resources and time, especially during the first stages for designing new drugs. The established analytical method is considered the first LC-MS/MS method for TEP estimation in the HLM matrix with its application to metabolic stability assessment.
1. Introduction
Cancer has a high mortality rate, accounting for approximately one-fourth of deaths internationally [1]. Lung cancer is considered one of the greatest widespread malignancies, as two million patients are diagnosed with it each year globally, causing 20% of all cancer deaths [2]. Among lung cancer cases, 90% are non-small cell lung carcinomas (NSCLC) that have numerous subtypes initiated by a range of activated oncogenes [3,4,5]. Although progress in developing a new drug series for cancer treatment has been slow, current molecular targeting strategies used in tumor suppressor genes and modulating oncogenes contributed to the improved prognosis of patients [6]. The treatment of NSCLC patients is significantly improved when such personalized targeted therapies are utilized [7,8]. For example, alterations in the mesenchymal–epithelial transition (MET) signaling pathway happen in 3–5% of NSCLC cases and are related to poor clinical prognosis and increased tumor aggressiveness [9,10]. Tepotinib (MSC2156119; TEP, see Figure 1) is a tyrosine kinase inhibitor that targets such pathways, and the FDA approved a breakthrough therapy designation to this drug for patients with NSCLC [11]. Previously published clinical evidence with TEP showed promising results in metastatic NSCLC [12] as one of two MET inhibitors (capmatinib and TEP) approved by the FDA for the treatment of NSCLC [13].
Figure 1.
Chemical structures of tepotinib and lapatinib (IS).
The metabolic stability of a drug or a chemical is its vulnerability to metabolism, and it is described as a clearance intrinsic [Clint] and an in vitro half-life [t1/2]. Half-life [t1/2] is known as the time needed for eliminating 50% of the parent drug. Intrinsic clearance [Clint] is the liver’s capacity to breakdown the drug in the blood through metabolic reactions. The two parameters were determined using the “in vitro half-life” methodology based on the “well-stirred” model [14,15] as it is the most frequently applied in in vitro drug metabolism expectation, the resulting factors can be utilized for extrapolation to numerous in vivo physiological parameters, involving the potential of accumulation and toxicity [16,17].
TEP treatment terminations were reported due to several adverse events, particularly peripheral edema, nausea, diarrhea, vomiting, fatigue and increased blood creatinine [18]. Consequently, metabolic stability (in silico and practical) experiments are required. Studying the metabolic stability of a TEP is a significant issue in developing drugs with improved metabolic stability profiles in the process of drug discovery [18]. It will be proposed that rapidly metabolized drugs cause a reduction in in vivo bioavailability, resulting in a smaller action duration [19]. TEP is mostly metabolized in the liver (by CYP3A4 and CYP2C8), with common drug–drug interactions with co-administered CYP3A modulators. Consequently, the dose is readjusted when the drug is taken with CYP3A inhibitors [20]. TEP has a rapid clearance rate from the human body if compared to earlier tested TKIs [21,22], with one of the contributing factors being its fast metabolism [23]. So as to determine TEP kinetics in vitro, this study is directed at establishing and validating an LC-MS/MS analytical method. A protein precipitation technique using ACN was used for TEP extraction from the HLM metabolic incubation matrix. All analytical parameters, for example recovery, calibration, precision and accuracy were estimated following the stated FDA guidelines. Finally, the established method was used for determination of TEP in the HLM matrix to calculate the in vitro metabolic stability that was guided by the results in the in silico software program (P450 model of StarDrop software; Cambridge, MA, USA).
2. Experimental
2.1. Materials
Pooled HLM matrix (M0567) from human livers (human male doners of different health states and mixed ages), formic acid (HCOOH), ammonium formate (NH4COOH) and acetonitrile (ACN) were purchased from Sigma-Aldrich (St. Louis, MO, USA). The HLM matrix was kept at −70 °C; the total protein content of the HLM pool was 20 mg/mL in 250 mM sucrose. Tepotinib (purity: 99.87%) and lapatinib (purity: 99.83%) were procured from MedChemExpress (Princeton, NJ, USA). Milli-Q plus water filtration system (Millipore company, Burlington, MA, USA) was utilized to produce water for HPLC.
2.2. In Silico TEP Metabolic Vulnerability Prediction
Identification of the TEP metabolism lability was performed using P450 model (StarDrop software) from the Optibrium Ltd. (Cambridge, MA, USA). The composite site lability (CSL) value in the metabolic landscape was utilized as an indication of TEP’s metabolic stability [24,25,26]. To estimate the susceptibility of TEP to metabolism, the site labilities of individual atoms can be collected to determine the CSL generating the overall metabolic lability of TEP. This is computed from the combined determined rates of metabolism for all sites on the molecule:
where kwater is the rate of water formation via the decoupling pathway. The value of kwater has been estimated experimentally, utilizing an intrinsic isotope effect method [27]. ktotal (ktotal = ∑all sites) is the sum of the predicted proportion of metabolism at all active sites.
CSL is estimated as a crucial feature in proposing the rate of metabolism for TEP before performing the in vitro metabolic experiments to verify the value of the study. The TEP (CN1CCC(COC2=CN=C(N=C2)C2=CC(CN3N=C(C=CC3=O)C3=CC(=CC=C3)C#N)=CC=C2)CC1) SMILES format of TEP was uploaded to the StarDrop software program for CSL prediction.
2.3. LC-MS/MS Method
The analytical parameters of the LC-MS/MS method were attuned to obtain a respectable separation of TEP and LTP (internal standard, IS) (Table 1). Rapid resolution liquid chromatorgraphy (RRLC) with Agilent 1200 instrument was performed using an eclipse plus C18 (reversed-phase) column and isocratic mobile phase (70% acetonitrile and 30% 10 mM NH4COOH in HPLC grade water, pH 3.5) at a flow rate of 0.15 mL/min. A triple quadrupole mass analyzer (Agilent 6410 QqQ) with an electrospray (ESI) ion source was used for mass analysis (ion detection and measurement) in positive ion mode. Nitrogen gas (11 L/min) was used for spray drying in the ESI source and for collision dissociation under pressure of 60 psi. A multiple-injector program was used for optimizing MS features to obtain the highest intensity of ion peaks. Capillary voltage of 4000 V and ESI temperature (T) of 350 °C were utilized. The Mass Hunter software program was used for LC-MS/MS system control, data analysis and data acquisition. Ion peak intensity was measured using MRM mode with mass transitions at m/z 493→112 for TEP (parent to fragment ions) and m/z 581→350 and 581→365 for LTP (SI), as presented in Figure 2. The collision energies and fragmentor voltages were 18 eV and 140 V for TEP and 32 eV and 145 V for LTP, respectively.

Table 1.
Analytical features of liquid chromatography and mass detection.
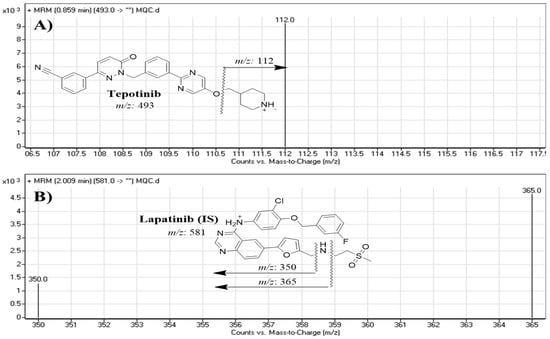
Figure 2.
MRM mass transitions of (A) tepotinib (TEP) and (B) lapatinib (IS).
2.4. TEP Stock Solutions
TEP and LTP showed a good solubility in the organic solvent (DMSO) at 11.11 mg/mL (22.56 mM; ultrasonic and warming and heat to 60 °C and 125 mg/mL), (215.12 mM; Need ultrasonic), respectively. Stock solutions of TEP and LTP (2 mg/mL) were made in dimethyl sulfoxide (DMSO). Three working solutions of TEP and LTP (WK1, 200 µg/mL, WK2, 20 µg/mL and WK3, 2 µg/mL) were made by sequential dilution in a mixture of DMSO and the mobile phase.
2.5. TEP Calibration Standards
DMSO quenched the in vitro metabolic enzymatic pathways even at a 0.2% concentration [28]. DMSO was utilized, as TEP and LTP are solvable in DMSO and for the deactivation of HLMs purposed at 2% conc with slight heating (at 50 °C for 5 min.) [29,30]. The HLM matrix was made by diluting 1 mg protein/1 mL deactivated HLMs (30 µL) to 1 mL with 0.1 M sodium phosphate buffer (pH 7.4) that contains 1 mM NADPH. Nine calibration levels, 5, 10, 30, 50, 100, 200, 300, 400 and 500 ng/mL, were utilized for generating the calibration line. The solutions were prepared from WK3 of TEP in 30 µL HLM matrix for each point. Four quality control (QCs) levels, lower limit of quantification (LLOQ), low (LQC), medium (MQC) and high (HQC) quality control levels, were chosen at (5, 15, 150, and 400 ng/mL, respectively). Fifty µL of internal standard WK3 (LTP) was then added to each concentration. The protein precipitation using organic solvent (ACN) was used for extraction of TEP and LTP [31,32,33]. Two ml of ACN was added to 1 mL of each calibration level and QC; they were vortex-mixed for 1 min and centrifuged at 14,000 rpm (at 4 °C for 12 min); then, the supernatant was filtered (0.22 µm syringe filter) into 1.5 mL HPLC vials, and 1 µL was injected into the LC-MS/MS instrument. A calibration plot was established by plotting the concentration of TEP (x-axis) against the peak area ratio of TEP to LTP (y-axis). The analytical method linearity was proven from the estimated linear regression parameters.
2.6. Method Validation
The validation of the established methodology was verified by calculating various analytical parameters. The details of the parameters were the same as those published in our previous work [34,35].
2.7. TEP Metabolic Stability
TEP metabolic stability was calculated by estimating the TEP remaining concentration after HLM pool incubation. One µM TEP was incubated with 30 µL HLM pool (containing 1 mg protein of microsomes) in phosphate buffer (100 mM) pH 7.4 (1 mL) and 3.3 mM magnesium chloride (MgCl2). Negative controls (absence of TEP or NADPH) were used. The incubation was performed at 37 °C for 10 min; then, the metabolic pathways were started by adding NADPH (1 mM). Fifty µL of internal standard WK3 (LTP) was added to the incubation mixture just before the metabolic pathway termination so as to avoid the metabolism effect on the IS concentration. The reaction was stopped at specific time points, 0, 0.5, 2.5, 7.5, 15, 30 and 50 min, by adding 2 mL acetonitrile. The TEP metabolic stability curve was developed from the experimental measurements using the established LC-MS/MS method. Negative controls were repeated (absence of NADPH or HLM) to confirm the data of the metabolic study. The % of the remaining TEP was plotted against incubation time. The linear portion of the plot was utilized to calculate the natural logarithm (ln) of % the remaining TEP against time (slope), which translates to the rate constant of TEP disappearance. This was used to determine in vitro t1/2, following the equation
Then, the Clint (mL/min/kg) of TEP was estimated [36] using the following equation
where a value of 45 mg of microsomal protein (mg HLMs) was considered in gram of liver tissue (g liver), and 26 g for liver tissue was considered in Kg of the body weight (Kg b.w.) [37,38].
3. Results and Discussions
3.1. In Silico TEP Metabolic Lability
The lability of each site on the TEP structure with respect to the activity of CYP3A4, which plays an important role in the TEP metabolism [20], is indicated by the metabolic landscape, which also allows for the prediction of the TEP metabolites to be confirmed experimentally. The obtained results indicated that the N-methyl piperidine (C1, C3, and C7) is the main reason of TEP metabolic instability, as shown by the CSL presented in Figure 3 (the value of 0.9983, revealing high lability to metabolism), which is in line with the experimental work (see below).
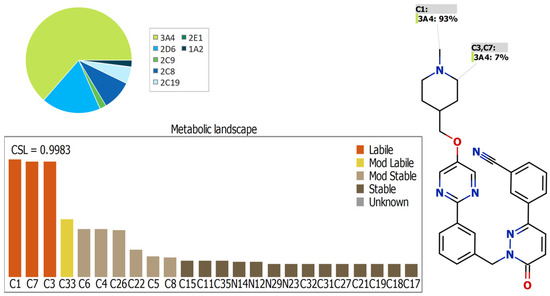
Figure 3.
Predicted metabolic lability of TEP predicted by StarDrop software package (P450 model).
3.2. Quantification of TEP with the Developed LC-MS/MS Method
LTP was selected as the IS for quantifying TEP in the HLM matrix in the in vitro metabolic incubation experiments due to the following reasons. First, TEP and LTP were extracted from the HLM matrix using the same method of extraction (protein precipitation), with a high yield for TEP (99.43 ± 2.21%) and LTP (96.9 ± 1.27%). Second, the chromatographic peaks of TEP (0.86 min) and LTP (2.0 min) were well-separated in 3 min., which resulted in a quickly established LC-MS/MS method, saving time and using less can, which resulted in a green chemistry method. Third, TEP and LTP are not administered together to the same patient. Therefore, the current LC-MS/MS could be used for the pharmacokinetic studies and therapeutic drug monitoring of TEP.
The chromatographic features (mobile phase, pH and nature of the stationary phase) that can affect the separation process were adjusted. The mobile phase was optimized to pH 3.5; increasing the pH of the mobile phase led to increased elution time and chromatographic peak dissymmetry (tailing). The organic/aqueous solvent ratio was fixed at 70: 30%; increasing the organic solvent led to overlapped peaks with low resolution, while a reduction in acetonitrile resulted in increased elution time with good resolution. Hydrophilic interaction liquid chromatography (HILIC) columns were tested as a stationary phase, which resulted in poor retention and low-quality separation (data not shown). Good separation was achieved using a C18 column. Under the final conditions, the elution times for TEP and LTP were 0.86 min and 2.0 min, respectively, with respectable chromatographic peak separation (Figure 4). The LC-MS/MS method run time was 3 min. No carryover of blank HLM were observed.
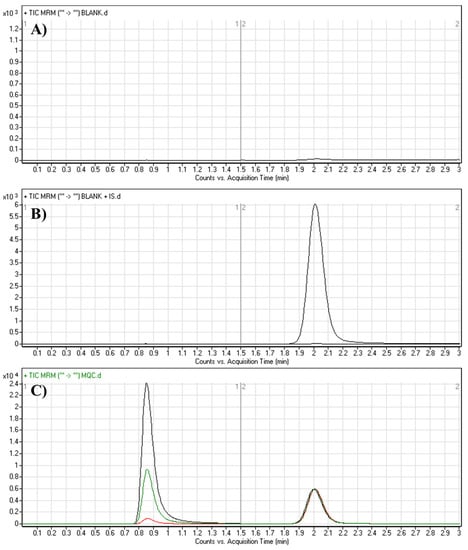
Figure 4.
MRM chromatograms of (A) blank HLM matrix, (B) blank HLM matrix with LTP and (C) QCs of TEP: LQC (15; red color), MQC (150; green color) and HQC (400; black color) revealing TEP peak (0.86 min) and LTP peak (2.0 min).
3.3. Validation Parameters
3.3.1. Specificity
The chromatograms in Figure 4 show respectable separation of TEP and LTP without interference from the blank HLM matrix constituents at the specific elution times of TEP and LTP. No carryover influence appeared in the resulting MRM chromatograms. The linearity range of the proposed method was 5–500 ng/mL with a correlation coefficient of R2 = 0.9999. The linear calibration equation was y = 0.5635714x + 0.4066 (Table 2). The LLQ and LOD peaks exhibited favorable signal-to-noise ratios (S/N) and respectable peak symmetries. The slope of the calibration graph was 0.5635714, whereas its relative error was 0.002105. The intercept of the line was 0.4066 and its relative error was 0.1123472. The LOD was 0.4759 ng/mL and the LOQ was 1.4421 ng/mL (Table 2). The recovery of TEP based on the calibration and QC concentrations in the HLM matrix confirmed the success of the proposed LC-MS/MS analytical method.
Table 2.
Analytical characteristics of the proposed LC-MS/MS analytical method (average data for six replicates).
3.3.2. Precision and Accuracy
The intra- and inter-day precisions of the adopted LC-MS/MS were 4.19% to 1.05% and 4.39% to 1.24%, respectively (Table 3). Intra- and inter-day accuracy and precision were in the range recommended by the FDA guidelines [32]. The relative error for spiked HLM (1.84% to −0.28) with a relative standard deviation of range 4.19% to 0.72% is presented in Table 4. LTP quality control samples show relative error range from 1.84% to −0.55%, whereas the RSD % was in the range from 4.19% to 1.05% (Table 4).
Table 3.
Intra-day (data for twelve replicates on one day) and inter-day (data for six replicates on three days) precision and accuracy of the proposed LC-MS/MS analytical method.
Table 4.
Back-calculated calibration levels of TEP (data for six replicates).
3.3.3. Extraction Recovery and Matrix Effects
The extraction recovery of TEP in spiked HLM matrix was 100.58 ± 0.97%, and the relative standard deviation was (RSD) < 4.18%. The results are displayed in Table 5. The matrix effect (ME) of the developed method for TEP or LTP was estimated by preparing two sets of HLM batches that were analyzed and injected into the LC-MS/MS system, Set 1 and 2, which were spiked with the LOQ concentration of TEP in addition to 100 ng/mL of IS (LTP). The matrix effects for TEP and LTP were calculated according to the following equations:
Table 5.
TEP extraction recovery (data for six replicates).
The HLM matrix containing TEP and LTP exhibited matrix effects of 101.46 ± 1.2% and 98.05 ± 2.1%, respectively. The IS normalized matrix effect was computed using the next equation:
The IS normalized ME was 1.04, well within the satisfactory range [39]. The normalized factor indicates that HLM has no clear influence on the ionization of either TEP or LTP.
3.4. Metabolic Stability
One µM TEP was metabolically incubated with HLM (1 mg/mL); the concentration was less than the Michaelis–Menten constant to achieve the linearity between the time of the metabolic incubation and the metabolic rate. The microsomal protein concentration (1 mg/mL) was used to ensure minimal protein binding. The concentration of TEP in the incubation samples was recorded using the peak area ratio based on the pre-constructed calibration curve against incubation time. The TEP metabolic stability curve (Figure 5) was established by recording the remaining concentration of TEP (percentage compared to zero time) against the time of metabolic incubation. From this curve, time points in the linear range of 0–15 min were selected to plot the natural logarithm (ln) of the percentage of the remaining TEP against time. The results (Table 6) exhibit that the linear regression equation of the straight line portion of the curve was y = −0.0306x + 4.5813, with R2 = 0.9914, which can be utilized to estimate the in vitro t1/2, utilizing the equation:
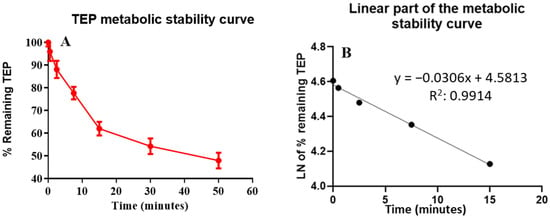
Figure 5.
(A) The curve of TEP metabolic stability and (B) the linear portion of TEP metabolic stability curve.
Table 6.
Parameters of TEP metabolic stability (data for three replicates).
The slope was 0.0306.
The clearance intrinsic to TEP was computed using the in vitro t1/2 method [19], so the Clint of TEP was 35.79 mL/min/kg [38]:
In vitro t1/2 and Clint were 22.65 min and 35.79 mL/min/kg, respectively. Using the scoring published by McNaney et al. [36], it is proposed that TEP has an intermediate clearance ratio character. By using other in silico software (the simulation and Cloe PK software programs; Framingham, MA, USA), these results could also be used to predict the in vivo TEP pharmacokinetics [40,41].
4. Conclusions
The in silico metabolic experiment was performed using the P450 metabolism model, which was expressed in the form of CSL (09983), which revealed the high lability of TEP to metabolism. A reliable LC-MS/MS method was established for the TEP estimation. The method was based on liquid chromatography separation with an isocratic mobile phase and a reversed C18 stationary phase in conjunction with mass detection using an MRM with an ESI source. Protein precipitation was applied for the extraction of TEP from the HLM matrix. The established LC-MS/MS method was adjusted, validated using the FDA guidelines and applied in a practical metabolic stability evaluation of TEP in an HLM matrix. The reproducibility of the LC-MS/MS method (inter- and intra-day accuracies and precisions) was within 4.39%, and linearity was established over a wide range (5–500 ng/mL). The metabolic stability assessment of TEP showed intermediate Clint (35.79 mL/min/kg) and in vitro t1/2 (22.65 min), indicating the moderate clearance character of TEP and the predicted optimum in vivo bioavailability. The metabolic experimental data of TEP agreed with the outcomes of the in silico software that revealed the value of in silico metabolic experiments before performing the practical work so as to save effort and time. The established LC-MS/MS method could be used for therapeutic drug monitoring (TDM) or pharmacokinetic studies for TEP after reoptimizing the extraction procedure, utilizing the same optimized chromatographic features.
Author Contributions
M.W.A. and G.A.E.M. conceived and designed the research. M.W.A. and H.A. conducted the experimental work and wrote the main manuscript. H.A. provided new reagents and analytical tools. M.W.A. revised the first draft of the manuscript and helped in preparing figures. A.A.K. helped in designing the methodology and software application. All authors reviewed, read and approved the manuscript. All experimental data were generated in house and no paper mill was used. All authors have read and agreed to the published version of the manuscript.
Funding
The authors extend their appreciation to the Deputyship for Research & Innovation, Ministry of Education in Saudi Arabia for funding this research work through the project no. (IFKSUOR3-252-1).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
The use of HLMs that were purchased from Sigma company releases it from the necessity of ethical approval.
Data Availability Statement
All data are available within the manuscript.
Acknowledgments
The authors extend their appreciation to the Deputyship for Research and Innovation, “Ministry of Education” in Saudi Arabia for funding this research (IFKSUOR3-252-1).
Conflicts of Interest
The authors declare no conflict of interest for the current work.
References
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Murray, T.; Thun, M.J. Cancer statistics, 2008. CA A Cancer J. Clin. 2008, 58, 71–96. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Siegel, R.L.; Jemal, A. Lung cancer statistics. In Lung Cancer and Personalized Medicine; Springer: Berlin/Heidelberg, Germany, 2016; pp. 1–19. [Google Scholar]
- Chmielecki, J.; Foo, J.; Oxnard, G.R.; Hutchinson, K.; Ohashi, K.; Somwar, R.; Wang, L.; Amato, K.R.; Arcila, M.; Sos, M.L. Optimization of dosing for EGFR-mutant non–small cell lung cancer with evolutionary cancer modeling. Sci. Transl. Med. 2011, 3, 90ra59. [Google Scholar] [CrossRef]
- Katayama, R.; Khan, T.M.; Benes, C.; Lifshits, E.; Ebi, H.; Rivera, V.M.; Shakespeare, W.C.; Iafrate, A.J.; Engelman, J.A.; Shaw, A.T. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc. Natl. Acad. Sci. USA 2011, 108, 7535–7540. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Yu, X.; Wu, S.; Wu, X.; Wang, Q.; Cheng, W.; Hu, W.; Kang, C.; Yang, W.; Li, Y.; et al. Instability Mechanism of Osimertinib in Plasma and a Solving Strategy in the Pharmacokinetics Study. Front. Pharmacol. 2022, 13, 928983. [Google Scholar] [CrossRef]
- Shenouda, S.K.; Alahari, S.K. MicroRNA function in cancer: Oncogene or a tumor suppressor? Cancer Metastasis Rev. 2009, 28, 369. [Google Scholar] [CrossRef]
- Sechler, M.; Cizmic, A.D.; Avasarala, S.; Van Scoyk, M.; Brzezinski, C.; Kelley, N.; Bikkavilli, R.K.; Winn, R.A. Non-small-cell lung cancer: Molecular targeted therapy and personalized medicine–drug resistance, mechanisms, and strategies. Pharm. Pers. Med. 2013, 6, 25. [Google Scholar]
- Cheng, L.; Alexander, R.E.; MacLennan, G.T.; Cummings, O.W.; Montironi, R.; Lopez-Beltran, A.; Cramer, H.M.; Davidson, D.D.; Zhang, S. Molecular pathology of lung cancer: Key to personalized medicine. Mod. Pathol. 2012, 25, 347–369. [Google Scholar] [CrossRef]
- Reungwetwattana, T.; Liang, Y.; Zhu, V.; Ou, S.-H.I. The race to target MET exon 14 skipping alterations in non-small cell lung cancer: The why, the how, the who, the unknown, and the inevitable. Lung Cancer 2017, 103, 27–37. [Google Scholar] [CrossRef]
- Kunii, K.; Davis, L.; Gorenstein, J.; Hatch, H.; Yashiro, M.; Di Bacco, A.; Elbi, C.; Lutterbach, B. FGFR2-amplified gastric cancer cell lines require FGFR2 and Erbb3 signaling for growth and survival. Cancer Res. 2008, 68, 2340–2348. [Google Scholar] [CrossRef]
- Abdelhameed, A.S.; Attwa, M.W.; Kadi, A.A. Identification of Iminium Intermediates Generation in the Metabolism of Tepotinib Using LC-MS/MS: In Silico and Practical Approaches to Bioactivation Pathway Elucidation. Molecules 2020, 25, 5004. [Google Scholar] [CrossRef] [PubMed]
- Paik, P.K.; Veillon, R.; Cortot, A.B.; Felip, E.; Sakai, H.; Mazieres, J.; Griesinger, F.; Horn, L.; Senellart, H.; Meerbeeck, J.P.V.; et al. Phase II study of tepotinib in NSCLC patients with METex14 mutations. J. Clin. Oncol. 2019, 37, 9005. [Google Scholar] [CrossRef]
- Ruiz-Cordero, R.; Devine, W.P. Targeted Therapy and Checkpoint Immunotherapy in Lung Cancer. Surg. Pathol. Clin. 2020, 13, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Houston, J.B. Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem. Pharm. 1994, 47, 1469–1479. [Google Scholar] [CrossRef] [PubMed]
- Obach, R.S.; Baxter, J.G.; Liston, T.E.; Silber, B.M.; Jones, B.C.; MacIntyre, F.; Rance, D.J.; Wastall, P. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J. Pharmacol. Exp. Ther. 1997, 283, 46–58. [Google Scholar]
- Nichols, J.W.; Schultz, I.R.; Fitzsimmons, P.N. In vitro–in vivo extrapolation of quantitative hepatic biotransformation data for fish: I. A review of methods, and strategies for incorporating intrinsic clearance estimates into chemical kinetic models. Aquat. Toxicol. 2006, 78, 74–90. [Google Scholar] [CrossRef]
- Pelkonen, O.; Turpeinen, M. In vitro–in vivo extrapolation of hepatic clearance: Biological tools, scaling factors, model assumptions and correct concentrations. Xenobiotica 2007, 37, 1066–1089. [Google Scholar] [CrossRef]
- Malapelle, U.; Muscarella, L.A.; Pisapia, P.; Rossi, A. Targeting emerging molecular alterations in the treatment of non-small cell lung cancer: Current challenges and the way forward. Expert Opin. Investig. Drugs 2020, 29, 363–372. [Google Scholar] [CrossRef]
- Baranczewski, P.; Stańczak, A.; Sundberg, K.; Svensson, R.; Wallin, A.; Jansson, J.; Garberg, P.; Postlind, H. Introduction to in vitro estimation of metabolic stability and drug interactions of new chemical entities in drug discovery and development. Pharmacol. Rep. 2006, 58, 453–472. [Google Scholar]
- Li, F.; MacKenzie, K.R.; Nyshadham, P.; Kerlec, K.A.; Matzuk, M.M. Identifying Metabolic Pathways of c-MET Tyrosine Kinase Inhibitor Tepotinib in Human and Mouse Liver Microsomes. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Attwa, M.W.; Kadi, A.A.; Abdelhameed, A.S. Detection and characterization of olmutinib reactive metabolites by LC–MS/MS: Elucidation of bioactivation pathways. J. Sep. Sci. 2020, 43, 708–718. [Google Scholar] [CrossRef]
- Attwa, M.W.; Kadi, A.A.; Abdelhameed, A.S.; Alhazmi, H.A. Metabolic Stability Assessment of New PARP Inhibitor Talazoparib Using Validated LC-MS/MS Methodology: In silico Metabolic Vulnerability and Toxicity Studies. Drug Des. Dev. Ther. 2020, 14, 783–793. [Google Scholar] [CrossRef]
- Vansteenkiste, J.F.; Van De Kerkhove, C.; Wauters, E.; Van Mol, P. Capmatinib for the treatment of non-small cell lung cancer. Expert Rev. Anticancer. Ther. 2019, 19, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Alrabiah, H.; Kadi, A.A.; Attwa, M.W.; Abdelhameed, A.S. A simple liquid chromatography-tandem mass spectrometry method to accurately determine the novel third-generation EGFR-TKI naquotinib with its applicability to metabolic stability assessment. RSC Adv. 2019, 9, 4862–4869. [Google Scholar] [CrossRef]
- Kadi, A.A.; Darwish, H.W.; Abuelizz, H.A.; Alsubi, T.A.; Attwa, M.W. Identification of reactive intermediate formation and bioactivation pathways in Abemaciclib metabolism by LC-MS/MS: In vitro metabolic investigation. R. Soc. Open Sci. 2019, 6, 181714. [Google Scholar] [CrossRef]
- Attwa, M.W.; AlRabiah, H.; Alsibaee, A.M.; Abdelhameed, A.S.; Kadi, A.A. An UPLC–ESI–MS/MS Bioanalytical Methodology for the Quantification of Gilteritinib in Human Liver Microsomes: Application to In Vitro and In Silico Metabolic Stability Estimation. Separations 2023, 10, 278. [Google Scholar] [CrossRef]
- Korzekwa, K.R.; Trager, W.F.; Gillette, J.R. Theory for the observed isotope effects from enzymatic systems that form multiple products via branched reaction pathways: Cytochrome P-450. Biochemistry 1989, 28, 9012–9018. [Google Scholar] [CrossRef] [PubMed]
- Busby, W.F.; Ackermann, J.M.; Crespi, C.L. Effect of methanol, ethanol, dimethyl sulfoxide, and acetonitrile on in vitro activities of cDNA-expressed human cytochromes P-450. Drug Metab. Dispos. 1999, 27, 246–249. [Google Scholar]
- Taniguchi-Takizawa, T.; Shimizu, M.; Kume, T.; Yamazaki, H. Benzydamine N-oxygenation as an index for flavin-containing monooxygenase activity and benzydamine N-demethylation by cytochrome P450 enzymes in liver microsomes from rats, dogs, monkeys, and humans. Drug Metab. Pharmacokinet. 2015, 30, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Fouin-Fortunet, H.; Tinel, M.; Descatoire, V.; Letteron, P.; Larrey, D.; Geneve, J.; Pessayre, D. Inactivation of cytochrome P-450 by the drug methoxsalen. J. Pharmacol. Exp. Ther. 1986, 236, 237–247. [Google Scholar]
- Attwa, M.W.; Kadi, A.A.; Darwish, H.W.; Amer, S.M.; Al-Shakliah, N.S. Identification and characterization of in vivo, in vitro and reactive metabolites of vandetanib using LC-ESI-MS/MS. Chem. Cent. J. 2018, 12, 99. [Google Scholar] [CrossRef]
- Alrabiah, H.; Kadi, A.A.; Aljohar, H.I.; Attwa, M.W.; Al-Shakliah, N.S.; Attia, S.M.; Mostafa, G.A.E. A new validated HPLC-MS/MS method for quantification and pharmacokinetic evaluation of dovitinib, a multi-kinase inhibitor, in mouse Plasma. Drug Des. Dev. Ther. 2020, 14, 407–415. [Google Scholar] [CrossRef]
- Attwa, M.W.; Darwish, H.W.; Alhazmi, H.A.; Kadi, A.A. Investigation of metabolic degradation of new ALK inhibitor: Entrectinib by LC-MS/MS. Clin. Chim. Acta 2018, 485, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Alanazi, M.M.; Alkahtani, H.M.; Almehizia, A.A.; Attwa, M.W.; Bakheit, A.H.; Darwish, H.W. Validated liquid chromatography tandem mass spectrometry for simultaneous quantification of foretinib and lapatinib, and application to metabolic stability investigation. RSC Adv. 2019, 9, 19325–19332. [Google Scholar] [CrossRef] [PubMed]
- Attwa, M.; Kadi, A.A.; Darwish, H.W.; Abdelhameed, A.S. Investigation of the metabolic stability of olmutinib by validated LC-MS/MS: Quantification in human plasma. RSC Adv. 2018, 8, 40387–40394. [Google Scholar] [CrossRef] [PubMed]
- McNaney, C.A.; Drexler, D.M.; Hnatyshyn, S.Y.; Zvyaga, T.A.; Knipe, J.O.; Belcastro, J.V.; Sanders, M. An automated liquid chromatography-mass spectrometry process to determine metabolic stability half-life and intrinsic clearance of drug candidates by substrate depletion. Assay Drug Dev. Technol. 2008, 6, 121–129. [Google Scholar] [CrossRef]
- Słoczyńska, K.; Gunia-Krzyżak, A.; Koczurkiewicz, P.; Wójcik-Pszczoła, K.; Żelaszczyk, D.; Popiół, J.; Pękala, E. Metabolic stability and its role in the discovery of new chemical entities. Acta Pharm. 2019, 69, 345–361. [Google Scholar] [CrossRef]
- Manzo, A.; Montanino, A.; Costanzo, R.; Sandomenico, C.; Palumbo, G.; Schettino, C.; Daniele, G.; Morabito, A.; Perrone, F.; Piccirillo, M.C. Chapter 33-EGFR Mutations: Best Results from Second- and Third-Generation Tyrosine Kinase Inhibitors. In Oncogenomics; Dammacco, F., Silvestris, F., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 477–486. [Google Scholar] [CrossRef]
- Kadian, N.; Raju, K.S.R.; Rashid, M.; Malik, M.Y.; Taneja, I.; Wahajuddin, M. Comparative assessment of bioanalytical method validation guidelines for pharmaceutical industry. J. Pharm. Biomed. Anal. 2016, 126, 83–97. [Google Scholar] [CrossRef]
- Leahy, D.E. Integrating in vitro ADMET data through generic physiologically based pharmacokinetic models. Expert Opin. Drug Metab. Toxicol. 2006, 2, 619–628. [Google Scholar] [CrossRef]
- Attwa, M.W.; Abdelhameed, A.S.; Alsibaee, A.M.; Kadi, A.A. A Rapid and Sensitive UPLC-MS/MS Method for Quantifying Capmatinib in Human Liver Microsomes: Evaluation of Metabolic Stability by In Silico and In Vitro Analysis. Separations 2023, 10, 247. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).