



Multi-Dimensional Liquid Chromatography of Pulse Triacylglycerols with Triple Parallel Mass Spectrometry


Abstract
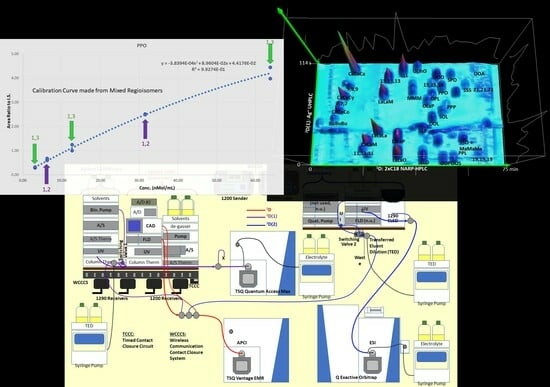
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
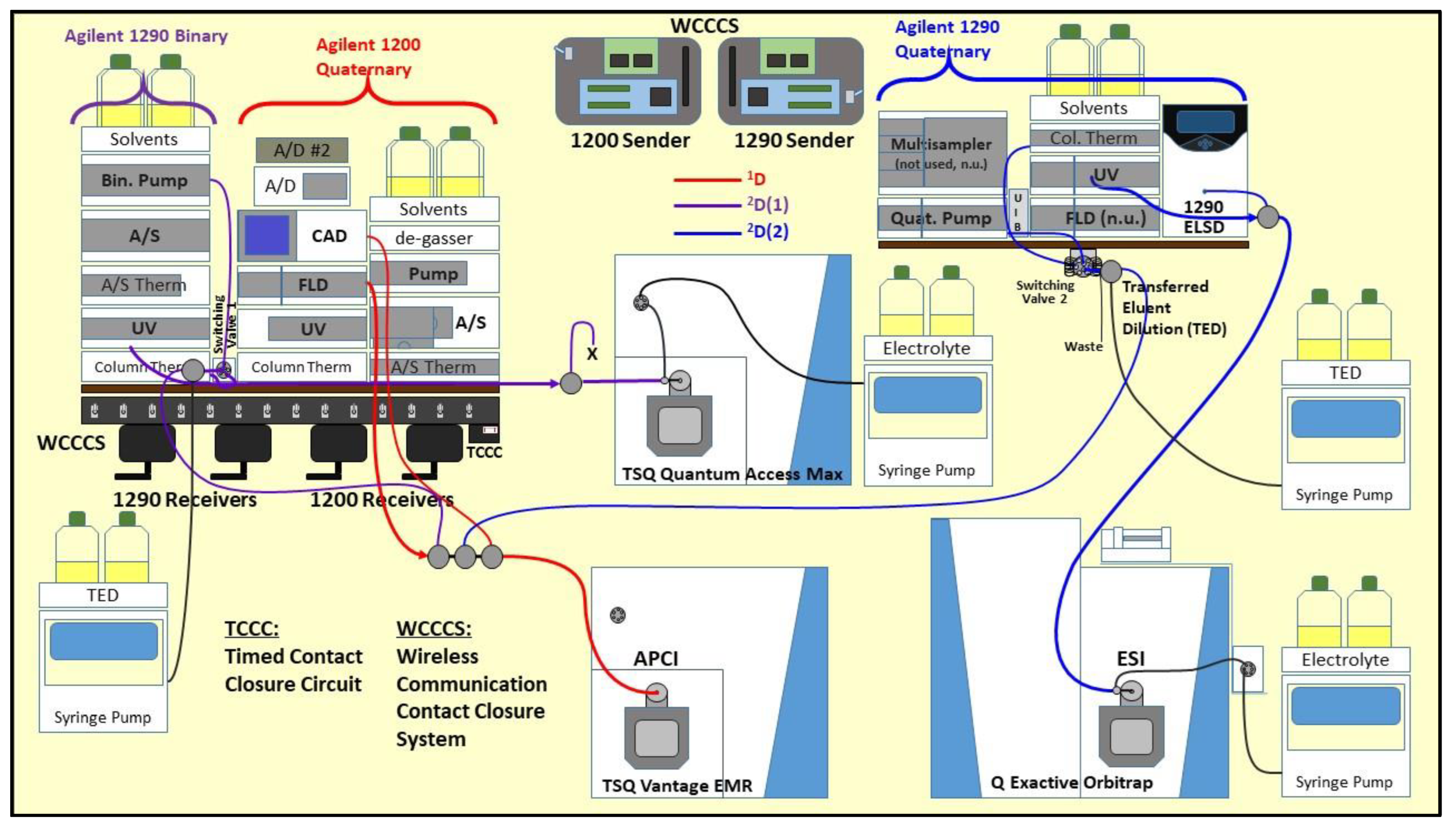
2.2. Liquid Chromatography
2.3. Mass Spectrometry
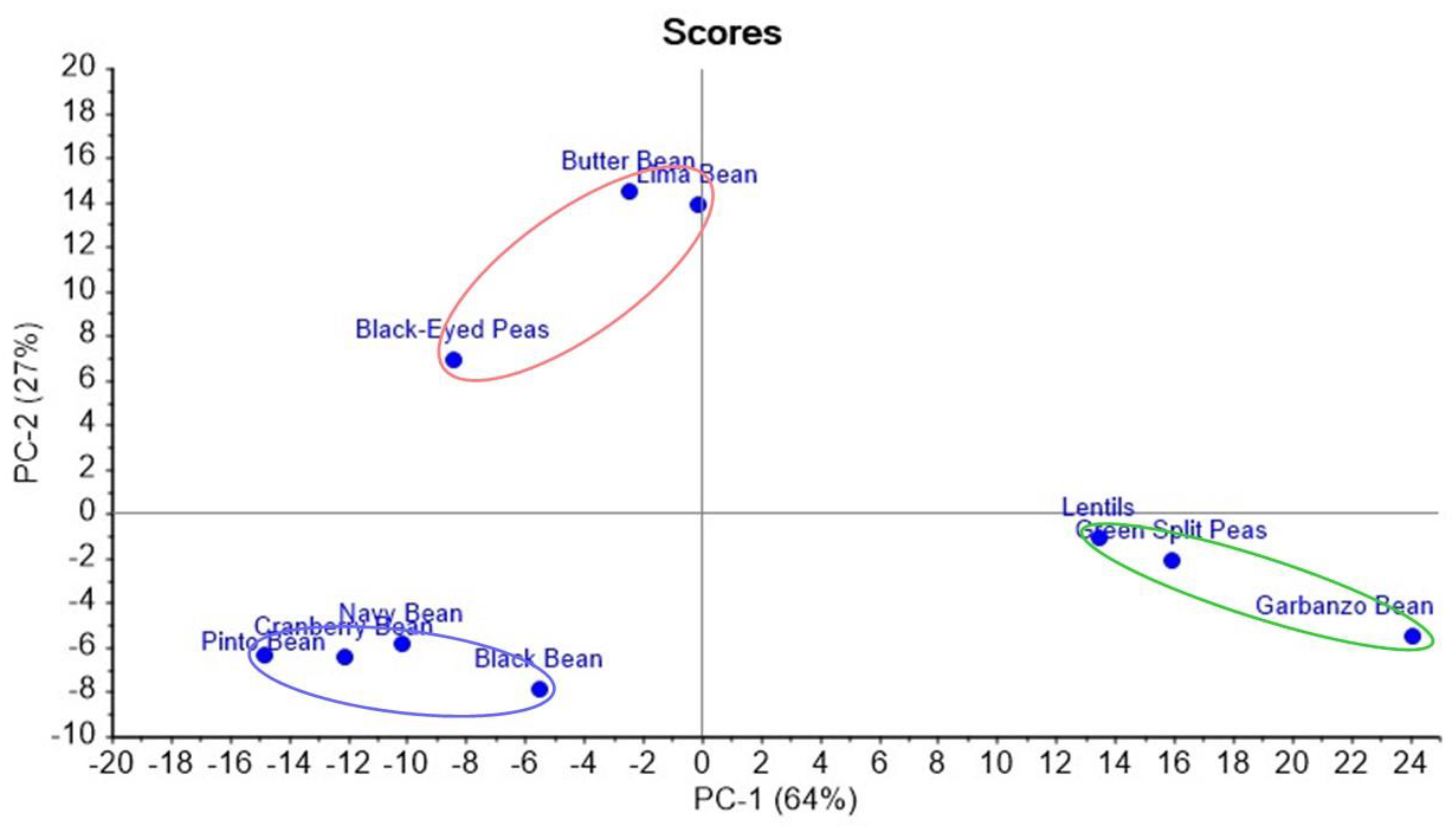
2.4. Calculations, Statistical Analysis
2.5. Abbreviations
3. Results
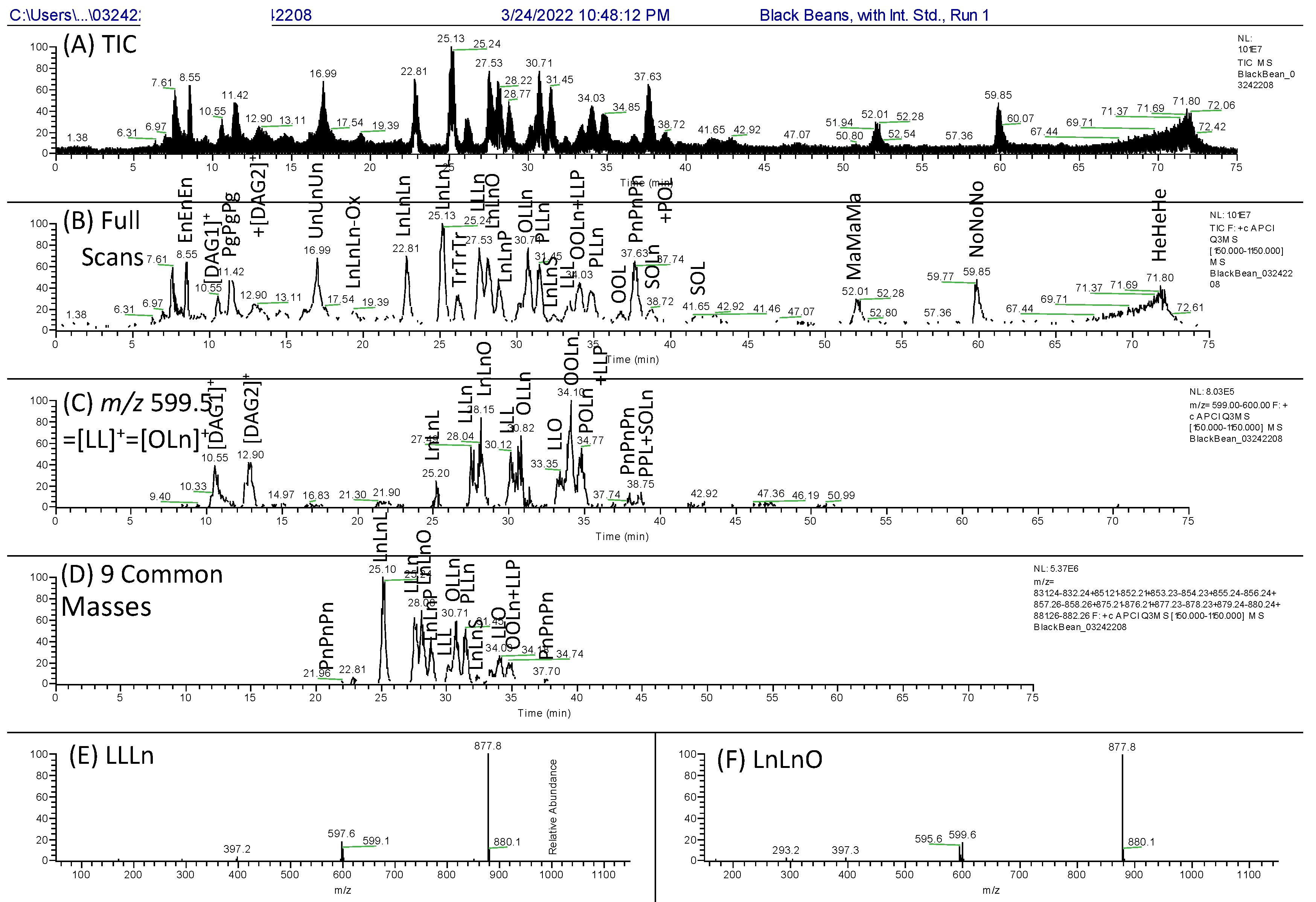
3.1. The First Dimension, 1D: NARP-HPLC
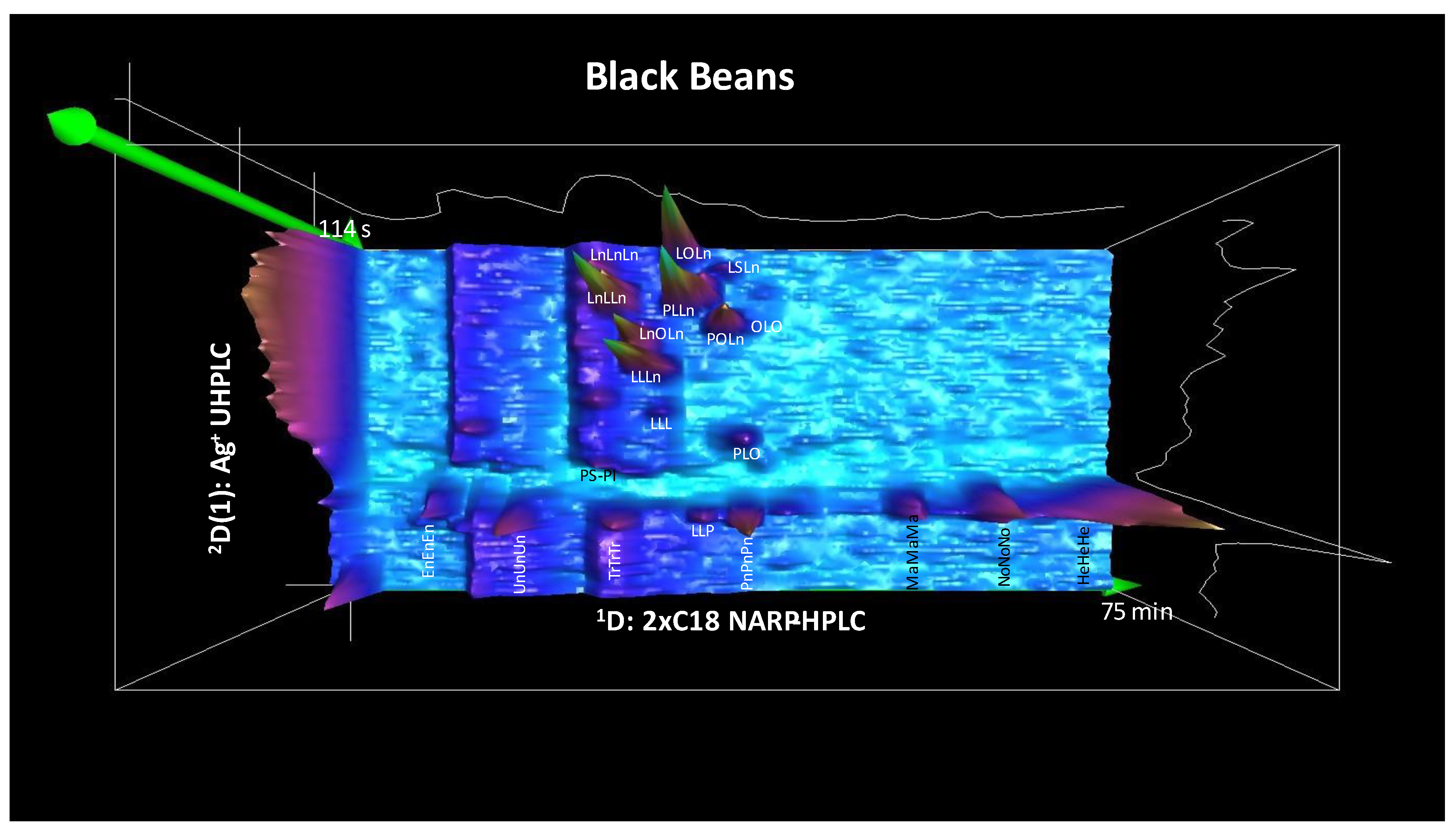
3.2. The First Second Dimension 2D(1): Argentation UHPLC
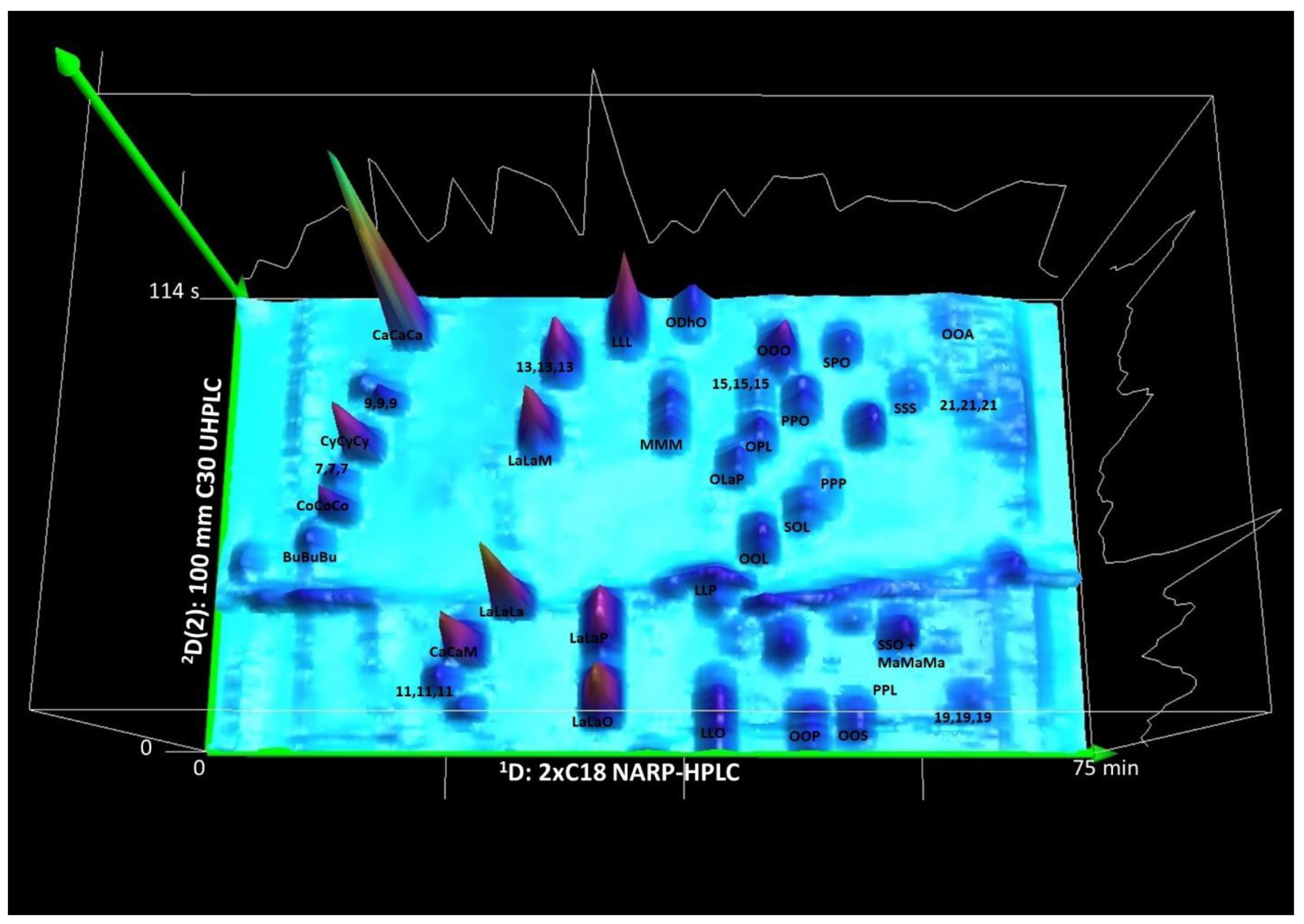
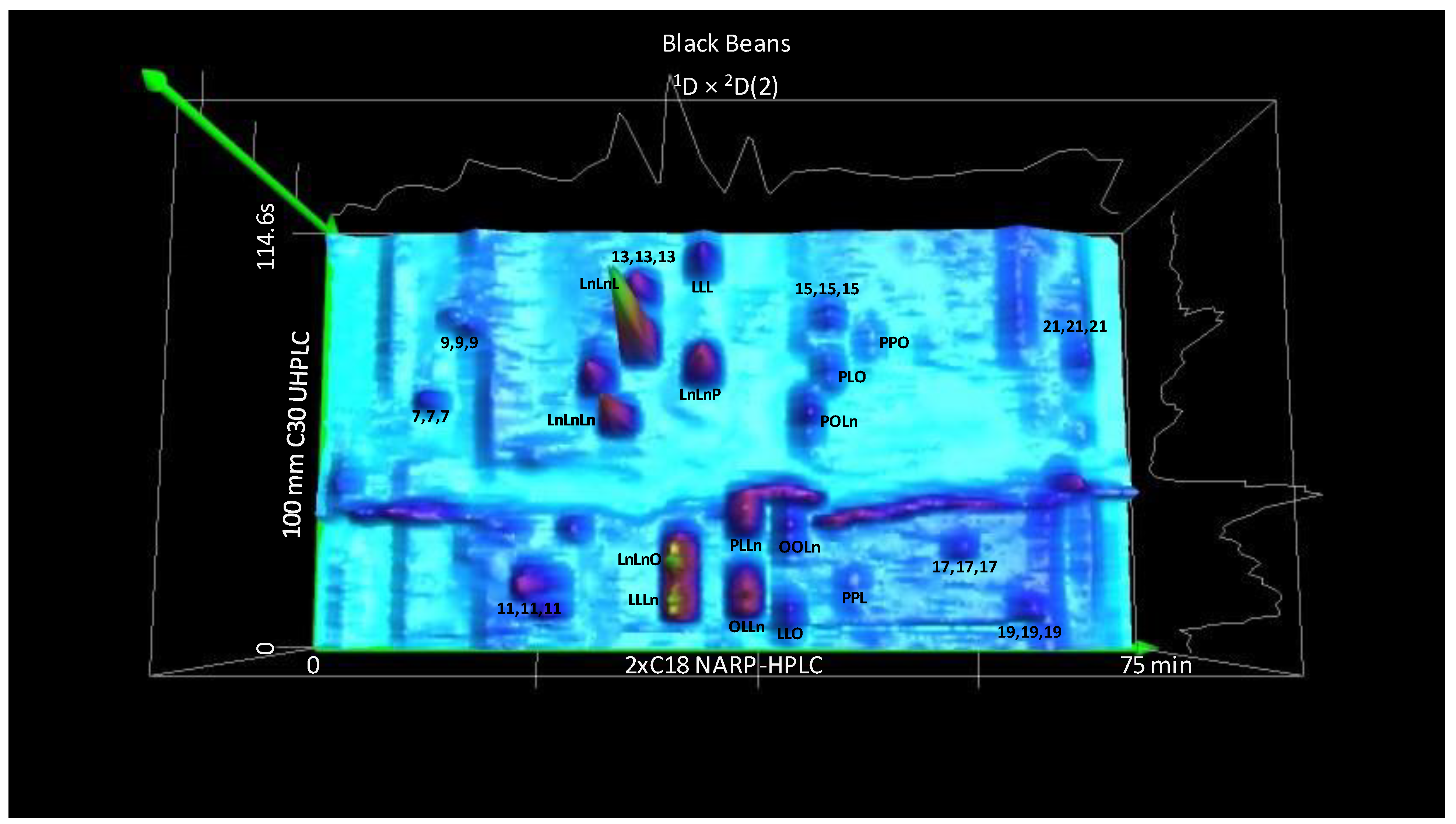
3.3. The Second Second Dimension 2D(2): C30 NARP-UHPLC

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
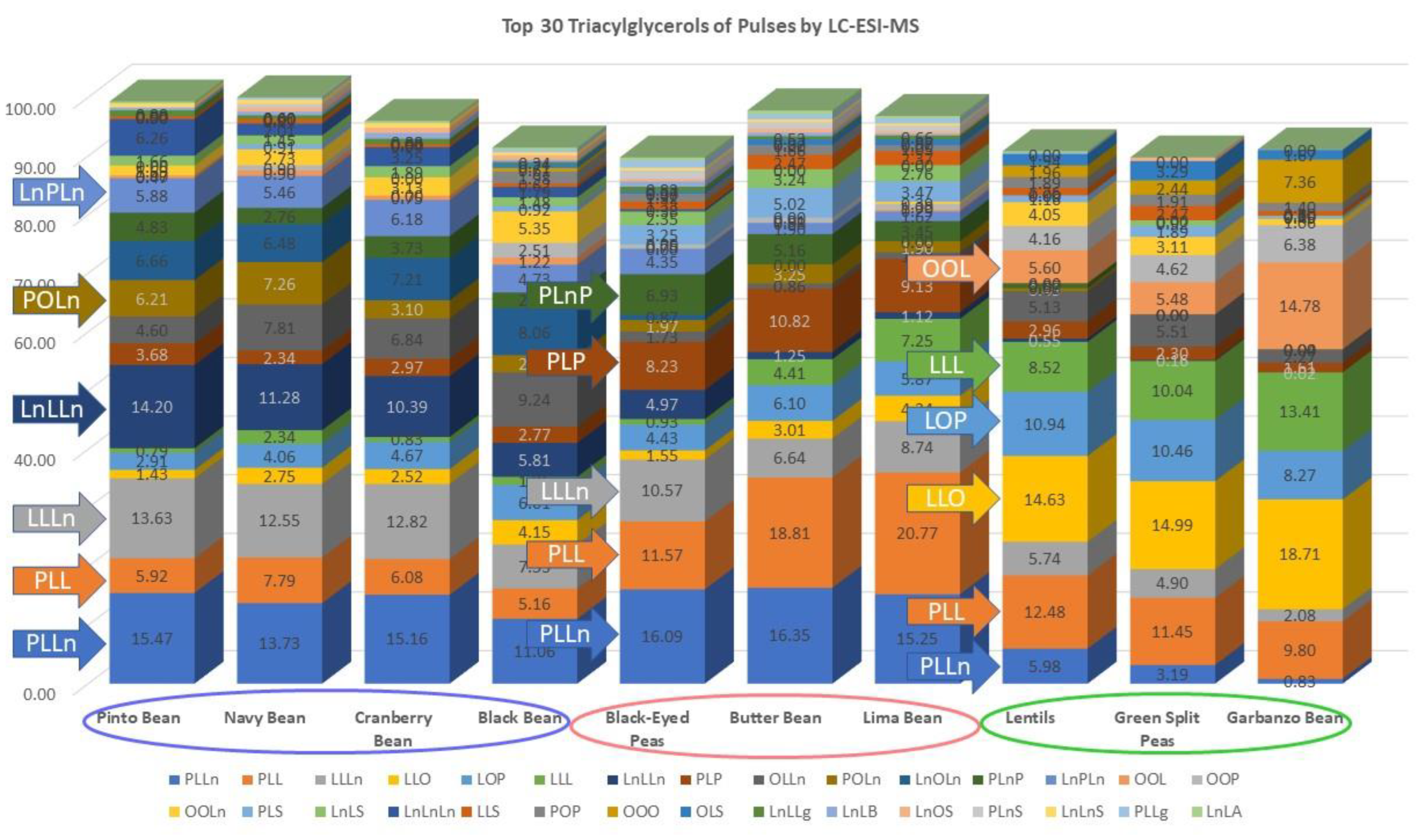
| TAG a | RT b | Pinto Bean | Navy Bean | Cran- Berry Bean | Black Bean | Black- Eyed Peas | Butter Bean | Lima Bean | Lentils | Green Split Peas | Gar- Banzo Bean |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PLLn c | 38.38 | 15.47 | 13.73 | 15.16 | 11.06 | 16.09 | 16.35 | 15.25 | 5.98 | 3.19 | 0.83 |
| PLL c | 42.12 | 5.92 | 7.79 | 6.08 | 5.16 | 11.57 | 18.81 | 20.77 | 12.48 | 11.45 | 9.80 |
| LLLn c | 32.47 | 13.63 | 12.55 | 12.82 | 7.53 | 10.57 | 6.64 | 8.74 | 5.74 | 4.90 | 2.08 |
| LLO c | 41.64 | 1.43 | 2.75 | 2.52 | 4.15 | 1.55 | 3.01 | 4.34 | 14.63 | 14.99 | 18.71 |
| LOP c | 46.52 | 2.91 | 4.06 | 4.67 | 6.01 | 4.43 | 6.10 | 5.87 | 10.94 | 10.46 | 8.27 |
| LLL | 35.83 | 0.79 | 2.34 | 0.83 | 1.36 | 0.93 | 4.41 | 7.25 | 8.52 | 10.04 | 13.41 |
| LnLLn c | 29.77 | 14.20 | 11.28 | 10.39 | 5.81 | 4.97 | 1.25 | 1.12 | 0.55 | 0.16 | 0.02 |
| PLP c | 47.47 | 3.68 | 2.34 | 2.97 | 2.77 | 8.23 | 10.82 | 9.13 | 2.96 | 2.30 | 1.61 |
| OLLn c | 37.67 | 4.60 | 7.81 | 6.84 | 9.24 | 1.73 | 0.86 | 1.08 | 5.13 | 5.51 | 2.27 |
| POLn c | 44.18 | 5.66 | 4.67 | 6.21 | 7.26 | 3.10 | 2.93 | 1.97 | 3.25 | 1.90 | 0.49 |
| LnOLn d | 32.59 | 6.66 | 6.48 | 7.21 | 8.06 | 0.87 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| PLnP c | 44.68 | 4.83 | 2.76 | 3.73 | 2.65 | 6.93 | 5.16 | 3.45 | 0.87 | 0.00 | 0.04 |
| LnPLn c | 34.08 | 5.88 | 5.46 | 6.18 | 4.73 | 4.35 | 1.90 | 1.62 | 0.00 | 0.00 | 0.00 |
| OOL c | 45.94 | 0.47 | 0.90 | 0.75 | 1.22 | 0.00 | 0.00 | 0.29 | 5.60 | 5.48 | 14.78 |
| OOP c | 49.35 | 0.00 | 0.90 | 0.00 | 2.51 | 0.75 | 0.91 | 1.00 | 4.16 | 4.62 | 6.38 |
| OOLn c | 41.97 | 1.69 | 2.73 | 3.13 | 5.35 | 0.00 | 0.00 | 0.39 | 4.05 | 3.11 | 1.06 |
| PLS c | 50.43 | 0.00 | 0.91 | 0.00 | 0.92 | 3.25 | 5.02 | 3.47 | 1.16 | 1.89 | 0.45 |
| LnLS c | 44.42 | 1.66 | 1.45 | 1.89 | 1.48 | 2.35 | 3.24 | 2.76 | 0.00 | 0.92 | 0.10 |
| LnLnLn | 26.08 | 6.26 | 2.01 | 3.25 | 1.79 | 0.38 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| LLS c | 46.65 | 0.50 | 0.81 | 0.54 | 0.62 | 1.33 | 2.47 | 2.37 | 1.26 | 2.47 | 0.80 |
| POP | 50.45 | 0.00 | 0.00 | 0.00 | 1.98 | 1.32 | 1.66 | 1.05 | 1.89 | 1.91 | 1.40 |
| OOO | 48.81 | 0.00 | 0.00 | 0.00 | 0.61 | 0.00 | 0.00 | 0.00 | 1.96 | 2.44 | 7.36 |
| OLS c | 49.37 | 0.00 | 0.00 | 0.00 | 0.71 | 0.37 | 0.97 | 0.97 | 1.94 | 3.29 | 1.67 |
| LnLLg c | 53.51 | 0.99 | 0.60 | 0.88 | 0.34 | 0.82 | 0.53 | 0.66 | 0.00 | 0.00 | 0.00 |
| LnLB c | 50.44 | 0.50 | 0.64 | 0.98 | 0.36 | 0.91 | 0.60 | 0.27 | 0.23 | 0.00 | 0.00 |
| LnOS c | 47.60 | 0.00 | 0.70 | 0.90 | 0.78 | 0.45 | 0.65 | 0.43 | 0.00 | 0.62 | 0.00 |
| PLnS c | 47.91 | 0.00 | 0.63 | 0.00 | 0.00 | 1.39 | 1.17 | 0.97 | 0.00 | 0.00 | 0.00 |
| LnLnS c | 39.83 | 0.67 | 0.72 | 0.76 | 0.51 | 0.49 | 0.46 | 0.40 | 0.00 | 0.00 | 0.00 |
| PLLg d | 59.03 | 0.00 | 0.50 | 0.00 | 0.48 | 1.19 | 0.85 | 0.84 | 0.00 | 0.00 | 0.07 |
| LnLA c | 47.65 | 0.33 | 0.75 | 0.38 | 0.32 | 0.56 | 0.67 | 0.40 | 0.26 | 0.00 | 0.00 |
| OOS d | 52.59 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 2.16 | 1.13 |
| MOL d | 42.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 1.78 | 0.82 | 0.50 |
| LLLg d | 56.21 | 0.00 | 0.37 | 0.00 | 0.23 | 0.73 | 0.55 | 0.00 | 0.42 | 0.27 | 0.13 |
| PLA d | 53.98 | 0.00 | 0.00 | 0.00 | 0.00 | 0.91 | 0.85 | 0.47 | 0.00 | 0.30 | 0.19 |
| LOB | 56.18 | 0.00 | 0.00 | 0.00 | 0.54 | 0.28 | 0.00 | 0.00 | 0.72 | 0.55 | 0.30 |
| LLB d | 53.21 | 0.00 | 0.27 | 0.00 | 0.14 | 0.87 | 0.00 | 0.32 | 0.33 | 0.00 | 0.19 |
| POS c | 53.91 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.47 | 0.00 | 1.21 | 0.47 |
| PoLLn c | 32.43 | 0.72 | 0.00 | 0.65 | 0.68 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.04 |
| LOA c | 53.19 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.64 | 0.68 | 0.60 |
| PLnLg d | 57.52 | 0.00 | 0.40 | 0.00 | 0.32 | 0.65 | 0.24 | 0.24 | 0.00 | 0.00 | 0.00 |
| LOLg c | 58.89 | 0.00 | 0.00 | 0.00 | 0.27 | 0.27 | 0.00 | 0.24 | 0.49 | 0.35 | 0.18 |
| LLA c | 49.56 | 0.00 | 0.00 | 0.00 | 0.00 | 0.38 | 0.00 | 0.29 | 0.26 | 0.23 | 0.32 |
| PLB d | 56.92 | 0.00 | 0.00 | 0.00 | 0.00 | 0.93 | 0.00 | 0.32 | 0.00 | 0.00 | 0.10 |
| SSL d | 53.98 | 0.00 | 0.00 | 0.00 | 0.00 | 0.17 | 0.45 | 0.36 | 0.00 | 0.27 | 0.04 |
| PLnB d | 54.90 | 0.00 | 0.00 | 0.00 | 0.37 | 0.88 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| OLG c | 48.96 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.39 | 0.37 | 0.62 |
| LLG d | 45.84 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.53 | 0.44 | 0.22 |
| LnLnA c | 44.80 | 0.25 | 0.00 | 0.27 | 0.17 | 0.20 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| LPG d | 49.34 | 0.00 | 0.00 | 0.00 | 0.00 | 0.51 | 0.26 | 0.00 | 0.31 | 0.00 | 0.00 |
| LnOB d | 53.31 | 0.00 | 0.40 | 0.00 | 0.51 | 0.22 | 0.00 | 0.00 | 0.06 | 0.00 | 0.00 |
| OOA c | 56.15 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.45 | 0.43 |
| LnLG c | 41.96 | 0.00 | 0.00 | 0.00 | 0.00 | 0.39 | 0.00 | 0.00 | 0.50 | 0.00 | 0.00 |
| LnLnB c | 48.08 | 0.30 | 0.00 | 0.00 | 0.17 | 0.25 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| OPoL c | 41.79 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.34 |
| LnPTc c | 56.24 | 0.00 | 0.00 | 0.00 | 0.00 | 0.59 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| LSB d | 59.03 | 0.00 | 0.06 | 0.00 | 0.05 | 0.21 | 0.15 | 0.07 | 0.00 | 0.00 | 0.01 |
| OOB c | 58.88 | 0.00 | 0.00 | 0.00 | 0.21 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.21 |
| OOG c | 51.66 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.40 |
| LnOA c | 50.38 | 0.00 | 0.00 | 0.00 | 0.32 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.01 |
| LgOLn d | 56.22 | 0.00 | 0.17 | 0.00 | 0.18 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| LPoP d | 42.02 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.25 | 0.14 |
| LSA d | 57.29 | 0.00 | 0.00 | 0.00 | 0.00 | 0.12 | 0.00 | 0.11 | 0.00 | 0.00 | 0.02 |
| OPG d | 52.61 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.21 |
| LnSB d | 57.52 | 0.00 | 0.06 | 0.00 | 0.05 | 0.11 | 0.06 | 0.02 | 0.00 | 0.00 | 0.00 |
| LPoL c | 35.77 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.37 |
| LnLnLg c | 51.03 | 0.00 | 0.00 | 0.00 | 0.00 | 0.19 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| POA d | 57.39 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.14 |
| POB d | 59.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.13 |
| PnLO d | 44.79 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.15 |
| PLnG d | 46.96 | 0.00 | 0.00 | 0.00 | 0.00 | 0.16 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| LnPA d | 51.30 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.13 | 0.00 | 0.00 | 0.00 |
| LOTc c | 57.46 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.13 |
| LnSA d | 54.90 | 0.00 | 0.00 | 0.00 | 0.03 | 0.09 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| LLTc c | 54.84 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.08 |
| L20:2O d | 45.59 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.09 |
| LLPn d | 40.01 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.08 |
| SLnS d | 51.30 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.08 | 0.00 | 0.00 | 0.00 |
| LOMa d | 47.92 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.07 |
| LL20:2 c | 41.52 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.06 |
| LLMa c | 44.84 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.06 |
| LO21:0 c | 54.81 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.06 |
| POLg d | 60.90 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.04 |
| OPnO d | 47.93 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.04 |
| L17:1O c | 44.48 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.03 |
| SOS d | 57.39 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.03 |
| LO15:1 d | 40.01 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.01 |
| OSA d | 59.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.01 |
| L17:1P d | 44.79 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.01 |
| L19:1P d | 47.92 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.01 |
| L17:2P d | 40.01 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.01 |
| O17:1P d | 47.93 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| OSB d | 60.90 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Sum | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 |
3.4. Regioisomer Quantification
4. Discussion
4.1. NARP-HPLC: The First Dimension, 1D
4.2. Silver-Ion Chromatography: The First Second Dimension 2D(1)
4.3. Multi-Cycle Chromatography: The Second Second Dimension 2D(2)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Vos, J.; Stoll, D.; Buckenmaier, S.; Eeltink, S.; Grinias, J.P. Advances in ultra-high-pressure and multi-dimensional liquid chromatography instrumentation and workflows. Anal. Sci. Adv. 2021, 2, 171–192. [Google Scholar] [CrossRef]
- Cardoso, C.L.; de Moraes, M.C.; Cass, Q.B. The Versatility of Two-Dimensional Liquid Chromatography. J. Braz. Chem. Soc. 2023, 34, 1565–1580. [Google Scholar] [CrossRef]
- Caño-Carrillo, I.; Gilbert-López, B.; Montero, L.; Martínez-Piernas, A.B.; García-Reyes, J.F.; Molina-Díaz, A. Comprehensive and heart-cutting multidimensional liquid chromatography-mass spectrometry and its applications in food analysis. Mass Spectrom. Rev. 2023. [Google Scholar] [CrossRef]
- Montero, L.; Herrero, M. Two-dimensional liquid chromatography approaches for food authenticity. Curr. Opin. Food Sci. 2023, 51, 101041. [Google Scholar] [CrossRef]
- Papatheocharidou, C.; Samanidou, V. Two-Dimensional High-Performance Liquid Chromatography as a Powerful Tool for Bioanalysis: The Paradigm of Antibiotics. Molecules 2023, 28, 5056. [Google Scholar] [CrossRef] [PubMed]
- van den Hurk, R.S.; Pursch, M.; Stoll, D.R.; Pirok, B.W.J. Recent trends in two-dimensional liquid chromatography. TrAC—Trends Anal. Chem. 2023, 166, 117166. [Google Scholar] [CrossRef]
- Foster, S.W.; Parker, D.; Kurre, S.; Boughton, J.; Stoll, D.R.; Grinias, J.P. A review of two-dimensional liquid chromatography approaches using parallel column arrays in the second dimension. Anal. Chim. Acta 2022, 1228, 340300. [Google Scholar] [CrossRef] [PubMed]
- Lv, W.; Shi, X.; Wang, S.; Xu, G. Multidimensional liquid chromatography-mass spectrometry for metabolomic and lipidomic analyses. TrAC—Trends Anal. Chem. 2019, 120, 115302. [Google Scholar] [CrossRef]
- Duong, V.-A.; Park, J.-M.; Lee, H. Review of three-dimensional liquid chromatography platforms for bottom-up proteomics. Int. J. Mol. Sci. 2020, 21, 1524. [Google Scholar] [CrossRef] [PubMed]
- Abdulhussain, N.; Nawada, S.; Schoenmakers, P. Latest Trends on the Future of Three-Dimensional Separations in Chromatography. Chem. Rev. 2021, 121, 12016–12034. [Google Scholar] [CrossRef]
- Cacciola, F.; Dugo, P.; Mondello, L. Multidimensional liquid chromatography in food analysis. TrAC—Trends Anal. Chem. 2017, 96, 116–123. [Google Scholar] [CrossRef]
- Wu, Z.; Schoenmakers, P.; Marriott, P.J. Nomenclature and Conventions in Comprehensive Multidimensional Chromatography—An Update. LCGC Eur. 2012, 25, 272–275. [Google Scholar]
- Washburn, M.P.; Wolters, D.; Yates, J.R. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 2001, 19, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.W.; Jorgenson, J.W. Comprehensive Three-Dimensional Separation of Peptides Using Size Exclusion Chromatography/Reversed Phase Liquid Chromatography/Optically Gated Capillary Zone Electrophoresis. Anal. Chem. 1995, 67, 3456–3463. [Google Scholar] [CrossRef] [PubMed]
- Fairchild, J.N.; Horváth, K.; Guiochon, G. Theoretical advantages and drawbacks of on-line, multidimensional liquid chromatography using multiple columns operated in parallel. J. Chromatogr. A 2009, 1216, 6210–6217. [Google Scholar] [CrossRef]
- Opiteck, G.J.; Ramirez, S.M.; Jorgenson, J.W.; Moseley, M.A., III. Comprehensive two-dimensional high-performance liquid chromatography for the isolation of overexpressed proteins and proteome mapping. Anal. Biochem. 1998, 258, 349–361. [Google Scholar] [CrossRef]
- Wagner, K.; Racaityte, K.; Unger, K.K.; Miliotis, T.; Edholm, L.E.; Bischoff, R.; Marko-Varga, G. Protein mapping by two-dimensional high performance liquid chromatography. J. Chromatogr. A 2000, 893, 293–305. [Google Scholar] [CrossRef]
- Venkatramani, C.J.; Zelechonok, Y. An automated orthogonal two-dimensional liquid chromatograph. Anal. Chem. 2003, 75, 3484–3494. [Google Scholar] [CrossRef]
- Wang, H.; Lhotka, H.R.; Bennett, R.; Potapenko, M.; Pickens, C.J.; Mann, B.F.; Haidar Ahmad, I.A.; Regalado, E.L. Introducing online multicolumn two-dimensional liquid chromatography screening for facile selection of stationary and mobile phase conditions in both dimensions. J. Chromatogr. A 2020, 1622, 460895. [Google Scholar] [CrossRef]
- Byrdwell, W.C.; Kotapati, H.K.; Goldschmidt, R.; Jakubec, P.; Nováková, L. Three-dimensional liquid chromatography with parallel second dimensions and quadruple parallel mass spectrometry for adult/infant formula analysis. J. Chromatogr. A 2022, 1661, 462682. [Google Scholar] [CrossRef]
- Themelis, T.; Amini, A.; De Vos, J.; Eeltink, S. Towards spatial comprehensive three-dimensional liquid chromatography: A tutorial review. Anal. Chim. Acta 2021, 1148, 238157. [Google Scholar] [CrossRef] [PubMed]
- Stoll, D.R.; Carr, P.W. Two-Dimensional Liquid Chromatography: A State of the Art Tutorial. Anal. Chem. 2017, 89, 519–531. [Google Scholar] [CrossRef]
- Byrdwell, W.C. Breaking the rules: Two- and three-dimensional liquid chromatography with four dimensions of mass spectrometry. Curr. Trends Mass Spectrom. 2021, 19, 6–11. [Google Scholar]
- Byrdwell, W.C. Comprehensive Dual Liquid Chromatography with Quadruple Mass Spectrometry (LC1MS2 × LC1MS2 = LC2MS4) for Analysis of Parinari Curatellifolia and Other Seed Oil Triacylglycerols. Anal. Chem. 2017, 89, 10537–10546. [Google Scholar] [CrossRef]
- Pól, J.; Kivilompolo, M.; Hyötyläinen, T. Comprehensive two-dimensional liquid chromatography (LC × LC): A review. LCGC Eur. 2011, 24, 232–243. [Google Scholar]
- Gritti, F.; Besner, S.; Cormier, S.; Gilar, M. Applications of high-resolution recycling liquid chromatography: From small to large molecules. J. Chromatogr. A 2017, 1524, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Aly, A.A.; Muller, M.; de Villiers, A.; Pirok, B.W.J.; Górecki, T. Parallel gradients in comprehensive multidimensional liquid chromatography enhance utilization of the separation space and the degree of orthogonality when the separation mechanisms are correlated. J. Chromatogr. A 2020, 1628, 461452. [Google Scholar] [CrossRef]
- Byrdwell, W.C. Dual parallel mass spectrometers for analysis of sphingolipid, glycerophospholipid and plasmalogen molecular species. Rapid Commun. Mass Spectrom. 1998, 12, 256–272. [Google Scholar] [CrossRef]
- Byrdwell, W.C. “Dilute-and-shoot” triple parallel mass spectrometry method for analysis of vitamin D and triacylglycerols in dietary supplements. Anal. Bioanal. Chem. 2011, 401, 3317–3334. [Google Scholar] [CrossRef]
- Byrdwell, W.C. Quadruple parallel mass spectrometry for analysis of vitamin D and triacylglycerols in a dietary supplement. J. Chromatogr. A 2013, 1320, 48–65. [Google Scholar] [CrossRef]
- Byrdwell, W.C.; Kotapati, H.K.; Goldschmidt, R. Fast chromatography of pulse triacylglycerols. J. Am. Oil Chem. Soc. 2022, 100, 25–43. [Google Scholar] [CrossRef]
- United Nations Food and Agriculture Organization; World Health Organization. Standard for certain pulses. Codex Aliment. 1989, CXS 171-1989. [Google Scholar]
- Hall, C.; Hillen, C.; Robinson, J.G. Composition, nutritional value, and health benefits of pulses. Cereal Chem. 2017, 94, 11–31. [Google Scholar] [CrossRef]
- Kotha, R.R.; Finley, J.W.; Luthria, D.L. Determination of Soluble Mono, Di, and Oligosaccharide Content in 23 Dry Beans (Phaseolus vulgaris L.). J. Agric. Food Chem. 2020, 68, 6412–6419. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Sloane-Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissue. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Byrdwell, W.C. A note on the use of workstation software programs for quantification. J. Liq. Chromatogr. Relat. Technol. 2019, 42, 570–574. [Google Scholar] [CrossRef]
- Byrdwell, W.C.; Emken, E.A. Analysis of triglycerides using atmospheric pressure chemical ionization mass spectrometry. Lipids 1995, 30, 173–175. [Google Scholar] [CrossRef]
- Byrdwell, W.C. Critical Ratios for structural analysis of triacylglycerols using mass spectrometry. Lipid Technol. 2015, 27, 258–261. [Google Scholar] [CrossRef]
- Byrdwell, W.C. The bottom-up solution to the triacylglycerol lipidome using atmospheric pressure chemical ionization mass spectrometry. Lipids 2005, 40, 383–417. [Google Scholar] [CrossRef]
- Byrdwell, W.C. The Updated Bottom Up Solution applied to mass spectrometry of soybean oil in a dietary supplement gelcap. Anal. Bioanal. Chem. 2015, 407, 5143–5160. [Google Scholar] [CrossRef]
- Mottram, H.R.; Evershed, R.P. Structure analysis of triacylglycerol positional isomers using atmospheric pressure chemical ionisation mass spectrometry. Tetrahedron Lett. 1996, 37, 8593–8596. [Google Scholar] [CrossRef]
- Laakso, P.; Voutilainen, P. Analysis of triacylglycerols by silver-ion high-performance liquid chromatography-atmospheric pressure chemical ionization mass spectrometry. Lipids 1996, 31, 1311–1322. [Google Scholar] [CrossRef]
- Byrdwell, W.C. Qualitative and Quantitative Analysis of Triacylglycerols by Atmospheric Pressure Ionization (APCI and ESI) Mass Spectrometry Techniques. In Modern Methods for Lipid Analysis by Liquid Chromatography/Mass Spectrometry and Related Techniques; Byrdwell, W.C., Ed.; AOCS Press: Champaign, IL, USA, 2005; pp. 298–412. ISBN 1-893997-75-8. [Google Scholar]
- Morris, L.J. Separations of lipids by silver ion chromatography. J. Lipid Res. 1966, 7, 717–732. [Google Scholar] [CrossRef]
- Dobson, G.; Christie, W.W.; Nikolova-Damyanova, B. Silver ion chromatography of lipids and fatty acids. J. Chromatogr. B Biomed. Sci. Appl. 1995, 671, 197–222. [Google Scholar] [CrossRef]
- Holčapek, M.; Lísa, M. Silver-Ion Liquid Chromatography—Mass Spectrometry. In Handbook of Advanced Chromatography/Mass Spectrometry Techniques; Holčapek, M., Byrdwell, W.C., Eds.; Elsevier/AOCS Press: Champaign, IL, USA, 2017; pp. 115–140. ISBN 978-0-12-811732-3. [Google Scholar]
- Byrdwell, W.C.; Emken, E.A.; Neff, W.E.; Adlof, R.O. Quantitative analysis of triglycerides using atmospheric pressure chemical ionization-mass spectrometry. Lipids 1996, 31, 919–935. [Google Scholar] [CrossRef] [PubMed]
- Byrdwell, W.C.; Neff, W.E.; List, G.R. Triacylglycerol analysis of potenial margarine base stocks by high-performance liquid chromatography with atmospheric pressure chemical ionization mass spectrometry and, flame ionization detection. J. Agric. Food Chem. 2001, 49, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Byrdwell, W.C. The Updated Bottom Up Solution Applied to Atmospheric Pressure Photoionization and Electrospray Ionization Mass Spectrometry. JAOCS J. Am. Oil Chem. Soc. 2015, 92, 1533–1547. [Google Scholar] [CrossRef]
- Holčapek, M.; Dvorakova, H.; Lísa, M.; Giron, A.J.; Sandra, P.; Cvacka, J. Regioisomeric analysis of triacylglycerols using silver-ion liquid chromatography-atmospheric pressure chemical ionization mass spectrometry: Comparison of five different mass analyzers. J. Chromatogr. A 2010, 1217, 8186–8194. [Google Scholar] [CrossRef]
- Liu, Q.; Xiao, J.; Yu, J.; Xie, Y.; Chen, X.; Yang, H. Improved enantioseparation via the twin-column based recycling high performance liquid chromatography. J. Chromatogr. A 2014, 1363, 236–241. [Google Scholar] [CrossRef]

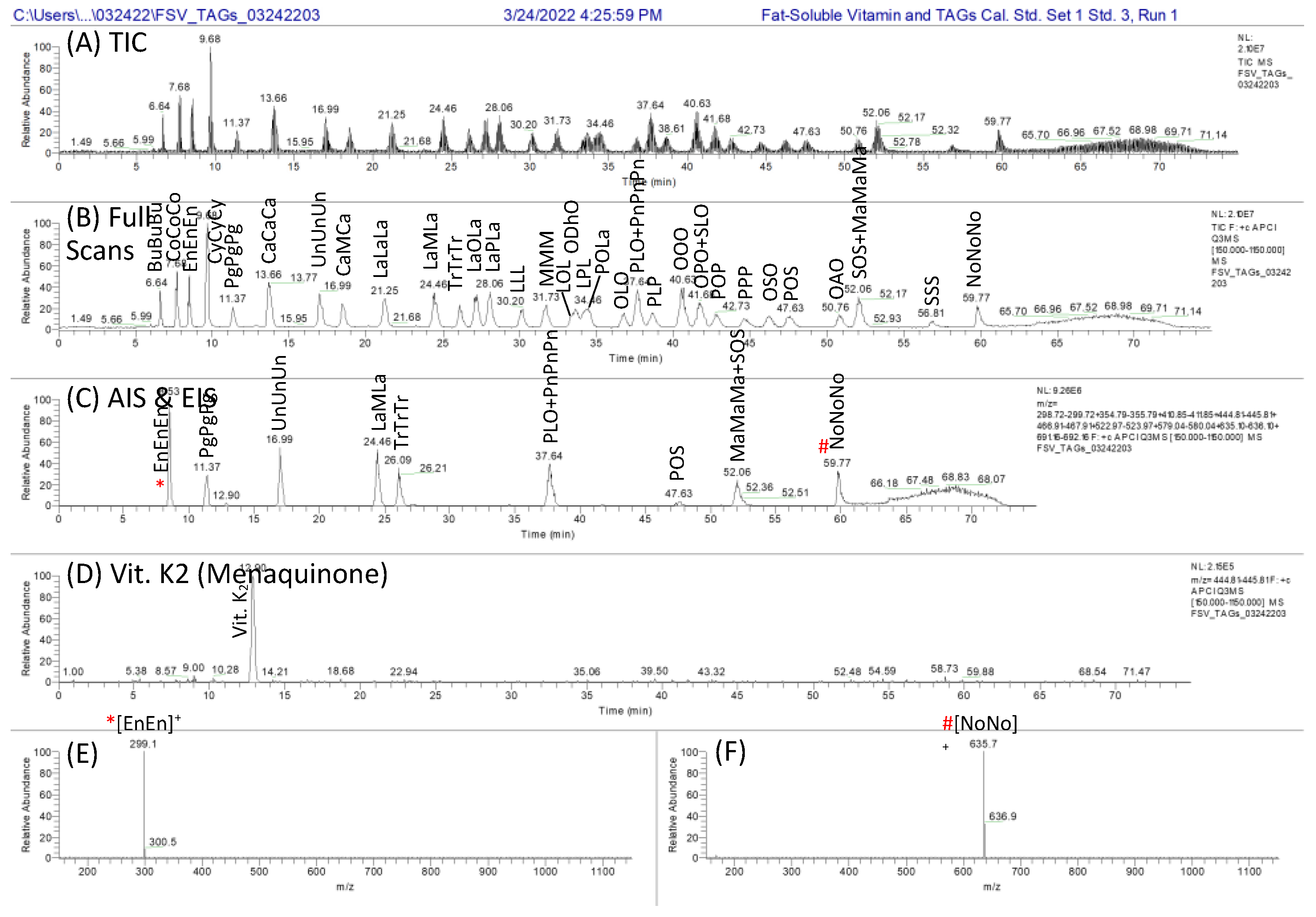
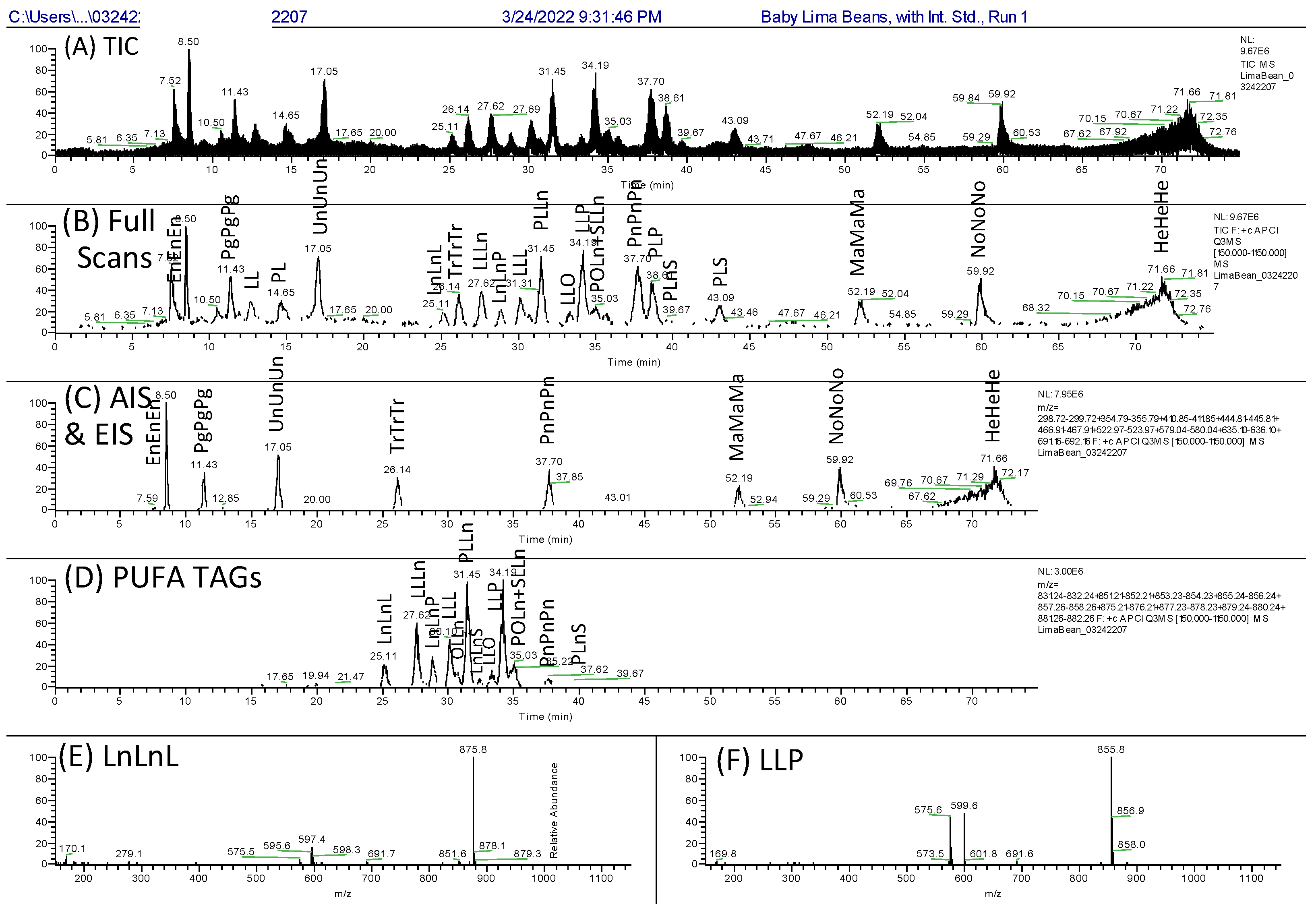


| Lipid Ion a | RT Min b | Black Bean | Black-Eyed Pea | Butter Bean | Cran- Berry Bean | Gar- Banzo Bean | Green Split | Lentils | Lima Bean | Navy Bean | Pinto Bean |
|---|---|---|---|---|---|---|---|---|---|---|---|
| C20H37NO2 + H | 8.61 | 0.00 | 0.05 | 0.00 | 0.38 | 0.06 | 0.00 | 0.05 | 0.39 | 15.28 | 0.04 |
| Cer(d18:1/16:0) + Na | 15.10 | 0.00 | 0.00 | 0.00 | 0.02 | 0.00 | 0.00 | 0.00 | 0.00 | 5.06 | 0.00 |
| Cer(d18:0/18:2) + Na | 15.03 | 0.00 | 0.00 | 0.00 | 0.06 | 0.00 | 0.00 | 0.00 | 0.05 | 6.66 | 0.00 |
| Cer(d18:1/18:2) + Na | 12.97 | 0.00 | 0.00 | 0.00 | 0.13 | 0.00 | 0.00 | 0.00 | 0.07 | 10.93 | 0.00 |
| Cer(d18:1/18:3) + Na | 12.82 | 0.00 | 0.00 | 0.00 | 0.06 | 0.00 | 0.00 | 0.00 | 0.00 | 4.96 | 0.00 |
| Cer(d18:1/18:3) + H | 15.10 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 5.06 | 0.00 |
| DGDG(18:2/18:2) + NH4 | 11.06 | 0.38 | 0.06 | 0.06 | 0.31 | 0.17 | 0.81 | 0.14 | 0.06 | 0.76 | 0.14 |
| DGDG(18:2/18:3) + NH4 | 10.88 | 0.28 | 0.14 | 0.08 | 0.29 | 0.05 | 0.62 | 0.16 | 0.02 | 0.72 | 0.21 |
| DGDG(18:3/18:3) + NH4 | 10.75 | 0.83 | 0.31 | 0.26 | 0.75 | 0.00 | 0.29 | 0.10 | 0.13 | 3.16 | 0.77 |
| LPC(16:0) + H | 10.38 | 1.84 | 1.98 | 0.18 | 5.93 | 0.77 | 0.53 | 0.58 | 3.04 | 8.50 | 2.75 |
| LPC(18:1) + H | 10.38 | 2.70 | 0.34 | 0.03 | 3.50 | 2.87 | 1.55 | 1.65 | 0.50 | 3.86 | 1.11 |
| LPC(18:2) + H | 10.32 | 2.60 | 3.49 | 0.40 | 8.38 | 3.85 | 2.41 | 2.56 | 6.67 | 7.38 | 3.42 |
| LPC(18:3) + H | 9.33 | 1.89 | 1.31 | 0.10 | 6.33 | 0.08 | 0.15 | 0.41 | 1.37 | 4.05 | 3.90 |
| LPE(16:0) + H | 13.12 | 0.50 | 0.33 | 0.06 | 1.00 | 0.08 | 0.13 | 0.07 | 0.74 | 2.41 | 0.56 |
| MGDG(18:1/18:3) + NH4 | 12.99 | 0.22 | 0.04 | 0.26 | 0.25 | 0.02 | 0.14 | 0.02 | 0.00 | 0.73 | 0.07 |
| MGDG(18:2/18:3) + NH4 | 12.81 | 0.09 | 0.05 | 0.22 | 0.15 | 0.00 | 0.02 | 0.00 | 0.00 | 0.44 | 0.06 |
| MGDG(18:3/18:3) + NH4 | 10.84 | 0.24 | 0.14 | 0.98 | 0.32 | 0.00 | 0.02 | 0.00 | 0.03 | 1.60 | 0.23 |
| PC(16:0/18:1) + H | 18.82 | 17.73 | 3.82 | 2.42 | 10.62 | 16.35 | 12.37 | 10.80 | 2.71 | 3.16 | 6.36 |
| PC(16:0/18:2) + H | 17.91 | 20.27 | 34.49 | 37.50 | 15.81 | 27.71 | 27.68 | 18.99 | 41.55 | 6.80 | 25.44 |
| PC(16:1/18:2) + H | 16.32 | 9.51 | 8.04 | 5.00 | 9.35 | 0.53 | 0.59 | 1.82 | 4.81 | 1.61 | 14.16 |
| PC(18:0/18:1) + H | 19.88 | 0.26 | 0.08 | 0.06 | 0.07 | 0.38 | 1.66 | 0.19 | 0.00 | 0.00 | 0.09 |
| PC(18:0/18:2) + H | 18.83 | 10.04 | 3.04 | 3.43 | 4.00 | 17.89 | 18.63 | 11.26 | 3.62 | 1.46 | 2.37 |
| PC (36:4) | 16.03 | 16.07 | 25.29 | 26.39 | 14.12 | 24.05 | 24.50 | 38.56 | 19.93 | 2.64 | 16.65 |
| PC (36:5) | 15.14 | 9.70 | 12.62 | 10.53 | 14.60 | 1.87 | 1.73 | 9.74 | 9.43 | 2.19 | 15.45 |
| PC (36:6) | 14.27 | 1.61 | 1.06 | 0.52 | 2.50 | 0.00 | 0.08 | 0.22 | 0.80 | 0.21 | 5.11 |
| PE(16:0/18:2) + H | 20.34 | 2.56 | 2.98 | 9.29 | 0.65 | 1.40 | 2.74 | 1.25 | 3.44 | 0.38 | 0.73 |
| PE(18:1/18:2) + H | 19.52 | 0.23 | 0.03 | 0.12 | 0.06 | 0.91 | 1.03 | 0.37 | 0.00 | 0.00 | 0.08 |
| PE(18:2/18:2) + H | 18.92 | 0.45 | 0.30 | 2.14 | 0.36 | 0.99 | 2.33 | 1.05 | 0.61 | 0.00 | 0.29 |
| Sum | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Byrdwell, W.C.; Kotapati, H.K. Multi-Dimensional Liquid Chromatography of Pulse Triacylglycerols with Triple Parallel Mass Spectrometry. Separations 2023, 10, 594. https://doi.org/10.3390/separations10120594
Byrdwell WC, Kotapati HK. Multi-Dimensional Liquid Chromatography of Pulse Triacylglycerols with Triple Parallel Mass Spectrometry. Separations. 2023; 10(12):594. https://doi.org/10.3390/separations10120594
Chicago/Turabian StyleByrdwell, William C., and Hari Kiran Kotapati. 2023. "Multi-Dimensional Liquid Chromatography of Pulse Triacylglycerols with Triple Parallel Mass Spectrometry" Separations 10, no. 12: 594. https://doi.org/10.3390/separations10120594
APA StyleByrdwell, W. C., & Kotapati, H. K. (2023). Multi-Dimensional Liquid Chromatography of Pulse Triacylglycerols with Triple Parallel Mass Spectrometry. Separations, 10(12), 594. https://doi.org/10.3390/separations10120594