
Poikilodermatous Plaque-like Hemangioma: Case Presentation and Literature Review

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
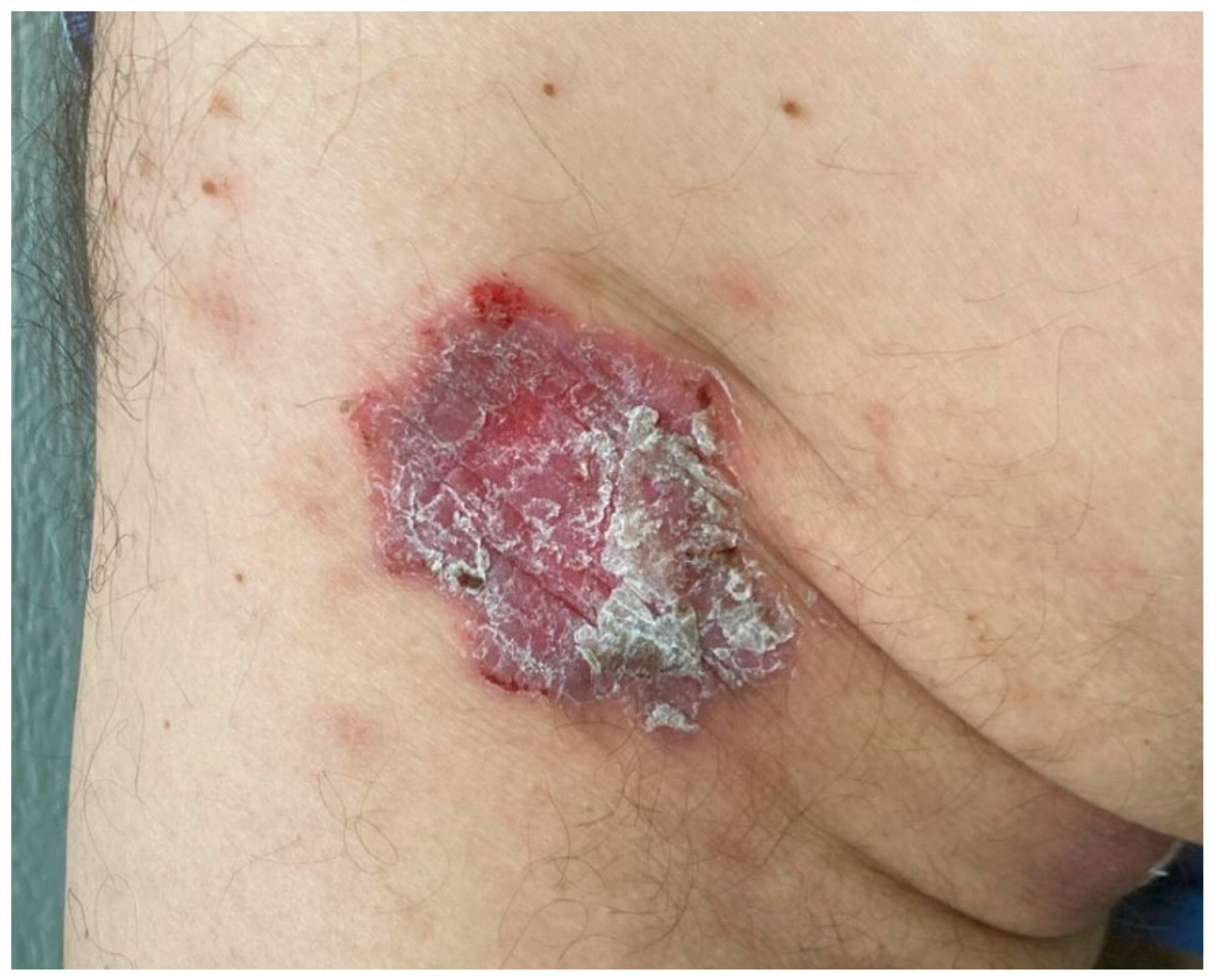
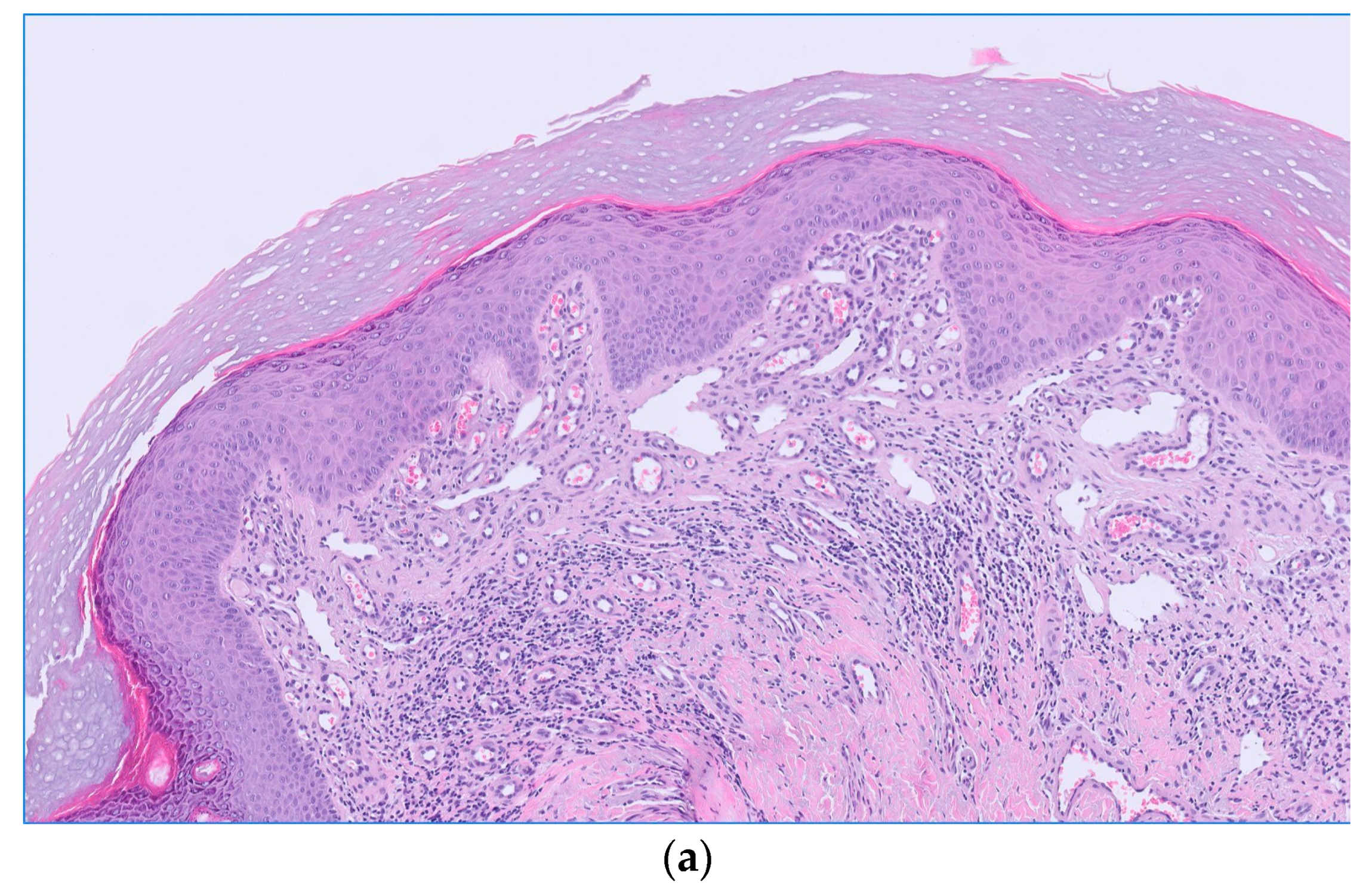
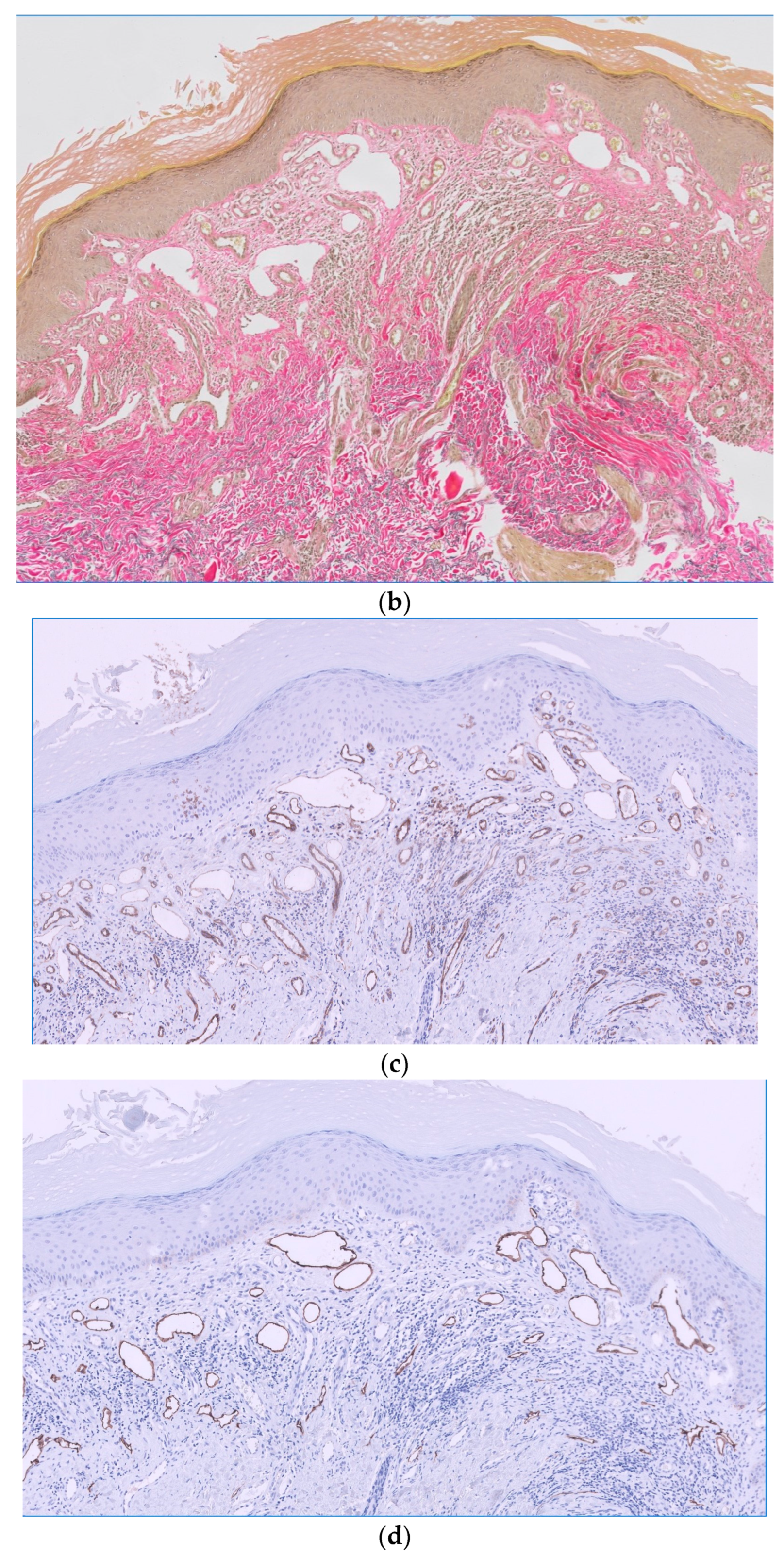
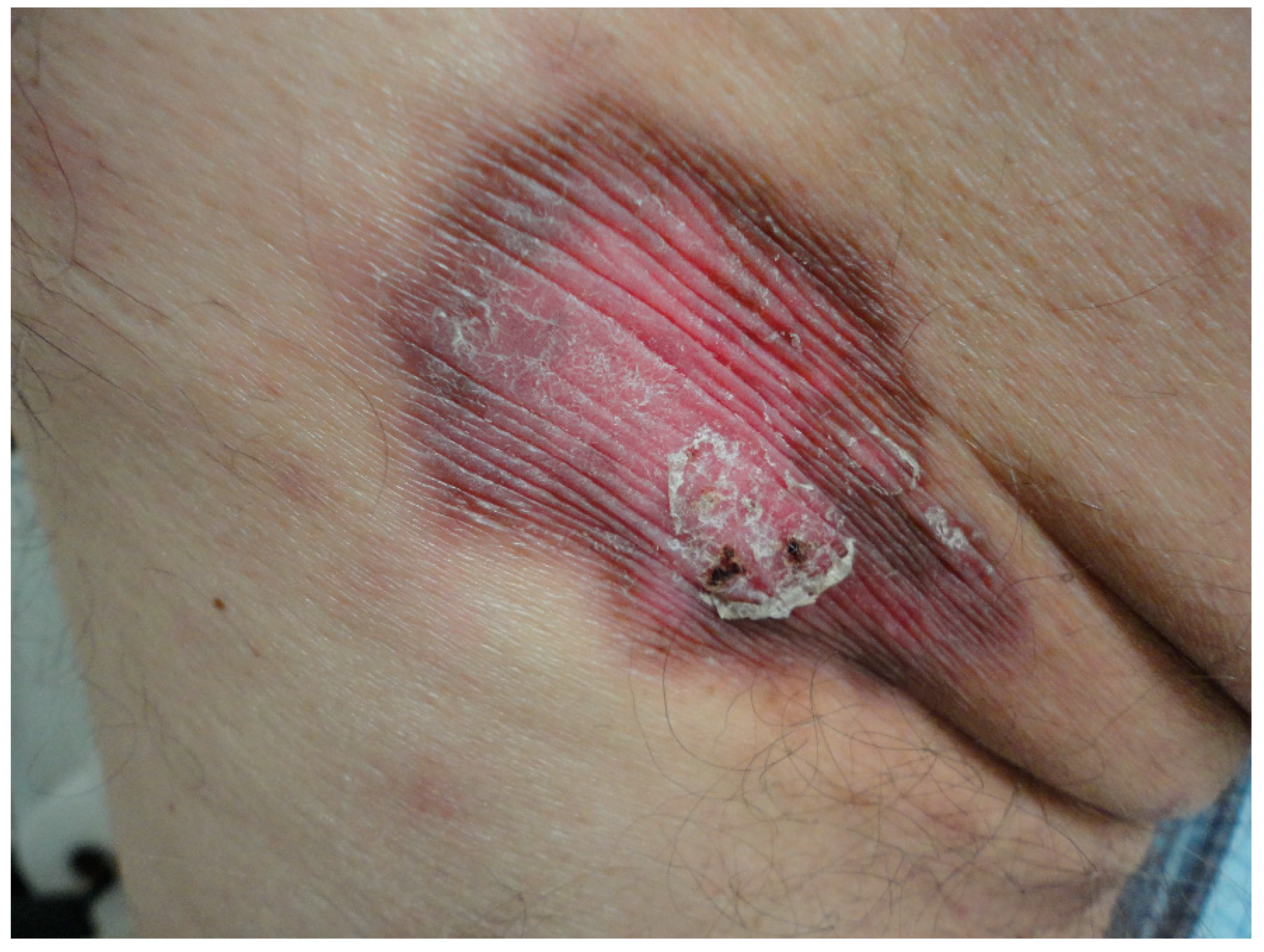
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Semkova, K.; Carr, R.; Grainger, M.; Green, R.; Hafejee, A.; Makrygeorgou, A.; Melly, L.; Motta, L.; Newsham, J.; Owen, C.; et al. Poikilodermatous plaque-like hemangioma: Case series of a newly defined entity. J. Am. Acad. Dermatol. 2019, 81, 1257–1270. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.; Hartman, R.; Mahalingam, M. Poikilodermatous plaque-like hemangioma: A benign vasoformative entity with reproducible histopathologic and clinical features. J. Cutan. Pathol. 2020, 47, 950–953. [Google Scholar] [CrossRef] [PubMed]
- Salgüero Fernández, I.; Hospital Gil, M.; Nájera Botello, L.; Roustan Gullón, G. Not All Is Infantile Hemangioma: An Erythematous Plaque in an Adult. Actas Dermosifiliogr. 2022, 113, 717–718. [Google Scholar] [CrossRef] [PubMed]
- Que, S.K.T.; Zwald, F.O.; Schmults, C.D. Cutaneous squamous cell carcinoma: Incidence, risk factors, diagnosis, and staging. J. Am. Acad. Dermatol. 2018, 78, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, P.; Asgari, M.M.; Green, A.C.; Guhan, S.M.; Arron, S.T.; Proby, C.M.; Rollison, D.E.; Harwood, C.A.; Toland, A.E. Keratinocyte Carcinomas: Current Concepts and Future Research Priorities. Clin. Cancer Res. 2019, 25, 2379–2391. [Google Scholar] [CrossRef] [PubMed]
- Jawed, S.I.; Myskowski, P.L.; Horwitz, S.; Moskowitz, A.; Querfeld, C. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): Part I. Diagnosis: Clinical and histopathologic features and new molecular and biologic markers. J. Am. Acad. Dermatol. 2014, 70, 205.e1–205.e16. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; John, A.M.; Handler, M.Z.; Schwartz, R.A. Fixed Drug Eruptions: An Update, Emphasizing the Potentially Lethal Generalized Bullous Fixed Drug Eruption. Am. J. Clin. Dermatol. 2020, 21, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Çaytemel, C.; Baykut, B.; Ağırgöl, Ş.; Caf, N.; Demir, F.T.; Türkoğlu, Z.; Uzuner, E.G. Pigmented purpuric dermatosis: Ten years of experience in a tertiary hospital and awareness of mycosis fungoides in differential diagnosis. J. Cutan. Pathol. 2021, 48, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.J.; Hunter, G.R.; Kekes-Szabo, T.; Snyder, S.; Treuth, M.S. Regional fat distribution in women and risk of cardiovascular disease. Am. J. Clin. Nutr. 1997, 65, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Hahnel, E.; Blume-Peytavi, U.; Trojahn, C.; Kottner, J. Associations between skin barrier characteristics, skin conditions and health of aged nursing home residents: A multi-center prevalence and correlational study. BMC Geriatr. 2017, 17, 263. [Google Scholar] [CrossRef] [PubMed]
- Hahnel, E.; Lichterfeld, A.; Blume-Peytavi, U.; Kottner, J. The epidemiology of skin conditions in the aged: A systematic review. J. Tissue Viability 2017, 26, 20–28. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz-Calvillo, P.; Vílchez-Márquez, F.; Ramos-Pleguezuelos, F.M.; Arias-Santiago, S. Poikilodermatous Plaque-like Hemangioma: Case Presentation and Literature Review. Dermatopathology 2024, 11, 147-153. https://doi.org/10.3390/dermatopathology11020015
Díaz-Calvillo P, Vílchez-Márquez F, Ramos-Pleguezuelos FM, Arias-Santiago S. Poikilodermatous Plaque-like Hemangioma: Case Presentation and Literature Review. Dermatopathology. 2024; 11(2):147-153. https://doi.org/10.3390/dermatopathology11020015
Chicago/Turabian StyleDíaz-Calvillo, Pablo, Francisco Vílchez-Márquez, Francisco Manuel Ramos-Pleguezuelos, and Salvador Arias-Santiago. 2024. "Poikilodermatous Plaque-like Hemangioma: Case Presentation and Literature Review" Dermatopathology 11, no. 2: 147-153. https://doi.org/10.3390/dermatopathology11020015
APA StyleDíaz-Calvillo, P., Vílchez-Márquez, F., Ramos-Pleguezuelos, F. M., & Arias-Santiago, S. (2024). Poikilodermatous Plaque-like Hemangioma: Case Presentation and Literature Review. Dermatopathology, 11(2), 147-153. https://doi.org/10.3390/dermatopathology11020015