Abstract
A drug-excipient interaction impurity associated with the degradation process of pramipexole was isolated. The impurity was detected during the stability study of pramipexole extended-release tablets. It was found at a relative retention time of 0.88 with respect to pramipexole, using the pramipexole gradient HPLC-UV detection method described in the USP. The structure of the impurity was identified and fully characterized using high resolution mass spectrometry, IR and NMR techniques, as presented herein. The degradation impurity was identified as (S)-N2-(methoxymethyl)-N6-propyl-4,5,6,7-tetrahydro-1,3-benzothiazole-2,6-diamine. The drug-excipient interaction mechanism of its formation was proposed. An efficient and straightforward synthetic approach was developed to prepare the degradation impurity to confirm its proposed degradation pathway and structure.
1. Introduction
The pramipexole dihydrochloride monohydrate is a non-ergot dopamine agonist [1] mainly utilized to cure symptoms of Parkinson’s disease (PD) [2]. Pramipexole is approved as a medication to treat PD based on its ability to stimulate dopamine receptors in the striatum [3]. Additionally, pramipexole is used for restless legs syndrome (RLS). It has both dopaminergic and neuroprotective effects; hence it is used as an antidepressant as well [4,5]. Pramipexole was approved in the USA in 1997 and sold under many brands, such as Mirapex® (Boehringer Ingelheim Pharmceuticals, Inc., Rigefield, CT, USA). Its chemical name is (S)-2-amino-4,5,6,7-tetrahydro-6-(propylamino) benzothiazole dihydrochloride monohydrate (C10H17N3S•2HCl•H2O) with a molecular weight of 302.72 Da. Pramipexole dihydrochloride monohydrate powder is white, tasteless and crystalline. Pramipexole is a chiral compound with one chiral center (Figure 1). Its melting point ranges from 296 °C to 301 °C with decomposition.
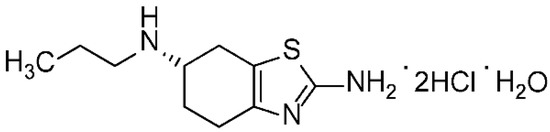
Figure 1.
Chemical structure of pramipexole dihydrochloride monohydrate.
Stability testing is critical when developing pharmaceutical products. When drugs degrade over time, even under suitable storage conditions, their efficacy and safety might be affected. Hence, impurity identification and profiling are given special attention by regulatory authorities. The impurities presented in pharmaceuticals must be identified, quantified and controlled. Many recent articles [6,7,8,9] describe and design approaches to isolating and identifying the process-related impurities of many drugs using different techniques.
Several studies have been published describing different methods for identifying and evaluating the organic impurities and related compounds of the pramipexole active pharmaceutical ingredient (API) and its corresponding formulated drug products. These methods are mainly based on the use of reversed-phase high-pressure liquid chromatography (HPLC) coupled with UV detection [10,11,12,13,14,15]. The synthesis of some of the pramipexole related impurities was also described in several of these published studies. Generally, pramipexole impurities are produced during the synthesis of pramipexole API, either from the starting materials, from the intermediates or as a by-product of the API manufacturing process. Impurities can also result from product degradation during drug product preparation/manufacturing due to exposure to unfavorable processing conditions or during drug product storage. The originator reported seventeen impurities in the quality inspection standards for pramipexole dihydrochloride tablets; twelve impurities are known as non-pharmacopeial pramipexole-related compounds (Figure 2), and the remaining five impurities (Table 1) are identified in the United State Pharmacopeia (USP) and European Pharmacopeia (Ph. Eur.) [2].
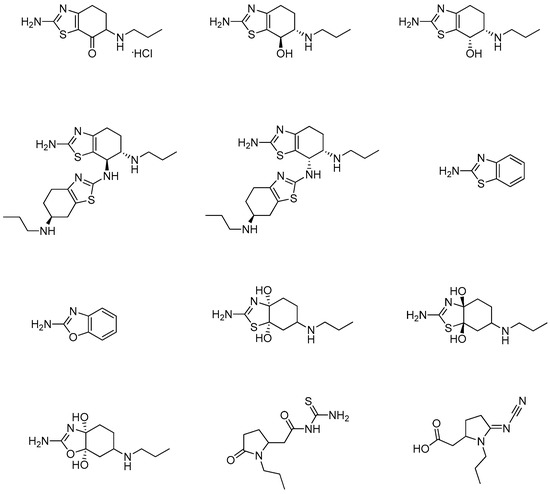
Figure 2.
Chemical structures of non-pharmacopeial pramipexole related compounds [2].

Table 1.
Structures of pharmacopeial pramipexole related compounds.
The USP and Ph. Eur. monographs for the API and the British Pharmacopeia (BP) monograph for the drug product use the same chromatographic method to determine the related organic impurities of pramipexole. The compendial methods describe the use of a reversed-phase C18 column with an ion-paired mobile phase (gradient) coupled to a UV detector.
During the development of a generic formula for pramipexole extended released tablet, stability studies were conducted to ensure the drug product’s safety and quality and to determine potential degradation products. When the USP method of analysis was implemented, a degradation impurity was evident at the relative retention time (RRT) of 0.88, with limits exceeding the acceptable limit for unidentified impurities as per the requirements of the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) Q3B(R2) Guideline [16].
This work describes, for the first time, the isolation, structure elucidation and retro-synthesis of the RRT 0.88 degradation impurity of pramipexole. This will help the pharmaceutical labs worldwide to prepare and use this impurity as a standard in the analytical studies and quality control protocols of the pramipexole drug product.
2. Materials and Methods
The pramipexole dihydrochloride (>98%) was purchased from Sigma-Aldrich (Steinheim, Germany). Pramipexole dihydrochloride extended-release tablets were manufactured by the Resonance Research Lab (Amman, Jordan). HPLC-grade acetonitrile (99.99%), sodium 1-octanesulfonate and phosphoric acid (85%, w/w) were bought from Merck (Darmstadt, Germany); potassium dihydrogen phosphate was purchased from Quality Reagent Chemicals (QReC, Auckland, New Zealand). Formalin 37% (stabilized with 10–15% methanol), methanol and dichloromethane (DCM) were purchased from TEDIA (Fairfield, OH, USA). The reaction was monitored using thin-layer chromatography (TLC) on silica gel plates (60 F254) from Merck (Darmstadt, Germany). Column chromatography was performed using Merck’s silica gel “Geduran Si 60” (0.063–0.200 mm). The ultrapure water used in the preparation of the HPLC mobile phases (resistivity, 18.2 MΩ cm) was generated by the Millipore water purification system (Molsheim, France).
1H, 13C and 2D-NMR spectra were obtained using a Bruker DPX-500 (Bruker Avance-III) (Bruker, Germany). Samples were dissolved in DMSO-d6 (Merck (Darmstadt, Germany), 99.8%), and TMS was used as an internal standard.
High-resolution mass spectra (HRMS) were obtained utilizing an APEX-IV (7 Tesla) instrument (Bruker, Switzerland) in the positive ESI mode and chloroform-methanol as solvent.
IR spectra were recorded on a Nicolet Nexus 670 FTIR spectrometer (Thermo Nicolet, Thermo Fisher Scientific Inc., Waltham, MA, USA) using a reflection sampling accessory (ATR).
The HPLC separation was performed by applying the USP method for the pramipexole drug product using a Dionex UltiMate 3000 HPLC system (Dionex, Idstein, Germany) supplied with an UltiMate 3000 Pump with the flow set to 1.5 mL/min, a 3000 Autosampler and a column compartment set at 40 °C. The volume of injection was 5 µL. The separation was carried out using an InertSil ODS-2 C18 column (250 mm × 4.6 mm, 5 µm) purchased from GL Sciences (Tokyo, Japan). Chromeleon software (version 7.1) was used to process and analyze the data. Mobile phase A was a mixture of phosphate buffer (66.9 mM) and sodium 1-octanesulfonate (21.3 mM) with pH adjusted to 3.0. Mobile phase B was a mixture of acetonitrile and mobile phase A (1:1, v/v). Each mobile phase was degassed for 10 min. after filtering it using a nylon membrane filter (0.22 mm) purchased from Agilent Technologies (Waldbronn, Germany). The mobile phase B ratio was changed from 40–80% B in 15 min, and then the column was re-equilibrated with the initial B ration (40%) for 5 min.
The LC-MS analysis was carried out using an HPLC system from Agilent 1290 Infinity coupled with a TOF/Q-TOF mass spectrometer from Agilent Technologies 6540 UHD (USA). The chromatographic separation was accomplished using a Zorbax Eclipse Plus C18, (50 mm × 2.1 mm × 1.8 µm) column obtained from Agilent. The method of our impurity identification was developed in-house and utilized mobile phase A: 0.1% aqueous formic acid solution and mobile phase B: 0.1% formic acid in acetonitrile. The flow rate was set at 0.6 mL/min., and %B was kept constant at 5% during 0–4 min before a linear gradient was applied from 4–5.1 min to increase the %B from 5% to 98%. The following parameters were applied for MS: +ve ESI; capillary voltage: 3500 V; N2 flow: 8 L/min; nebulizer pressure: 35 psi and capillary temperature 320 °C.
2.1. Pramipexole Extended-Release Tablet Formulation and Sample Preparation
The unit composition of pramipexole dihydrochloride extended-release tablets of 0.375, 0.75, 1.5, 2.25, 3.0, 3.75 and 4.5 mg is listed in Table S1.
Ten pramipexole 0.375 mg tablets were crushed and mixed; then an amount of the powder was transferred to a volumetric flask to prepare a 1.5 mg/mL solution of pramipexole using a mixture of 20:80 (EtOH:Mobile phase A) as a solvent. All samples were filtrated using a 0.45 µm syringe nylon filter (Millipore, Bedford, MA, USA) before being injected into the HPLC system.
2.2. Synthesis of (S)-N2-(Methoxymethyl)-N6-Propyl-4,5,6,7-Tetrahydro-1,3-Benzothiazole-2,6-Diamine
In a 100 mL beaker, (S)-2-amino-4,5,6,7-tetrahydro-6-(propylamino) benzothiazole dihydrochloride monohydrate (pramipexole API) (2.5 g, 8.3 mmol) was dissolved in 9.0 mL of water, and the resulting solution was basified using 2.0 N NaOH (pH 10.0) to form a white slurry material. The crude mixture was then extracted three times with a mixture of (30 mL DCM and 1 mL MeOH). The combined organic phases were collected and allowed to concentrate under reduced pressure to furnish a white solid material (2.1 g).
The resulting pramipexole-free base was dissolved in 25.0 mL of MeOH. Formalin solution (1.0 mL, 37% in water) was added dropwise over 10 min, and then 0.5 mL of acetic acid was added, and the reaction mixture was stirred for 10 hrs at room temperature. The reaction was followed by HPLC and by TLC. In TLC, the Rf value of the starting free base was 0.29, while the product Rf value was 0.54 in DCM: MeOH (5:1). At the beginning, a spot with a lower Rf value than the starting material appeared, but then a second spot with a higher Rf value gradually formed, and it was identified as the target compound. The clear reaction mixture was evaporated under reduced pressure, keeping the water bath at 30 °C. The resulting crude product was then subjected to column chromatography using silica gel and an eluent mixture of DCM: MeOH (10:1, changed gradually to 5:1). The target compound (Figure 3) was finally obtained as a white solid (1.0 g, yield = 47%).
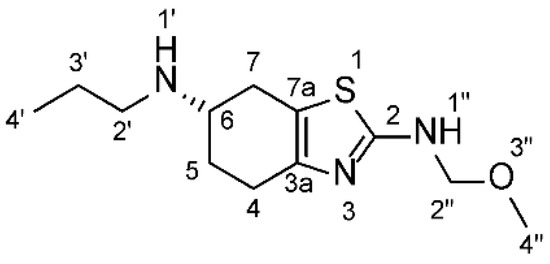
Figure 3.
Chemical structure of the synthesized impurity ((S)-N2-(Methoxymethyl)-N6-propyl-4,5,6,7-tetrahydro-1,3-benzothiazole-2,6-diamine.
H NMR (DMSO-d6, 500 MHz): δ = 0.92 (t, 3H, H-4′), 1.63 (m, 2H, H-3′), 1.84, 2.18, 2.56, 2.68, 3.02 (m, 6H, H-4, H-5, H-7), 2.90 (m, 2H, H-2′), 3.16 (bs, 3H, H-4″), 3.41 (1H, H-6), 4.54 (m, 2H, H-2″), 8.30 (m, 1H, NH-1″), 8.89 (bs, 1H, NH-1′) ppm.
13C NMR (DMSO-d6, 125 MHz): δ = 11.11 (C-4′), 19.14 (C-3′), (24.34, 25.14, 25.22 (C-4, C-5, C-7)), 45.85 (C-2′), 53.32 (C-6), 54.42 (C-4″, (OCH3)), 75.71 (C-2″, (OCH2)), 111.62 (C-7a), 144.46 (C-3a), 166.07 (C-2) ppm.
HRMS: calcd. for C12H22N3OS [M+H]+ 256.14781; found = 256.14863.
IR (neat): 3167, 2941, 2815, 2727, 2513, 2423, 1560, 1531, 1458, 1418, 1377, 1280, 1128, 1077, 1038, 1019, 983, 912, 854, 759, 714, 605 cm−1.
3. Results and Discussion
3.1. Detection of the Degradation Impurity
A significant degradation impurity with a relative retention time (RRT) 0.88 (Figure 4) with respect to pramipexole was detected during the stability study of the drug product using the organic impurity test described in the USP monograph for pramipexole drug products. The compendial HPLC method uses a reversed-phase C18 column with an ion-paired mobile phase (gradient) coupled to a UV detector [17]. The level of the target degradation impurity in the final drug product formulation increased from less than 0.1% relative to the drug substance at the initial testing point to more than 1.5% after 12 months when the drug product was stored at 30 °C/65% RH conditions (long term storage), which is more than the acceptable level for unidentified impurities according to the ICH guidelines [16]. According to the ICH, the acceptable level for unidentified impurities is 0.5%, calculated based on a 1.5 mg total daily intake (TDI) [4]. Therefore, it was necessary to identify the chemical structure of this degradation impurity.
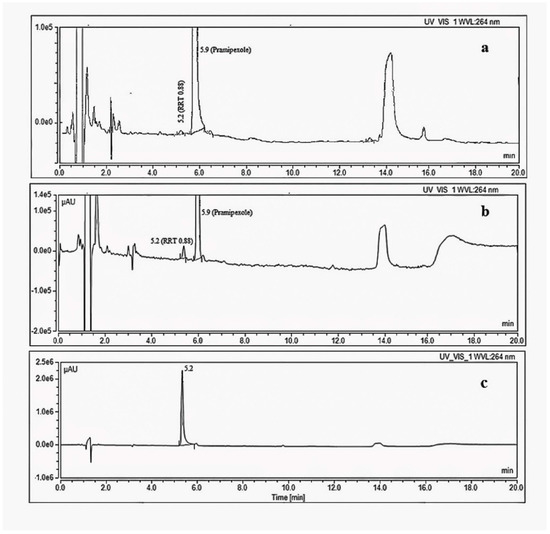
Figure 4.
HPLC-UV chromatograms using organic impurity test method in USP monograph of pramipexole (a) Initial stability sample, 1.5 mg·mL−1 (b) stability sample stored for 12 months at 30 °C/65%RH, 1.5 mg·mL−1 (c) 0.5 mg/mL solution of the synthesized degradation impurity.
3.2. Separation and Retrosynthesis of the Degradation Impurity (RRT 0.88) from Pramipexole API
The pramipexole degradation impurity at RRT 0.88 was isolated from sample solution using HPLC. This was followed by applying a new LC-MS method specifically developed to identify this impurity. The USP test procedure calls for using an ion-pair reagent that is unsuitable for LC-MS analysis due to the high buffer content in the mobile phase. Therefore, a new analysis method compatible with LC-MS was developed, and the impurity at RRT 0.88 was analyzed and identified using the newly developed LC-MS method (Figure 5a). The mass spectrum of the degradation impurity was obtained using LC-MS-QTOF (Figure 5b). A molecular ion peak [M+H]+ at m/z 256.1483 is consistent with the molecular formula C12H22N3OS for [M+H]+.
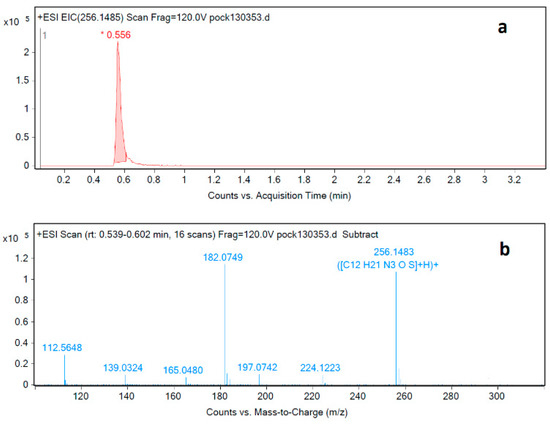
Figure 5.
LC-MS-QTOF (a) chromatogram and (b) MS spectrum corresponding to injection of degradation impurity identification solution.
By analyzing the LC-MS mass spectrum of the target impurity, a new methoxymethyl group was recognized as part of the overall impurity structure. A full literature review revealed that a synthetic method for producing similar compounds has been reported by Ji et al. [18]. The reported synthetic approach shows that the N-methoxymethyl moiety can be synthesized from its corresponding amine by the condensation of the amine with formalin and methanol, which are considered as solvents and reactants (the source of methoxymethyl group) at the same time (Figure S1). The reaction conditions reported by Ji et al. for the synthesis of N-methoxymethyl moieties were applied in the case of pramipexole hydrochloride. In this case, pramipexole is considered the source of the primary amine group needed for such reaction.
To confirm the suitability of the approach employed by Ji et al., pramipexole-free base samples were stressed in the presence of methanol and formalin at room temperature. An increase in the intensity of the impurity at RRT 0.88 was observed. As a result, the target impurity was thought to be an N-methoxymethyl pramipexole impurity.
To scale-up the target impurity for further analysis and assessment, the reaction described by Ji et al. was taken further. The impurity was synthesized and purified using the pramipexole-free base as the starting material; the free base pramipexole was reacted with formalin (formalin 37% stabilized with 10–15% methanol), and acetic acid was present as a catalyst (Figure 6). The reaction resulted in the target degradation impurity at a yield of 40%. The synthesized impurity was injected into HPLC and analyzed according to the USP method. The relative retention time matched the expected RRT of 0.88. The synthesized impurity was then characterized based on IR, HRMS, 1H/13C NMR, DEPT135 and 2D NMR spectra (Figures S2–S11).
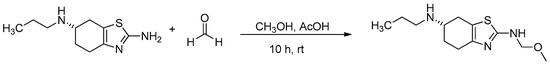
Figure 6.
Synthesis of (S)-N2-(methoxymethyl)-N6-propyl-4,5,6,7-tetrahydro-1,3-benzothiazole-2,6-diamine.
3.3. Proposed Source and Mechanism of Formation of Pramipexole N-Methoxymethyl Degradation Impurity (RRT 0.88) in Pramipexole Tablets
It is known that the poor stability of the prior art compositions of pramipexole dihydrochloride can be attributed to the presence of celluloses or any other excipients that may contain trace levels of formalin. Formalin can catalyze the conversion of pramipexole dihydrochloride to its corresponding imine (Figure 7) [19,20]. The specific cellulose used as an excipient in the tablet formulation analyzed in this research is hydroxypropyl methyl cellulose (HPMC), a well-known source for the methoxy group adding to the formed imine intermediate. Taking all excipient and formulation related information into consideration, the proposed degradation pathway is described in Figure 8. It should be noted that the tablet formula includes other excipients, as described in Table S1. Apart from HPMC, none of these excipients contribute to the identified reaction.
Figure 7.
Chemical structure of the pramipexole imine related compound.
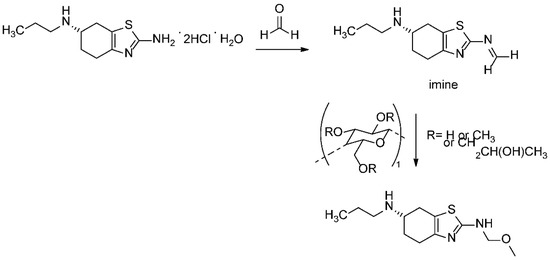
Figure 8.
The proposed formation mechanism of pramipexole N-methoxymethyl degradation impurity.
To prove the proposed mechanism, a solution of HPMC and pramipexole was prepared, and then formalin was added; the reaction mixture was then set at 120 °C for 1 h, and the aliquot of the reaction mixture was injected into the HPLC using the same USP method. The pramipexole N-methoxymethyl degradation impurity (RRT 0.88) was isolated and observed (Figure S12).
3.4. Structure Elucidation of the Degradation Impurity at RRT 0.88
The pramipexole 1H-NMR and 13C-NMR spectrum analysis of the degradation product reveals two new peaks in each spectrum over the 1H-NMR and 13C-NMR spectrum of the corresponding pramipexole API. In 1H-NMR, two peaks were at 3.16 ppm, which represents 3H of (OCH3) of the added methoxy group, and at 4.54 ppm which represents 2H of (OCH2) of the added methoxy methyl group. The 13C-NMR new peaks related to the degradation product were observed at 54.42 ppm, which corresponds to CH3 according to DEPT135, and 75.71 ppm, which corresponds to CH2 according to DEPT135.
The 2D-NMR experiments HMBC, HMQC and COSY were also employed in this work to further confirm the proposed structure of the RRT 0.88 degradation impurity. The NMR spectra of both pramipexole and the degradation products are given in the supporting information (Figures S3–S12). The structure elucidation results collected from the HRMS and NMR experiments indicate that the isolated degradation impurity at RRT 0.88 is (S)-N2-(methoxymethyl)-N6-propyl-4,5,6,7-tetrahydro-1,3-benzothiazole-2,6-diamine (Figure 5a). To the authors’ knowledge, the identification and synthesis of this degradation impurity have not been previously reported in the literature.
4. Conclusions
A degradation impurity of pramipexole was identified, synthesized and fully characterized using different techniques. The RRT 0.88 degradant was observed during product storage under different temperature and humidity conditions as part of the finished dosage form stability program. The degradation impurity was identified as (S)-N2-(methoxymethyl)-N6-propyl-4,5,6,7-tetrahydro-1,3-benzothiazole-2,6-diamine. The pramipexole N-methoxymethyl degradation impurity was synthesized, and the root cause for its formation and its degradation pathway were identified.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/separations10010007/s1, Figure S1: Synthesis of N-(methoxymethyl)benzothiazole reported by Z. Ji et al./Bioorg. Med. Chem. Lett. 25 (2015) 4065–4068; Figure S2: High-Resolution MS spectrum of the synthesized impurity; Figure S3: 1H-NMR spectrum (in DMSO-d6) of the synthesized impurity; Figure S4: 1H-NMR spectrum (in DMSO-d6) of the pramipexole API; Figure S5: 13C NMR spectrum (in DMSO-d6) of the synthesized impurity; Figure S6: 13C NMR spectrum (in DMSO-d6) of the pramipexole API; Figure S7: DEPT-135 spectrum of the synthesized impurity; Figure S8: DEPT-135 spectrum of the pramipexole API; Figure S9: HMQC spectrum of the synthesized impurity; Figure S10: COSY spectrum of the synthesized impurity; Figure S11: HMBC spectrum of the synthesized impurity; Figure S12: HPLC-UV chromatograms using organic impurity test method in USP monograph of pramipexole corresponding to injection of reaction mixture of HPMC and pramipexole in presence of formaldehyde; temperature: 120 °C; time: 2 h; Table S1: Unit composition of pramipexole dihydrochloride extended-release tablets.
Author Contributions
A.A. and F.D. contributed to the design and implementation of the research; N.A.-R. and A.R. to the analysis of the results and to the writing of the manuscript; B.S., J.Z. and S.M. contributed by editing the manuscript and providing needed resources; A.A. and F.D. conceived the original idea and supervised the project. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Deanship of scientific research at German Jordanian University, Grant number 01/2018 and the Deanship of scientific research at Zarqa University.
Data Availability Statement
Not applicable.
Acknowledgments
Thanks to Huda Kafafi for assistance with the organic synthesis as a research assistant from German Jordanian University.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Krishna, G.V.; Kumar, S.K.; Reddy, A.M.; Rajeswari, J. A new validated stability indicating ion-pair hplc method for evaluation of impurities of pramipexole from low dose extended release formulation. Asian J. Chem. 2017, 29, 923–928. [Google Scholar] [CrossRef]
- Hu, T.; Yang, F.; Jiang, T.; Chen, W.; Zhang, J.; Li, J.; Jiang, X.; Shen, J. Synthesis of Impurities of Pramipexole Dihydrochloride. Org. Process Res. Dev. 2016, 20, 1899–1905. [Google Scholar] [CrossRef]
- Pawar, D.S.; Dole, M.N.; Sawant, S.D. Application of stability indicating high performance thin layer chromatographic method for quantitation of pramipexole in pharmaceutical dosage form. Int. J. Res. Pharm. Sci. 2013, 4, 183–186. [Google Scholar] [CrossRef]
- Biglan, K.M.; Holloway, R.G. A review of pramipexole and its clinical utility in Parkinson’s disease. Expert Opin. Pharmacother. 2002, 3, 197–210. [Google Scholar] [PubMed]
- Aiken, C.B. Pramipexole in psychiatry: A systematic review of the literature. J. Clin. Psychiatry 2007, 68, 1231–1237. [Google Scholar] [CrossRef]
- Zhao, M.; Wu, X.; Yu, Z.; Sun, Y.; Liu, Z.; Yuan, J.; Liu, H.; Jin, Y. Identification, synthesis and characterization of avanafil process impurities and determination by UPLC. RSC Adv. 2022, 12, 9256–9262. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wang, Y.; Wang, H.; Zheng, Z.; Meng, Z.; Xue, M.; Xu, Z. Separation and identification of an impurity from the istradefylline intermediate. RSC Adv. 2020, 10, 14493–14499. [Google Scholar] [CrossRef] [PubMed]
- Manoel, J.W.; Primieri, G.B.; Bueno, L.M.; Wingert, N.R.; Volpato, N.M.; Garcia, C.V.; Schapoval, E.E.S.; Steppe, M. The application of quality by design in the development of the liquid chromatography method to determine empagliflozin in the presence of its organic impurities. RSC Adv. 2020, 10, 7313–7320. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, X.; Yan, Y.; Tu, Y.; Feng, X.; Jiang, W.; Zheng, F. Identification and genotoxicity evaluation of two carbamate impurities in rasagiline. RSC Adv. 2016, 6, 106268–106274. [Google Scholar] [CrossRef]
- Malenović, A.; Jančić-Stojanović, B.; Vemić, A.; Ivanović, D.; Medenica, M. Validation of a Column Liquid Chromatographic Method for the Analysis of Pramipexole and Its Five Impurities. J. AOAC Int. 2010, 93, 1102–1112. [Google Scholar] [CrossRef]
- Jančić, B.; Medenica, M.; Ivanović, D.; Malenović, A. Experimental design in chromatographic analysis of pramipexole and its impurities. Acta Chim. Slov. 2007, 54, 49–54. [Google Scholar]
- Jančić, B.; Medenica, M.; Ivanović, D.; Malenović, A. Chromatographic determination of dissociation constants of pramipexole and its impurities. Chromatographia 2007, 65, 633–635. [Google Scholar] [CrossRef]
- Vemić, A.; Rakić, T.; Malenović, A.; Medenica, M. Chaotropic salts in liquid chromatographic method development for the determination of pramipexole and its impurities following quality-by-design principles. J. Pharm. Biomed. Anal. 2015, 102, 314–320. [Google Scholar] [CrossRef]
- Łaszcz, M.; Trzcińska, K.; Kubiszewski, M.; Kosmacińska, B.; Glice, M. Stability studies and structural characterization of pramipexole. J. Pharm. Biomed. Anal. 2010, 53, 1033–1036. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.H.; Stanic, M.; Molnár, I. In silico robustness testing of a compendial HPLC purity method by using of a multidimensional design space build by chromatography modeling—Case study pramipexole. J. Pharm. Biomed. Anal. 2014, 91, 97–107. [Google Scholar] [CrossRef]
- International Council on Harmonisation of Technical Requirements for Registration Pharmaceuticals Human Use.
- United State Pharmacopiea 1st Suppl. 2020, DocID: GUID-8F1C63DC-DE51-4104-9869-4D463A4E0B84_4.
- Ji, Z.; Zhou, F.; Wei, S. Synthesis and herbicidal activities of benzothiazole N,O-acetals. Bioorg. Med. Chem. Lett. 2015, 25, 4065–4068. [Google Scholar] [CrossRef] [PubMed]
- Lavudu, P.; Rani, A.P.; Sekaran, C.B.; Kumar, K.S.; Ramesh, A. Determination of pramipexole dihydrochloride in tablet dosage forms by visible spectrophotometric method using acetyl acetone-formaldehyde reagent. Chem. Sci. J. 2012, 2012, CSJ-49. [Google Scholar]
- Vibhute, S.D.; Agarwal, S.; Joshi, Y. Stable formulations of pramipexole hydrochloride. WO 2012/140604, 2012. [Google Scholar]













Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).