

Online Membrane Sampling for the Mass Spectrometric Analysis of Oil Sands Process Affected Water-Derived Naphthenic Acids in Real-World Samples

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples and Standards
2.2. Condensed Phase Membrane Introduction Mass Spectrometry
2.3. Peak Picking for Unit Mass Resolution Quantitation Method
2.4. SPE-Orbitrap
3. Results and Discussion
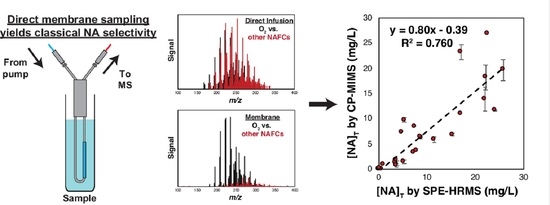
3.1. Evaluation of PDMS Permselectivity
3.2. Adaptation to Unit Mass Resolution Instrumentation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goverment of Alberta. Oil Sands Facts and Statistics; Goverment of Alberta: Edmonton, AB, Canada, 2021. [Google Scholar]
- Alberta Energy Regulator. State of Fluid Tailings Management for Mineable Oil Sands; Alberta Energy Regulator: Calgary, AB, Canada, 2020. [Google Scholar]
- Allen, E.W. Process water treatment in Canada’s oil sands industry: I. Target pollutants and treatment objectives. J. Environ. Eng. Sci. 2008, 7, 123–138. [Google Scholar] [CrossRef]
- Headley, J.V.; Peru, K.M.; Barrow, M.P. Advances in mass spectrometric characterization of naphthenic acids fraction compounds in oil sands environmental samples and crude oil--A review. Mass Spectrom. Rev. 2016, 35, 311–328. [Google Scholar] [CrossRef]
- Ripmeester, M.J.; Duford, D.A. Method for routine “naphthenic acids fraction compounds” determination in oil sands process-affected water by liquid-liquid extraction in dichloromethane and Fourier-Transform Infrared Spectroscopy. Chemosphere 2019, 233, 687–696. [Google Scholar] [CrossRef]
- Meshref, M.N.A.; Ibrahim, M.D.; Huang, R.; Yang, L.; How, Z.T.; Klamerth, N.; Chelme-Ayala, P.; Hughes, S.A.; Brown, C.; Mahaffey, A.; et al. Fourier transform infrared spectroscopy as a surrogate tool for the quantification of naphthenic acids in oil sands process water and groundwater. Sci. Total Environ. 2020, 734, 139191. [Google Scholar] [CrossRef]
- Krogh, E.T.; Gill, C.G. Condensed Phase Membrane Introduction Mass Spectrometry–Continuous, Direct and Online Measurements in Complex Samples. In Advances in the Use of Liquid Chromatography Mass Spectrometry (LC-MS)-Instrumentation Developments and Applications; Elsevier: Amsterdam, The Netherlands, 2018; pp. 173–203. [Google Scholar]
- Termopoli, V.; Piergiovanni, M.; Ballabio, D.; Consonni, V.; Cruz Muñoz, E.; Gosetti, F. Condensed Phase Membrane Introduction Mass Spectrometry: A Direct Alternative to Fully Exploit the Mass Spectrometry Potential in Environmental Sample Analysis. Separations 2023, 10, 139. [Google Scholar] [CrossRef]
- Crank, J. The Mathematics of Diffusion; Oxford University Press: London, UK, 1975. [Google Scholar]
- LaPack, M.A.; Tou, J.C.; Enke, C.G. Membrane mass spectrometry for the direct trace analysis of volatile organic compounds in air and water. Anal. Chem. 1990, 62, 1265–1271. [Google Scholar] [CrossRef]
- Vandergrift, G.W.; Monaghan, J.; Krogh, E.T.; Gill, C.G. Direct Analysis of Polyaromatic Hydrocarbons in Soil and Aqueous Samples Using Condensed Phase Membrane Introduction Tandem Mass Spectrometry with Low-Energy Liquid Electron Ionization. Anal. Chem. 2019, 91, 1587–1594. [Google Scholar] [CrossRef] [PubMed]
- Vandergrift, G.W.; Krogh, E.T.; Gill, C.G. Direct, Isomer-Specific Quantitation of Polycyclic Aromatic Hydrocarbons in Soils Using Membrane Introduction Mass Spectrometry and Chemical Ionization. Anal. Chem. 2020, 92, 15480–15488. [Google Scholar] [CrossRef]
- Vandergrift, G.W.; Lattanzio-Battle, W.; Krogh, E.T.; Gill, C.G. Condensed Phase Membrane Introduction Mass Spectrometry with In Situ Liquid Reagent Chemical Ionization in a Liquid Electron Ionization Source (CP-MIMS-LEI/CI). J. Am. Soc. Mass Spectrom. 2020, 31, 908–916. [Google Scholar] [CrossRef]
- Monaghan, J.; Jaeger, A.; Agua, A.R.; Stanton, R.S.; Pirrung, M.; Gill, C.G.; Krogh, E.T. A Direct Mass Spectrometry Method for the Rapid Analysis of Ubiquitous Tire-Derived Toxin N-(1,3-Dimethylbutyl)-N′-phenyl-p-phenylenediamine Quinone (6-PPDQ). ES T Lett. 2021, 8, 1051–1056. [Google Scholar] [CrossRef]
- Duncan, K.D.; Letourneau, D.R.; Vandergrift, G.W.; Jobst, K.; Reiner, E.; Gill, C.G.; Krogh, E.T. A semi-quantitative approach for the rapid screening and mass profiling of naphthenic acids directly in contaminated aqueous samples. J. Mass Spectrom. 2016, 51, 44–52. [Google Scholar] [CrossRef]
- Duncan, K.D.; Richards, L.C.; Monaghan, J.; Simair, M.C.; Ajaero, C.; Peru, K.M.; Friesen, V.; McMartin, D.W.; Headley, J.V.; Gill, C.G.; et al. Direct analysis of naphthenic acids in constructed wetland samples by condensed phase membrane introduction mass spectrometry. Sci. Total Environ. 2020, 716, 137063. [Google Scholar] [CrossRef] [PubMed]
- Monaghan, J.; Richards, L.C.; Vandergrift, G.W.; Hounjet, L.J.; Stoyanov, S.R.; Gill, C.G.; Krogh, E.T. Direct mass spectrometric analysis of naphthenic acids and polycyclic aromatic hydrocarbons in waters impacted by diluted bitumen and conventional crude oil. Sci. Total Environ. 2021, 765, 144206. [Google Scholar] [CrossRef]
- Rowland, S.J.; West, C.E.; Jones, D.; Scarlett, A.G.; Frank, R.A.; Hewitt, L.M. Steroidal aromatic ‘naphthenic acids’ in oil sands process-affected water: Structural comparisons with environmental estrogens. Environ. Sci. Technol. 2011, 45, 9806–9815. [Google Scholar] [CrossRef] [PubMed]
- Marentette, J.R.; Frank, R.A.; Bartlett, A.J.; Gillis, P.L.; Hewitt, L.M.; Peru, K.M.; Headley, J.V.; Brunswick, P.; Shang, D.; Parrott, J.L. Toxicity of naphthenic acid fraction components extracted from fresh and aged oil sands process-affected waters, and commercial naphthenic acid mixtures, to fathead minnow (Pimephales promelas) embryos. Aquat. Toxicol. 2015, 164, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Duncan, K.D.; Hawkes, J.A.; Berg, M.; Clarijs, B.; Gill, C.G.; Bergquist, J.; Lanekoff, I.; Krogh, E.T. Membrane Sampling Separates Naphthenic Acids from Biogenic Dissolved Organic Matter for Direct Analysis by Mass Spectrometry. Environ. Sci. Technol. 2022, 56, 3096–3105. [Google Scholar] [CrossRef] [PubMed]
- Vandergrift, G.W.; Krogh, E.T.; Gill, C.G. Polymer Inclusion Membranes with Condensed Phase Membrane Introduction Mass Spectrometry (CP-MIMS): Improved Analytical Response Time and Sensitivity. Anal. Chem. 2017, 89, 5629–5636. [Google Scholar] [CrossRef]
- Monaghan, J.; Xin, Q.; Aplin, R.; Jaeger, A.; Heshka, N.E.; Hounjet, L.J.; Gill, C.G.; Krogh, E.T. Aqueous Naphthenic Acids and Polycyclic Aromatic Hydrocarbons in a Meso-Scale Spill Tank Affected by Diluted Bitumen Analyzed Directly by Membrane Introduction Mass Spectrometry. J. Hazard. Mater. 2022, 440, 129798. [Google Scholar] [CrossRef]
- Hughes, S.A.; Mahaffey, A.; Shore, B.; Baker, J.; Kilgour, B.; Brown, C.; Peru, K.M.; Headley, J.V.; Bailey, H.C. Using ultrahigh-resolution mass spectrometry and toxicity identification techniques to characterize the toxicity of oil sands process-affected water: The case for classical naphthenic acids. Environ. Toxicol. Chem. 2017, 36, 3148–3157. [Google Scholar] [CrossRef]
- Rogers, V.V.; Liber, K.; MacKinnon, M.D. Isolation and characterization of naphthenic acids from Athabasca oil sands tailings pond water. Chemosphere 2002, 48, 519–527. [Google Scholar] [CrossRef]
- Ajaero, C.; Peru, K.M.; Simair, M.; Friesen, V.; O’Sullivan, G.; Hughes, S.A.; McMartin, D.W.; Headley, J.V. Fate and behavior of oil sands naphthenic acids in a pilot-scale treatment wetland as characterized by negative-ion electrospray ionization Orbitrap mass spectrometry. Sci Total Environ. 2018, 631–632, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Duncan, K.D.; McCauley, E.P.; Krogh, E.T.; Gill, C.G. Characterization of a condensed-phase membrane introduction mass spectrometry (CP-MIMS) interface using a methanol acceptor phase coupled with electrospray ionization for the continuous on-line quantitation of polar, low-volatility analytes at trace levels in complex aqueous samples. Rapid Commun. Mass Spectrom. 2011, 25, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, J.A.; D’Andrilli, J.; Agar, J.N.; Barrow, M.P.; Berg, S.M.; Catalán, N.; Chen, H.; Chu, R.K.; Cole, R.B.; Dittmar, T.; et al. An international laboratory comparison of dissolved organic matter composition by high resolution mass spectrometry: Are we getting the same answer? Limnology and Oceanography: Methods 2020, 18, 235–258. [Google Scholar] [CrossRef]
- Duncan, K.D.; Vandergrift, G.W.; Krogh, E.T.; Gill, C.G. Ionization suppression effects with condensed phase membrane introduction mass spectrometry: Methods to increase the linear dynamic range and sensitivity. J. Mass Spectrom. 2015, 50, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Vander Meulen, I.J.; Schock, D.M.; Parrott, J.L.; Simair, M.C.; Mundy, L.J.; Ajaero, C.; Pauli, B.D.; Peru, K.M.; McMartin, D.W.; Headley, J.V. Transformation of bitumen-derived naphthenic acid fraction compounds across surface waters of wetlands in the Athabasca Oil Sands region. Sci. Total Environ. 2022, 806, 150619. [Google Scholar] [CrossRef]
- Potter, D.W.; Pawliszyn, J. Detection of substituted benzenes in water at the pg/ml level using solid-phase microextraction and gas chromatography-ion trap mass spectrometry. J. Chromatogr. 1992, 625, 247–255. [Google Scholar] [CrossRef]
- Boscaini, E.; Alexander, M.L.; Prazeller, P.; Märk, T.D. Investigation of fundamental physical properties of a polydimethylsiloxane (PDMS) membrane using a proton transfer reaction mass spectrometer (PTRMS). Int. J. Mass Spectrom. 2004, 239, 179–186. [Google Scholar] [CrossRef]
- Peru, K.M.; Thomas, M.J.; Palacio Lozano, D.C.; McMartin, D.W.; Headley, J.V.; Barrow, M.P. Characterization of oil sands naphthenic acids by negative-ion electrospray ionization mass spectrometry: Influence of acidic versus basic transfer solvent. Chemosphere 2019, 222, 1017–1024. [Google Scholar] [CrossRef]
- Letourneau, D.R. Rapid Quantitative and Qualitative Screening of Naphthenic Acids in Contaminated Waters Using Condensed Phase Membrane Introduction Mass Spectrometry. Chemistry. Master’s Thesis, University of Victoria, Victoria, BC, Canada, 2016; pp. 1–136. [Google Scholar]
- Zarkovic, T.M.; Borden, S.A.; Krogh, E.T.; Gill, C.G. A passive membrane system for on-line mass spectrometry reagent addition. Rapid Commun. Mass Spectrom. 2023, 37, e9487. [Google Scholar] [CrossRef]
- Kovalchik, K.A.; MacLennan, M.S.; Peru, K.M.; Headley, J.V.; Chen, D.D.Y. Standard method design considerations for semi-quantification of total naphthenic acids in oil sands process affected water by mass spectrometry: A review. Front. Chem. Sci. Eng. 2017, 11, 497–507. [Google Scholar] [CrossRef]
- Chemspider. Royal Society of Chemistry. 2022. Available online: https://www.chemspider.com/DatasourceDetails.aspx?id=186 (accessed on 1 March 2023).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Extraction Date | Source | Total NAFCs (mg/L) |
|---|---|---|---|
| 2013 | Pre-2013 | -- | 5000 |
| 2015/1 | February 2015 | Shell | 8000 |
| 2015/2 | July 2015 | CNRL | 8050 |
| 2018 1 | February 2018 | Syncrude | 5300 |
| Compound Name | Formula | Log(KOW) 2 | Relative PE | IE 5 | log(K’pdms) | ||
|---|---|---|---|---|---|---|---|
| Within Group 3 | Global 4 | ||||||
| Small | 3-phenylpropanoic acid | C9H10O2 | 2.29 | 1.0 | 1.0 | 1.0 | −1.9 |
| 3-(4-hydroxyphenyl)propanoic acid | C9H10O3 | 1.81 | 0.0034 | 0.0034 | 0.5 | −4.0 | |
| 2-hydroxy-3-phenylpropanoic acid | C9H10O3 | 1.06 | 0.014 | 0.014 | 1.1 | −3.4 | |
| 3-(4-methylthiophenyl)propanoic acid | C10H12SO2 | 2.89 | 2.3 | 2.3 | 0.9 | −1.5 | |
| Medium | 4-phenylbutanoic acid | C10H12O2 | 2.78 | 1.0 | 6.2 | 0.8 | −0.8 |
| 2-hydroxy-4-phenylbutanoic acid | C10H12O3 | 1.30 | 0.02 | 0.1 | 1.2 | −2.8 | |
| 4-(4-hydroxyphenyl)butanoic acid | C10H12O3 | 2.30 | 0.001 | 0.006 | 0.8 | −3.7 | |
| 4-keto-4-phenylbutanoic acid | C10H10O3 | 1.42 | 0.019 | 0.12 | 0.2 | −2.7 | |
| 4-(4-methoxyphenyl)butanoic acid | C11H14O3 | 2.86 | 0.19 | 1.2 | 0.8 | −1.6 | |
| 4-(3,4-dimethoxyphenyl)butanoic acid | C12H16O4 | 2.42 | 0.029 | 0.18 | 0.7 | −2.5 | |
| Large | 6-phenylhexanoic acid | C12H16O2 | 3.76 | 1.0 | 22 | 1.0 | −0.7 |
| 4-pentylcyclohexanecarboxylic acid | C12H22O2 | 4.74 | 4.2 | 93 | 1.0 | 0.20 | |
| 4-heptylbenzoic acid | C14H20O2 | 5.37 | 8.2 | 180 | 0.7 | 0.40 | |
| 1-pyrenebutyric acid | C20H16O2 | 5.72 | 10 | 220 | 0.3 | 0.71 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monaghan, J.; Steenis, D.; Vander Meulen, I.J.; Peru, K.M.; Headley, J.V.; Gill, C.G.; Krogh, E.T. Online Membrane Sampling for the Mass Spectrometric Analysis of Oil Sands Process Affected Water-Derived Naphthenic Acids in Real-World Samples. Separations 2023, 10, 228. https://doi.org/10.3390/separations10040228
Monaghan J, Steenis D, Vander Meulen IJ, Peru KM, Headley JV, Gill CG, Krogh ET. Online Membrane Sampling for the Mass Spectrometric Analysis of Oil Sands Process Affected Water-Derived Naphthenic Acids in Real-World Samples. Separations. 2023; 10(4):228. https://doi.org/10.3390/separations10040228
Chicago/Turabian StyleMonaghan, Joseph, Dylan Steenis, Ian J. Vander Meulen, Kerry M. Peru, John V. Headley, Chris G. Gill, and Erik T. Krogh. 2023. "Online Membrane Sampling for the Mass Spectrometric Analysis of Oil Sands Process Affected Water-Derived Naphthenic Acids in Real-World Samples" Separations 10, no. 4: 228. https://doi.org/10.3390/separations10040228
APA StyleMonaghan, J., Steenis, D., Vander Meulen, I. J., Peru, K. M., Headley, J. V., Gill, C. G., & Krogh, E. T. (2023). Online Membrane Sampling for the Mass Spectrometric Analysis of Oil Sands Process Affected Water-Derived Naphthenic Acids in Real-World Samples. Separations, 10(4), 228. https://doi.org/10.3390/separations10040228