
Pros and Cons of Separation, Fractionation and Cleanup for Enhancement of the Quantitative Analysis of Bitumen-Derived Organics in Process-Affected Waters—A Review


Abstract
:1. Introduction
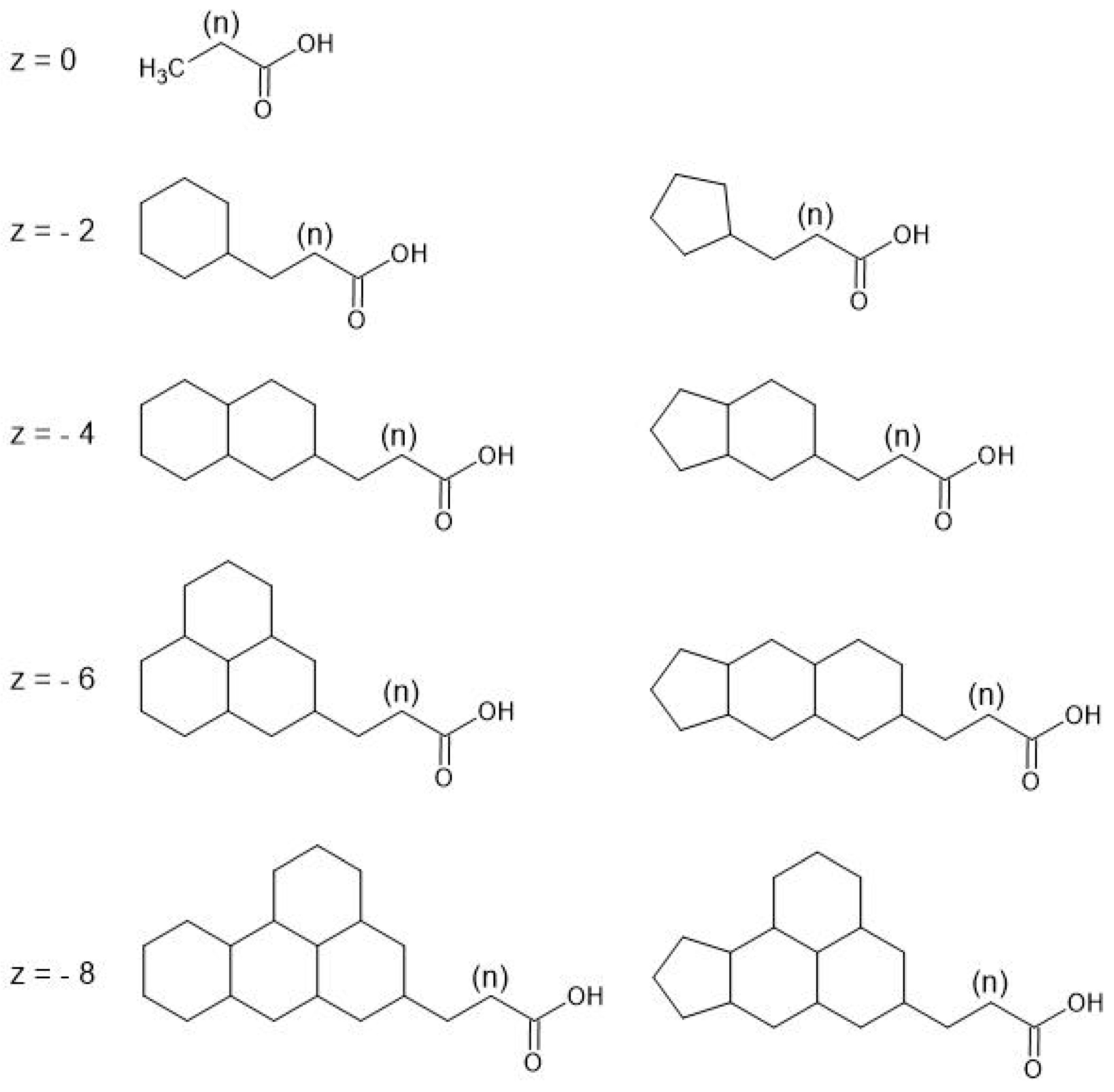
2. Critical Analytical Parameters
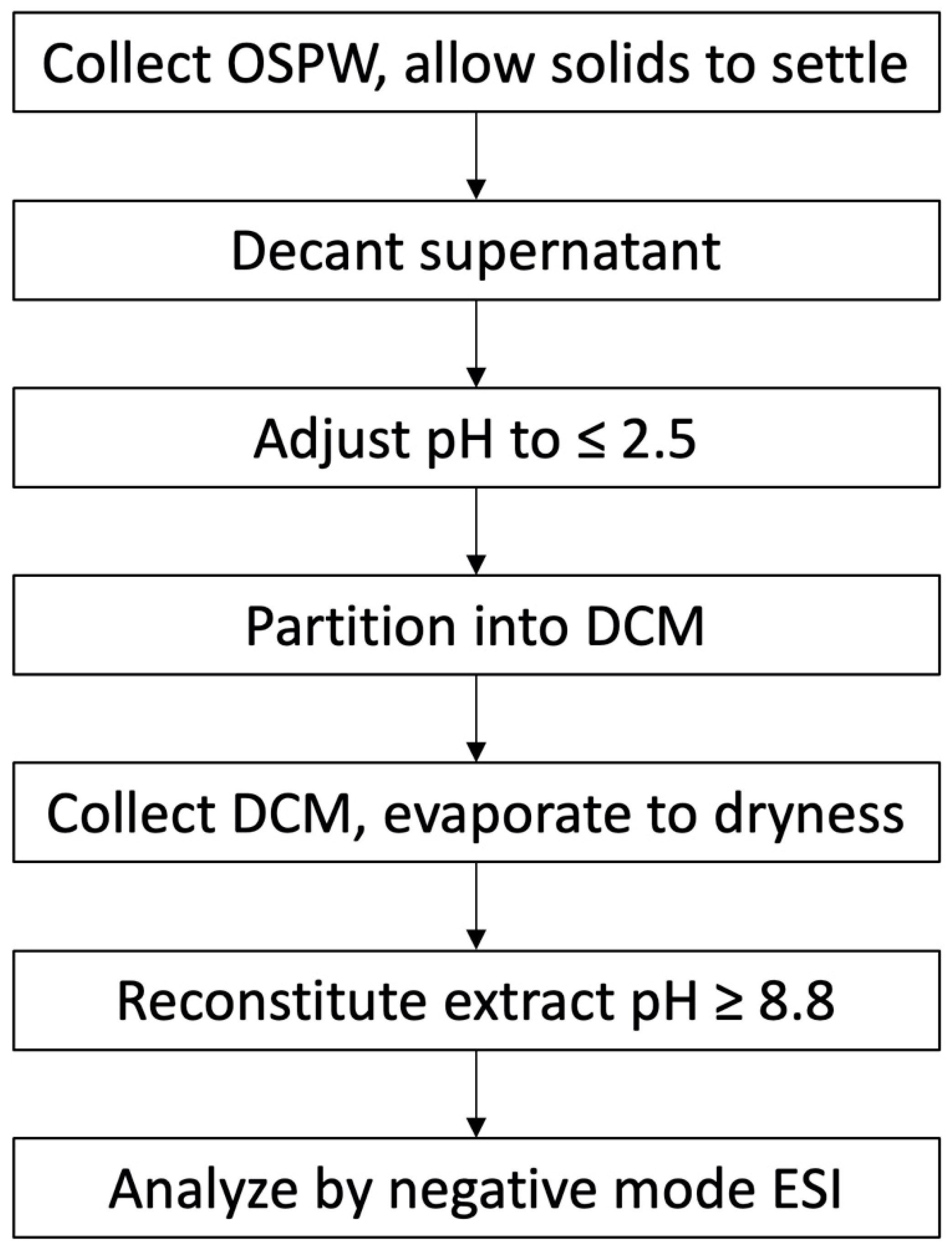
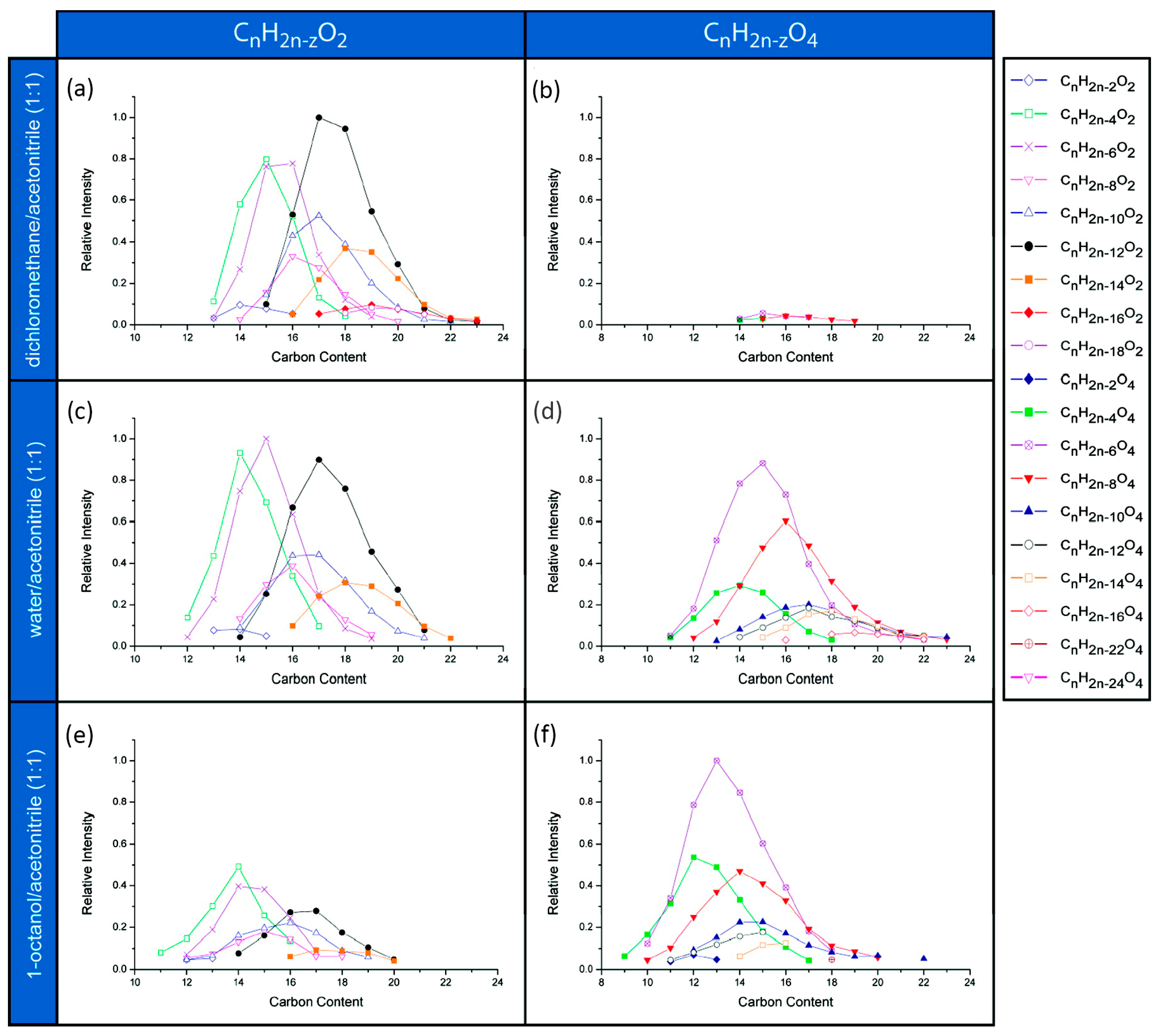
2.1. Choice of Extraction Solvent
2.2. Salt Content of the Sample
2.3. pH of Extraction
2.4. Sample Cleanup or No Cleanup
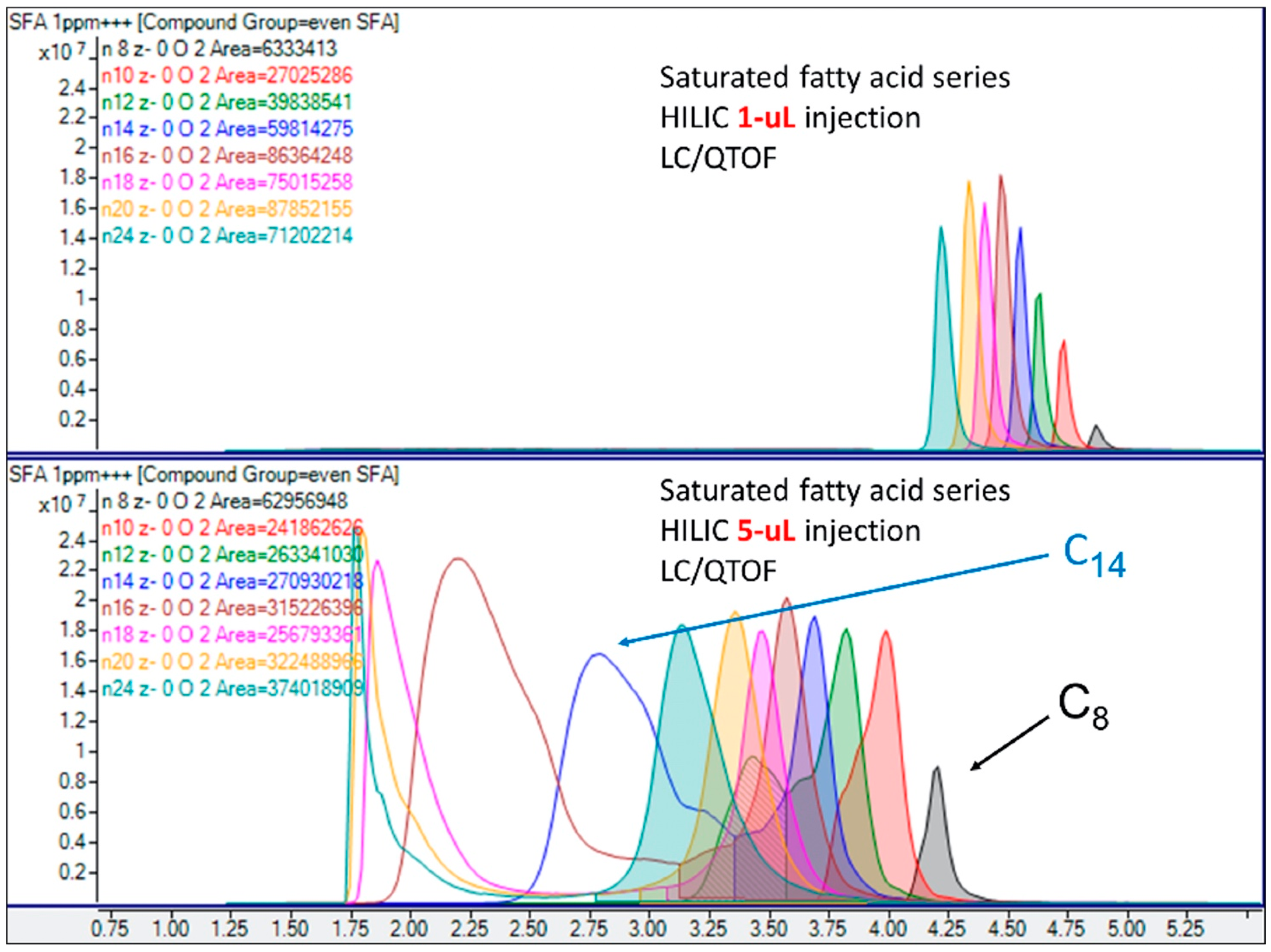
2.5. On-Line Chromatography vs. Injection or Infusion
2.6. Calibration Standards
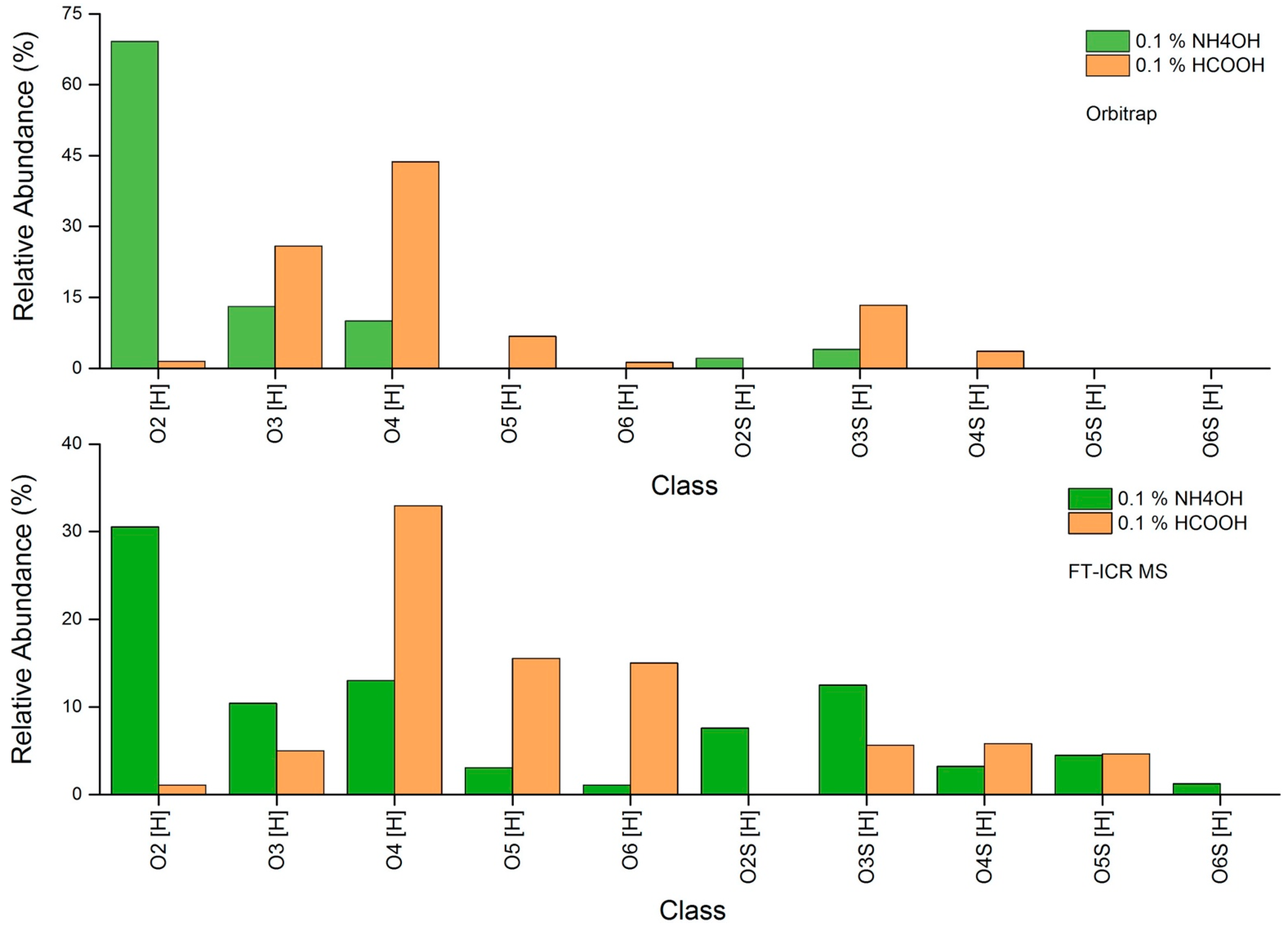
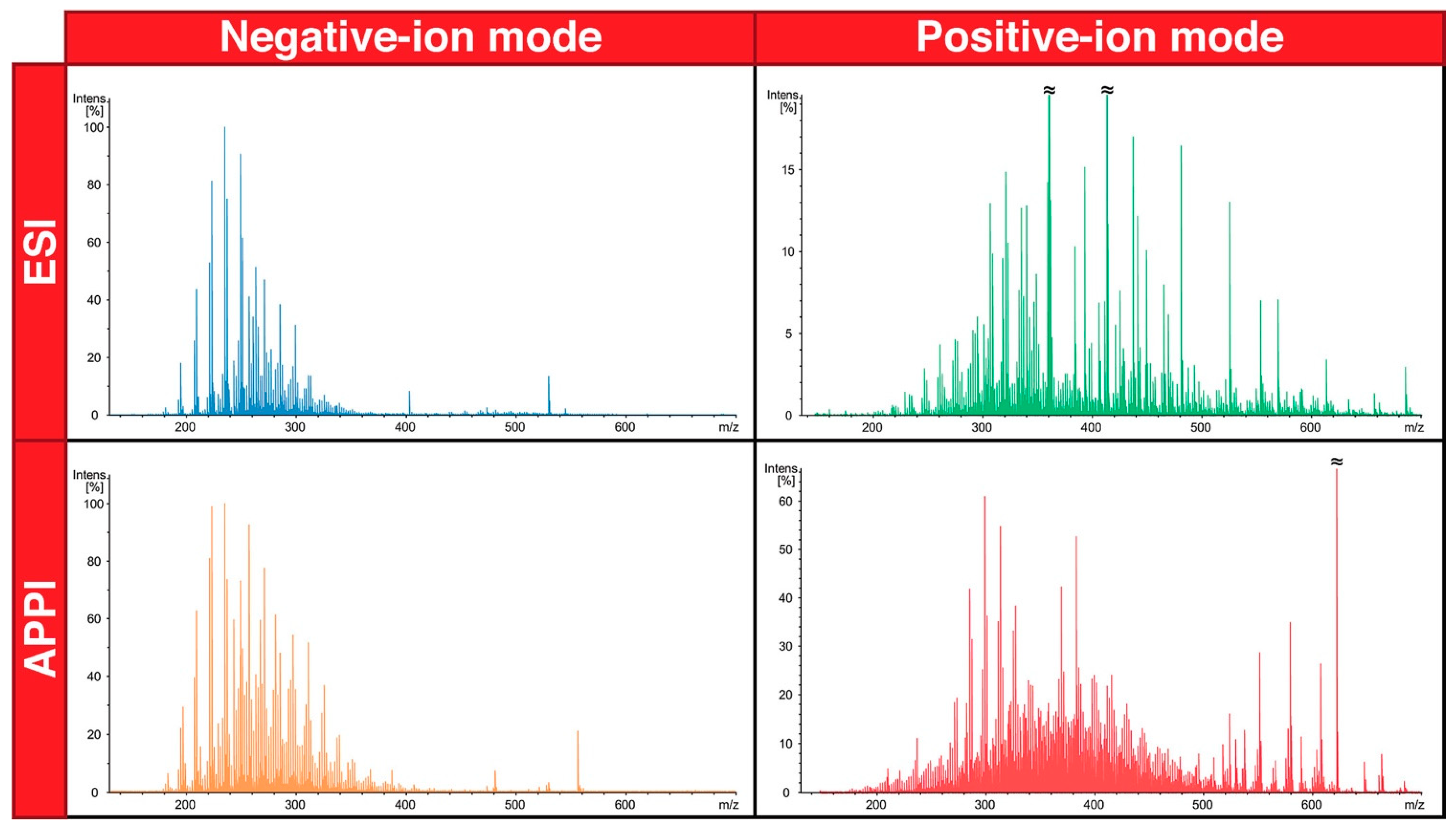
2.7. Ionization Mode for Detection of the Analytes
2.8. Naphthenic Acids vs. Acid Extractable Organics
2.9. Mass Resolution of the Mass Spectrometer Instrument
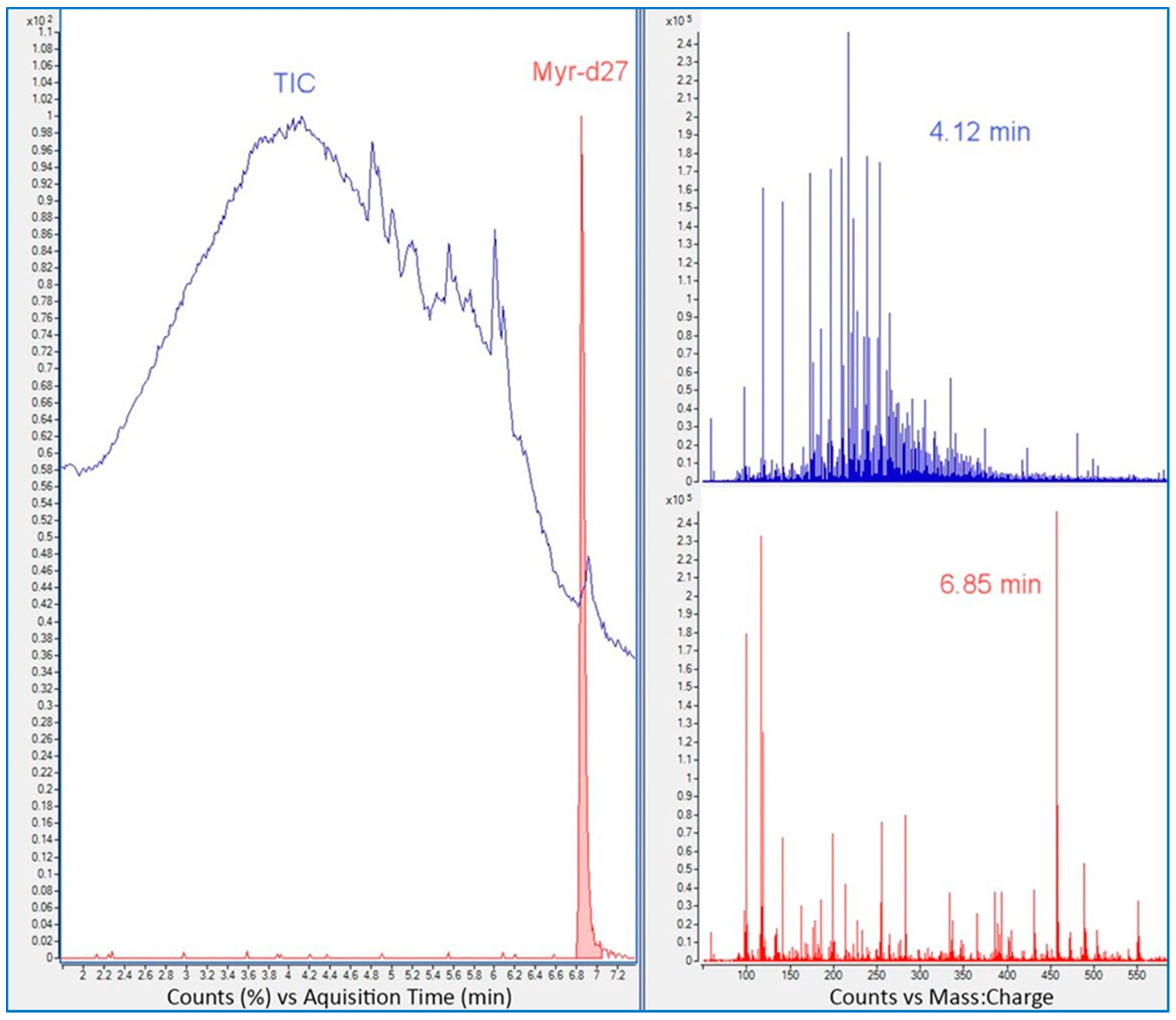
2.10. Use of Internal vs. External Standard
3. Conclusions and Recommendations
- Choice of extraction solvent: If using liquid–liquid extraction, adjust the aqueous pH to ≤1.5 and extract with DCM.
- Salt content of sample: Partition cNAs into a water-immiscible solvent such as DCM or use a wash step with SPE cleanup to avoid salt in the final extract. Use LC and discard the early eluant to waste before directing column eluant to the MS source.
- pH of extraction: Classical NAs have pKa values that range from 3.5 to 6.8. When partitioning into the organic phase, use pH ≤ 1.5. When partitioning into the aqueous phase, use pH ≥ 8.8. A narrow range will reduce the extraction of additional unwanted compounds.
- Sample cleanup: Perform LLE or SPE cleanup prior to MS analysis to reduce interferences and minimize ion suppression.
- On-line chromatography: Use LC separation to reduce interferences, minimize ion suppression, and add a time dimension to compound resolution.
- Calibration standards: Use Merichem® mixture or concentrated NAFC extract from OSPW in acetonitrile.
- Ionization mode: For cNAs, use electrospray negative-ion mode.
- NAs versus NAFCs: Classical NAs are a subset of NAFCs. Use method parameters to focus on recovering and measuring only cNAs.
- Mass resolution: The minimum resolution should be 25,000 when using an LC separation. The infusion should be 50,000 for previously characterized OSPW, and 100,000+ for unknowns.
- Use of internal standard: use an ISTD to monitor instrument performance.
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Canadian Association of Petroleum Producers. 2019 Crude Oil Forecast, Markets and Transportation; Canadian Association of Petroleum Producers: Calgary, AB, Canada, 2019; Volume 26. [Google Scholar]
- Monaghan, J.; Richards, L.C.; Vandergrift, G.W.; Hounjet, L.J.; Stoyanov, S.R.; Gill, C.G.; Krogh, E.T. Direct mass spectrometric analysis of naphthenic acids and polycyclic aromatic hydrocarbons in waters impacted by diluted bitumen and conventional crude oil. Sci. Total Environ. 2021, 765, 144206. [Google Scholar] [CrossRef]
- Allen, E.W. Process water treatment in Canada’s oil sands industry: I. Target pollutants and treatment objectives. J. Environ. Eng. Sci. 2008, 7, 123–138. [Google Scholar] [CrossRef]
- Clemente, J.S.; Fedorak, P.M. A review of the occurrence, analyses, toxicity, and biodegradation of naphthenic acids. Chemosphere 2005, 60, 585–600. [Google Scholar] [CrossRef]
- Morandi, G.D.; Wiseman, S.B.; Pereira, A.; Mankidy, R.; Gault, I.G.M.; Martin, J.W.; Giesy, J.P. Effects-directed analysis of dissolved organic compounds in oil sands process-affected water. Environ. Sci. Technol. 2015, 49, 12395–12404. [Google Scholar] [CrossRef]
- Scarlett, A.G.; West, C.E.; Jones, D.; Galloway, T.S.; Rowland, S.J. Predicted toxicity of naphthenic acids present in oil sands process-affected waters to a range of environmental and human endpoints. Sci. Total Environ. 2012, 425, 119–127. [Google Scholar] [CrossRef]
- Hughes, S.A.; Mahaffey, A.; Shore, B.; Baker, J.; Kilgour, B.; Brown, C.; Peru, K.M.; Headley, J.V.; Bailey, H.C. Using ultrahigh-resolution mass spectrometry and toxicity identification techniques to characterize the toxicity of oil sands process-affected water: The case for classical naphthenic acids. Environ. Toxicol. Chem. 2017, 36, 3148–3157. [Google Scholar] [CrossRef]
- Clemente, J.S.; Prasad, N.G.N.; MacKinnon, M.D.; Fedorak, P.M. A statistical comparison of naphthenic acids characterized by gas chromatography-mass spectrometry. Chemosphere 2003, 50, 1265–1274. [Google Scholar] [CrossRef]
- Headley, J.V.; Peru, K.M.; Mohamed, M.H.; Frank, R.A.; Martin, J.W.; Hazewinkel, R.R.O.; Humphries, D.; Gurprasad, N.P.; Hewitt, L.M.; Muir, D.C.G.; et al. Chemical fingerprinting of naphthenic acids and oil sands process waters—A review of analytical methods for environmental samples. J. Environ. Sci. Health Part A 2013, 48, 1145–1163. [Google Scholar] [CrossRef]
- Yue, S.; Ramsay, B.A.; Wang, J.; Ramsay, J.A. Biodegradation and detoxification of naphthenic acids in oil sands process affected waters. Sci. Total Environ. 2016, 572, 273–279. [Google Scholar] [CrossRef]
- Folwell, B.D.; McGenity, T.J.; Whitby, C. Diamondoids are not forever: Microbial biotransformation of diamondoid carboxylic acids. Microb. Biotechnol. 2020, 13, 495–508. [Google Scholar] [CrossRef]
- Cancelli, A.M.; Gobas, F.A.P.C. Treatment of naphthenic acids in oil sands process-affected waters with a surface flow treatment wetland: Mass removal, half-life, and toxicity-reduction. Environ. Res. 2022, 213, 113755. [Google Scholar] [CrossRef]
- Ajaero, C.; Meulen IVander Simair, M.C.; Roux MLe Parrott, J.; Peru, K.M.; McMartin, D.W.; Headley, J.V. Developments in molecular level characterization of naphthenic acid fraction compounds degradation in a constructed wetland treatment system. Environments 2020, 7, 89. [Google Scholar] [CrossRef]
- Leshuk, T.; Wong, T.; Linley, S.; Peru, K.M.; Headley, J.V.; Gu, F. Solar photocatalytic degradation of naphthenic acids in oil sands process-affected water. Chemosphere 2016, 144, 1854–1861. [Google Scholar] [CrossRef]
- Quinlan, P.J.; Tam, K.C. Water treatment technologies for the remediation of naphthenic acids in oil sands process-affected water. Chem. Eng. J. 2015, 279, 696–714. [Google Scholar] [CrossRef]
- Kovalchik, K.A.; MacLennan, M.S.; Peru, K.M.; Headley, J.V.; Chen, D.D.Y. Standard method design considerations for semi-quantification of total naphthenic acids in oil sands process affected water by mass spectrometry: A review. Front. Chem. Sci. Eng. 2017, 11, 497–507. [Google Scholar] [CrossRef]
- Peru, K.M.; Thomas, M.J.; Palacio Lozano, D.C.; McMartin, D.W.; Headley, J.V.; Barrow, M.P. Characterization of oil sands naphthenic acids by negative-ion electrospray ionization mass spectrometry: Influence of acidic versus basic transfer solvent. Chemosphere 2019, 222, 1017–1024. [Google Scholar] [CrossRef]
- Rogers, V.V.; Liber, K.; MacKinnon, M.D. Isolation and characterization of naphthenic acids from Athabasca oil sands tailings pond water. Chemosphere 2002, 48, 519–527. [Google Scholar] [CrossRef]
- Headley, J.V.; Peru, K.M.; Barrow, M.P.; Derrick, P.J. Characterization of naphthenic acids from Athabasca oil sands using electrospray ionization: The significant influence of solvents. Anal. Chem. 2007, 79, 6222–6229. [Google Scholar] [CrossRef]
- Ripmeester, M.J.; Duford, D.A. Method for routine “naphthenic acids fraction compounds” determination in oil sands process-affected water by liquid-liquid extraction in dichloromethane and Fourier-Transform Infrared Spectroscopy. Chemosphere 2019, 233, 687–696. [Google Scholar] [CrossRef]
- Headley, J.V.; Barrow, M.P.; Peru, K.; Derrick, P.J. Salting-out effects on the characterization of naphthenic acids from Athabasca oil sands using electrospray ionization. J. Environ. Sci. Health Part A 2011, 46, 844–854. [Google Scholar] [CrossRef]
- Celsie, A.; Parnis, J.M.; Mackay, D. Impact of temperature, pH, and salinity changes on the physico-chemical properties of model naphthenic acids. Chemosphere 2016, 146, 40–50. [Google Scholar] [CrossRef]
- Grewer, D.M.; Young, R.F.; Whittal, R.M.; Fedorak, P.M. Naphthenic acids and other acid-extractables in water samples from Alberta: What is being measured? Sci. Total Environ. 2010, 408, 5997–6010. [Google Scholar] [CrossRef]
- Headley, J.V.; Peru, K.M.; McMartin, D.W.; Winkler, M. Determination of dissolved naphthenic acids in natural waters by using negative-ion electrospray mass spectrometry. J. AOAC Int. 2002, 85, 182–187. [Google Scholar] [CrossRef]
- Huang, R.; Sun, N.; Chelme-Ayala, P.; McPhedran, K.N.; Changalov, M.; Gamal El-Din, M. Fractionation of oil sands-process affected water using pH-dependent extractions: A study of dissociation constants for naphthenic acids species. Chemosphere 2015, 127, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Barrow, M.P.; Witt, M.; Headley, J.V.; Peru, K.M. Athabasca oil sands process water: Characterization by atmospheric pressure photoionization and electrospray ionization Fourier Transform Ion Cyclotron Resonance mass spectrometry. Anal. Chem. 2010, 82, 3727–3735. [Google Scholar] [CrossRef]
- Pereira, A.S.; Bhattacharjee, S.; Martin, J.W. Characterization of oil sands process-affected waters by liquid chromatography orbitrap mass spectrometry. Environ. Sci. Technol. 2013, 47, 5504–5513. [Google Scholar] [CrossRef] [PubMed]
- Qin, R.; Lillico, D.; How, Z.T.; Huang, R.; Belosevic, M.; Stafford, J.; Gamal El-Din, M. Separation of oil sands process water organics and inorganics and examination of their acute toxicity using standard in-vitro bioassays. Sci. Total Environ. 2019, 695, 133532. [Google Scholar] [CrossRef]
- Marshall, A.G.; Blakney, G.T.; Chen, T.; Kaiser, N.K.; McKenna, A.M.; Rodgers, R.P.; Ruddy, B.M.; Xian, F. Mass resolution and mass accuracy: How much is enough? Mass Spectrom. 2013, 2, S0009. [Google Scholar] [CrossRef]
- Feehan, J.F.; Monaghan, J.; Gill, C.G.; Krogh, E.T. Direct measurement of acid dissociation constants of trace organic compounds at nanomolar levels in aqueous solution by condensed phase–membrane introduction mass spectrometry. Environ. Toxicol. Chem. 2019, 38, 1879–1889. [Google Scholar] [CrossRef]
- Duncan, K.D.; Richards, L.C.; Monaghan, J.; Simair, M.C.; Ajaero, C.; Peru, K.M.; Friesen, V.; McMartin, D.W.; Headley, J.V.; Gill, C.G.; et al. Direct analysis of naphthenic acids in constructed wetland samples by condensed phase membrane introduction mass spectrometry. Sci. Total Environ. 2020, 716, 137063. [Google Scholar] [CrossRef] [PubMed]
- Hindle, R.; Noestheden, M.; Peru, K.; Headley, J. Quantitative analysis of naphthenic acids in water by liquid chromatography–accurate mass time-of-flight mass spectrometry. J. Chromatogr. A 2013, 1286, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Brunswick, P.; Shang, D.; van Aggelen, G.; Hindle, R.; Hewitt, L.M.; Frank, R.A.; Haberl, M.; Kim, M. Trace analysis of total naphthenic acids in aqueous environmental matrices by liquid chromatography/mass spectrometry-quadrupole time of flight mass spectrometry direct injection. J. Chromatogr. A 2015, 1405, 49–71. [Google Scholar] [CrossRef]
- Hughes, S.A.; Huang, R.; Mahaffey, A.; Chelme-Ayala, P.; Klamerth, N.; Meshref, M.N.A.; Ibrahim, M.D.; Brown, C.; Peru, K.M.; Headley, J.V.; et al. Comparison of methods for determination of total oil sands-derived naphthenic acids in water samples. Chemosphere 2017, 187, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Hindle, R.; (Vogon Laboratory Services, Cochrane, AB, Canada). Manuscript in preparation. 2023.
- Canadian Council of Ministers of the Environment. Canadian Water Quality Guidelines for the Protection of Aquatic Life: Ammonia; Canadian Environmental Quality Guidelines; Canadian Council of Ministers of the Environment: Winnipeg, MB, Canada, 2010; Volume 8. [Google Scholar]
- Kind, T.; Fiehn, O. Metabolomic database annotations via query of elemental compositions: Mass accuracy is insufficient even at less than 1 ppm. BMC Bioinform. 2006, 7, 234. [Google Scholar] [CrossRef]
- Bowman, D.T.; Warren, L.A.; Slater, G.F. Isomer-specific monitoring of naphthenic acids at an oil sands pit lake by comprehensive two-dimensional gas chromatography–mass spectrometry. Sci. Total Environ. 2020, 746, 140985. [Google Scholar] [CrossRef] [PubMed]
- McEwen, C.N.; Larsen, B.S. Ionization mechanisms related to negative ion APPI, APCI, and DART. J. Am. Soc. Mass Spectrom. 2009, 20, 1518–1521. [Google Scholar] [CrossRef]
- Raffaelli, A.; Saba, A. Atmospheric pressure photoionization mass spectrometry. Mass Spectrom. Rev. 2003, 22, 318–331. [Google Scholar] [CrossRef]
- Brunswick, P.; Hewitt, L.M.; Frank, R.A.; Kim, M.; van Aggelen, G.; Shang, D. A traceable reference for direct comparative assessment of total naphthenic acid concentrations in commercial and acid extractable organic mixtures derived from oil sands process water. J. Environ. Sci. Health Part A 2017, 52, 274–280. [Google Scholar] [CrossRef]
- Hindle, R.; (Vogon Laboratory Services, Cochrane, AB, Canada). Manuscript in preparation. 2023.
- Huang, R.; Chen, Y.; Meshref, M.N.A.; Chelme-Ayala, P.; Dong, S.; Ibrahim, M.D.; Wang, C.; Klamerth, N.; Hughes, S.A.; Headley, J.V.; et al. Characterization and determination of naphthenic acids species in oil sands process-affected water and groundwater from oil sands development area of Alberta, Canada. Water Res. 2018, 128, 129–137. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Factor | Conclusions |
|---|---|---|
| 1 | Definition of total NAs | Use the classical definition of NAs |
| 2 | Extraction phase, pH, temperature | Liquid–liquid extraction at pH 2 and room temperature with DCM as organic phase, or use ENV + SPE |
| 3 | Use of surrogate standards | Use isotopically labelled model compounds as surrogate standards |
| 4 | Minimum resolving power of instrument | 50,000 at m/z 200, acknowledging that potential interferences contribute to method uncertainty |
| 5 | Use of derivatization | Do not utilize derivatization |
| 6 | Polarity and mode of ionization | Negative-ion-mode ESI |
| 7 | Suitable calibration standard and internal standard | Use commercially available Merichem NA mixture and at least one isotopically labelled internal standard |
| 8 | Use of on-line or off-line fractionation of sample | Employ on-line chromatography prior to MS detection |
| Compound | Additive | Intensity | Apparent pH |
|---|---|---|---|
| Biphenyl-4-carboxylic acid | 0.1% NH4OH | 5.34 × 1010 | 8.91 |
| C13H10O2 | 0.1% HCOOH | 4.79 × 108 | 3.52 |
| Anthraquinone-2-carboxylic acid | 0.1% NH4OH | 5.46 × 1010 | 9.19 |
| C15H8O4 | 0.1% HCOOH | 3.31 × 109 | 3.68 |
| Trimesic acid | 0.1% NH4OH | 2.81 × 1010 | 8.75 |
| C9H6O6 | 0.1% HCOOH | 1.86 × 1010 | 3.59 |
| Ion Formula | m/z (Calc) | Diff (ppm) | Resolution |
|---|---|---|---|
| C9H25N6O2S | 281.1765 | 0.8 | 1,406,000 |
| C16H25O4 | 281.1758 | −1.7 | 562,000 |
| C17H21N4 | 281.1772 | 3.1 | 312,000 |
| C11H27N3O3S | 281.1779 | 5.5 | 176,000 |
| C14H23N3O3 | 281.1745 | −6.4 | 156,000 |
| C19H23N O | 281.1785 | 7.8 | 128,000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hindle, R.; Headley, J.; Muench, D.G. Pros and Cons of Separation, Fractionation and Cleanup for Enhancement of the Quantitative Analysis of Bitumen-Derived Organics in Process-Affected Waters—A Review. Separations 2023, 10, 583. https://doi.org/10.3390/separations10120583
Hindle R, Headley J, Muench DG. Pros and Cons of Separation, Fractionation and Cleanup for Enhancement of the Quantitative Analysis of Bitumen-Derived Organics in Process-Affected Waters—A Review. Separations. 2023; 10(12):583. https://doi.org/10.3390/separations10120583
Chicago/Turabian StyleHindle, Ralph, John Headley, and Douglas G. Muench. 2023. "Pros and Cons of Separation, Fractionation and Cleanup for Enhancement of the Quantitative Analysis of Bitumen-Derived Organics in Process-Affected Waters—A Review" Separations 10, no. 12: 583. https://doi.org/10.3390/separations10120583
APA StyleHindle, R., Headley, J., & Muench, D. G. (2023). Pros and Cons of Separation, Fractionation and Cleanup for Enhancement of the Quantitative Analysis of Bitumen-Derived Organics in Process-Affected Waters—A Review. Separations, 10(12), 583. https://doi.org/10.3390/separations10120583