
Adenoviral Vector COVID-19 Vaccines: Process and Cost Analysis




Abstract
1. Introduction
1.1. Protein Subunit Vaccine Platform
1.2. Adenovirus Vector Vaccine Platform
- They are relatively thermostable [11].
1.3. mRNA Vaccine Platform
2. Materials and Methods
2.1. Software
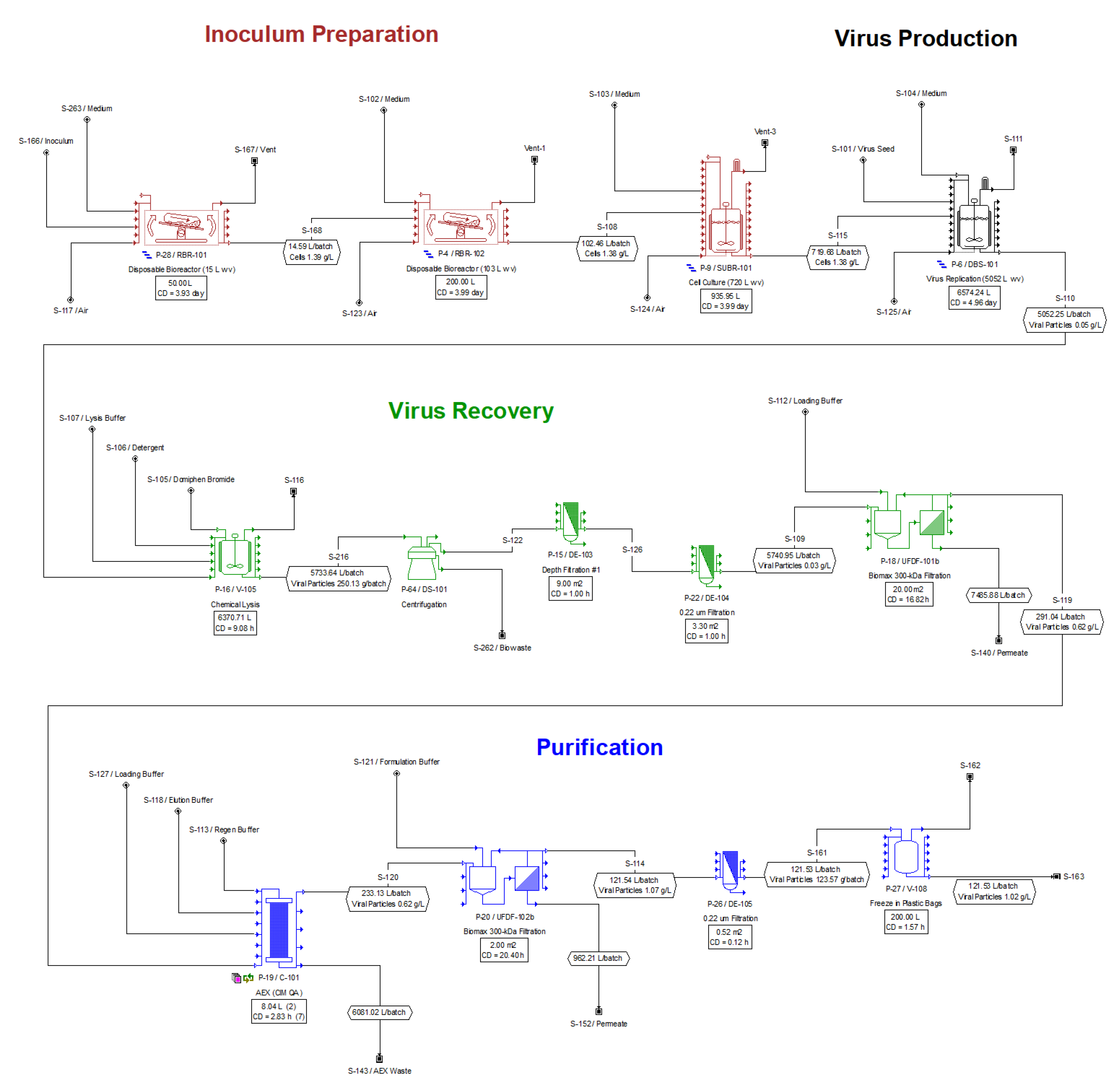
2.2. Process Description
- Batch Scenarios (B1 and B2): In the batch processes, the viral production stage includes a host cell growth phase prior to infection within the same stainless-steel stirred-tank bioreactor. The cell culture stage that comes immediately before the viral production stage is also performed in a stainless-steel stirred bioreactor. The fundamental difference between B1 and B2 is the virus titer achieved in the viral production step, with levels of 5 × 1010 viral particles (VP)/mL and 1 × 1011 VP/mL for B1 and B2, respectively. The batch processes were largely based on references [19,27,28].
- Perfusion Scenarios (P1 and P2): In the perfusion processes, two single-use bioreactors (SUBs) equipped with an external alternating tangential flow (ATF) microfiltration system are used in each run. The host cells are first expanded in the first bioreactor and then transferred into the second bioreactor for viral production. Both stages are operated in perfusion mode. The fundamental difference between P1 and P2 is the virus titer achieved in the viral production step, with levels of 1 × 1012 VP/mL and 2 × 1012 VP/mL for P1 and P2, respectively. These processes were largely based on references [18,29].
- Chemical Lysis
- DNA Precipitation
- Clarification (Centrifugation/Depth Filtration)
- Ultrafiltration-Diafiltration #1
- Anion-Exchange Chromatography
- Ultrafiltration-Diafiltration #2
- Sterile Filtration
2.2.1. Inoculum Preparation
Batch Processes (B1 and B2)
Perfusion Processes (P1 and P2)
- A perfusion phase that takes 6 days [18,29], leading to a cell density of approximately 50 × 106 cells/mL. The perfusion rate is set to 2 working volumes per day [18,29]. The microfiltration membrane is assumed to have a rejection coefficient (RC) of 1.00 for the cells, and the recovery percentage (Permeate/Feed) is assumed to be 99.5%. The perfusion phase follows a stoichiometric model with a conversion rate of 90%.
2.2.2. Virus Production
Batch Processes (B1 and B2)
- Cell Growth: This phase starts by adding a seed cell culture with a cell density of 1.4 × 106 cells/mL to 6 volumes of fresh medium resulting in a cell density of 0.2 × 106 cells/mL, identical to the cell expansion steps described earlier. Similarly, the cell growth phase is modeled by an exponential growth equation with the same doubling time and stoichiometry.
- Viral Replication: After 64 h, when the cell density reaches 0.9 × 106 cells/mL [19], the cell growth phase is deemed complete, and the viral replication phase is triggered by infecting the cell culture with a concentrated adenovirus suspension (1 × 1012 VP/mL). The volume of suspension is such that the number of VPs per cell (i.e., the multiplicity of infection (MOI)) is equal to 280. The viral replication phase takes 48 h [19,27,28,35] and is modeled by a stoichiometric reaction similar to that used to represent cell growth:
→ 116 CO2 + νVP Viral Particles + (30 − νVP) Cells + 30 Cells + 52 Water
Perfusion Processes (P1 and P2)
- Batch Viral Replication Phase: The high cell density culture obtained by perfusion is diluted in fresh medium so that the initial cell density is equal to 15 × 106 cells/mL [18,29]. After a brief mixing period (10 min), the culture is infected with a concentrated adenovirus seed, thus beginning the batch replication phase. The concentration of the virus seed and MOI are 1 × 1012 VP/mL, and 70 [18,29], respectively. Viral replication is carried out in batch mode for 5 h [18,29]; this phase is represented by the same stoichiometry as viral replication in the batch process, with a coefficient νVP equal to that of the perfusion phase (see below). The conversion rate of this phase is assumed to be 10%. At the end of the batch phase, the ATF perfusion system is turned on, starting the perfusion phase.
- Perfusion Viral Replication Phase: This phase takes 4 days [18,29] and is represented by the same stoichiometric model as the batch phase. The perfusion rate is 2 working volumes per day [18,29], and the conversion rate of this phase is 80%. The coefficient νVP is specified so that the final concentration of VPs is equal to 1.0 × 1012 VP/mL in scenario P1, and 2.0 × 1012 VP/mL in scenario P2. These values cover a range of values reported in the literature [18,29]. The ATF microfiltration membrane has a rejection coefficient of 1.00 for the cells and 0.98 for the VPs. This high VP rejection coefficient is required to minimize the loss of VPs through the microfiltration membrane [18,29] (under 5%), considering that VPs are represented as extracellular entities. The recovery percentage (Permeate/Feed) is assumed to be 99.5%.
2.2.3. Virus Recovery
Chemical Lysis
DNA Precipitation
Cell Lysate Clarification
Batch Processes (B1 and B2)
Perfusion Processes (P1 and P2)
Ultrafiltration-Diafiltration #1
2.2.4. Purification
Anion-Exchange Chromatography
Ultrafiltration-Diafiltration #2
Sterile Filtration
2.3. Analysis of Process Scale
2.4. Cost Analysis
3. Results and Discussion
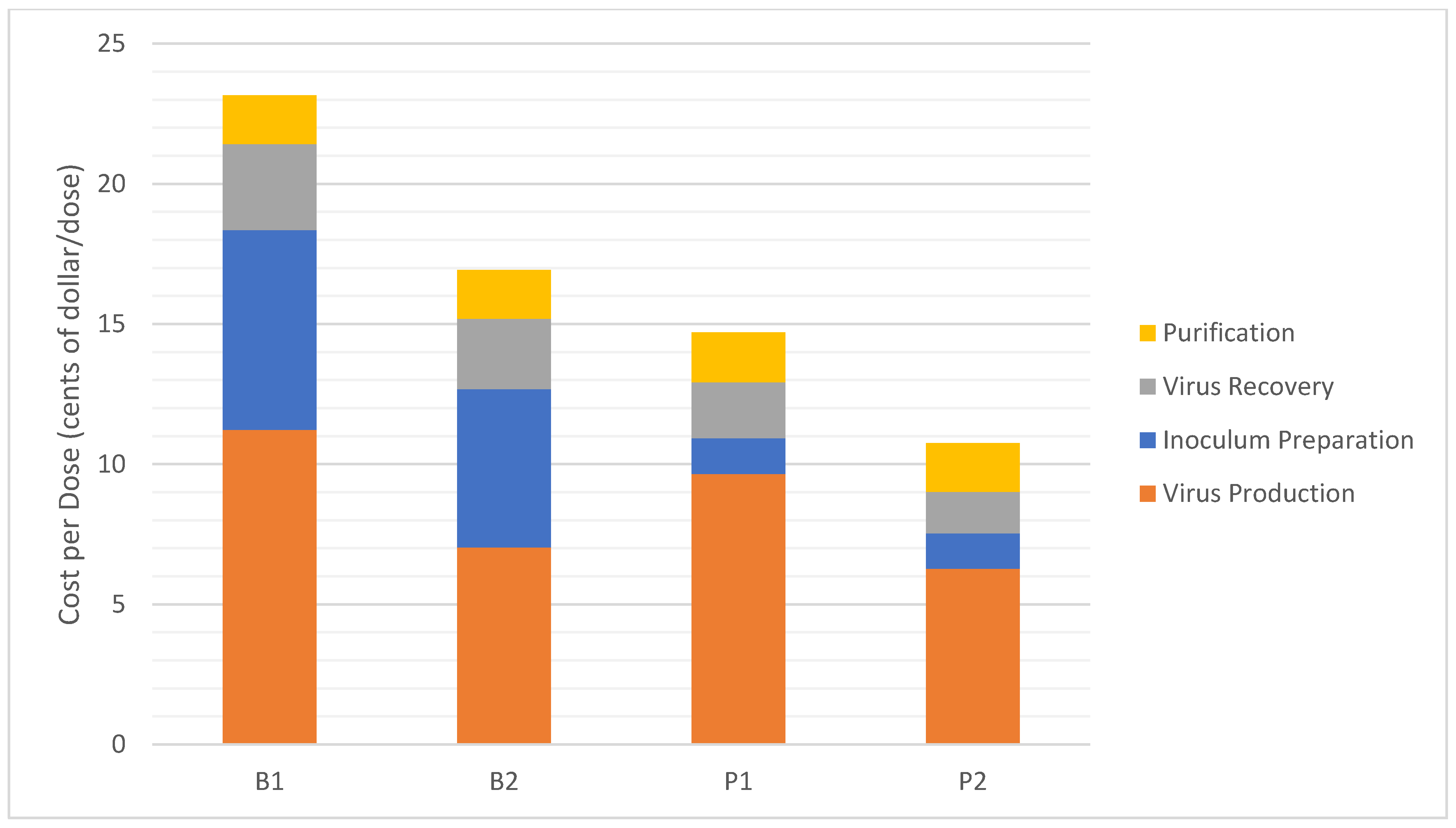
3.1. Cost Analysis
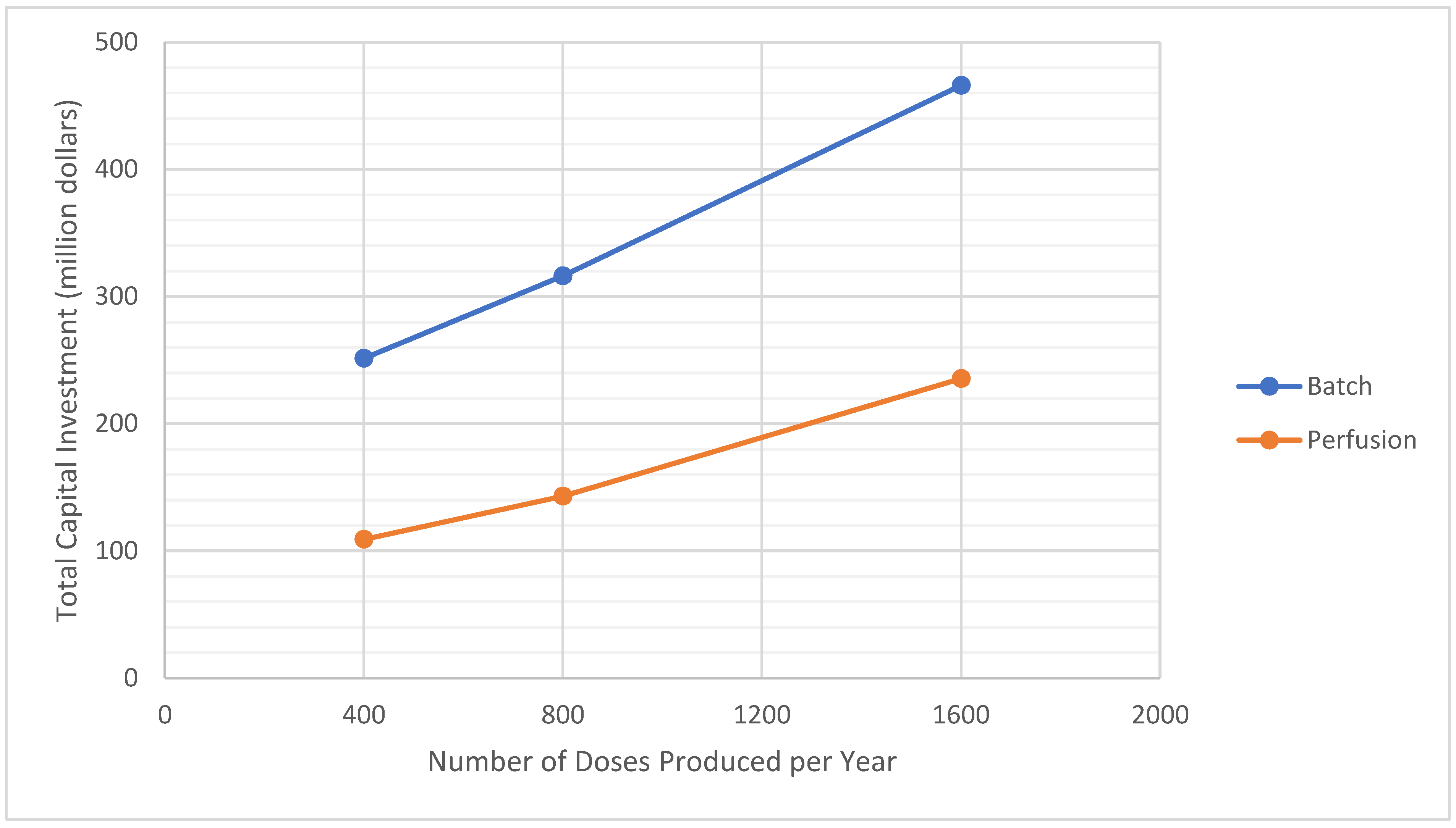
3.2. Analysis of Process Scale
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- The New York Times. Coronavirus Vaccine Tracker. Available online: https://www.nytimes.com/interactive/2020/science/coronavirus-vaccine-tracker.html (accessed on 26 November 2020).
- Huang, Y.; Yang, C.; Xu, X.-F.; Xu, W.; Liu, S.-W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Greaney, A.J.; Loes, A.N.; Gentles, L.E.; Crawford, K.H.; Starr, T.N.; Malone, K.D.; Chu, H.Y.; Bloom, J.D. Antibodies elicited by mRNA-1273 vaccination bind more broadly to the receptor binding domain than do those from SARS-CoV-2 infection. Sci. Transl. Med. 2021, 13, eabi9915. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, S.; Coyle, E.M.; Klenow, L.; Tang, J.; Grubbs, G.; Liu, S.; Wang, T.; Golding, H.; Khurana, S. Antibody signature induced by SARS-CoV-2 spike protein immunogens in rabbits. Sci. Transl. Med. 2020, 12, eabc3539. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zeng, H.; Gu, J.; Li, H.; Zheng, L.; Zou, Q. Progress and Prospects on Vaccine Development against SARS-CoV-2. Vaccines 2020, 8, 153. [Google Scholar] [CrossRef]
- Li, Y.; Tenchov, R.; Smoot, J.; Liu, C.; Watkins, S.; Zhou, Q. A Comprehensive Review of the Global Efforts on COVID-19 Vaccine Development. ACS Cent. Sci. 2021, 7, 512–533. [Google Scholar] [CrossRef]
- Kyriakidis, N.C.; López-Cortés, A.; González, E.V.; Grimaldos, A.B.; Prado, E.O. SARS-CoV-2 vaccines strategies: A comprehensive review of phase 3 candidates. NPJ Vaccines 2021, 6, 1–17. [Google Scholar] [CrossRef]
- Sharon, D.; Kamen, A. Advancements in the design and scalable production of viral gene transfer vectors. Biotechnol. Bioeng. 2018, 115, 25–40. [Google Scholar] [CrossRef]
- Rollier, C.S.; Reyes-Sandoval, A.; Cottingham, M.; Ewer, K.; Hill, A.V. Viral vectors as vaccine platforms: Deployment in sight. Curr. Opin. Immunol. 2011, 23, 377–382. [Google Scholar] [CrossRef]
- Kallel, H.; Kamen, A.A. Large-scale adenovirus and poxvirus-vectored vaccine manufacturing to enable clinical trials. Biotechnol. J. 2015, 10, 741–747. [Google Scholar] [CrossRef]
- Tatsis, N.; Ertl, H.C. Adenoviruses as vaccine vectors. Mol. Ther. 2004, 10, 616–629. [Google Scholar] [CrossRef]
- Alhashimi, M.; Elkashif, A.; Sayedahmed, E.; Mittal, S. Nonhuman Adenoviral Vector-Based Platforms and Their Utility in Designing Next Generation of Vaccines for Infectious Diseases. Viruses 2021, 13, 1493. [Google Scholar] [CrossRef]
- Fougeroux, C.; Holst, P.J. Future Prospects for the Development of Cost-Effective Adenovirus Vaccines. Int. J. Mol. Sci. 2017, 18, 686. [Google Scholar] [CrossRef] [PubMed]
- Voysey, M.; Clemens, S.A.C.; Madhi, S.A.; Weckx, L.Y.; Folegatti, P.M.; Aley, P.K.; Angus, B.; Baillie, V.L.; Barnabas, S.L.; Bhorat, Q.E.; et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: An interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet 2020, 397, 99–111. [Google Scholar] [CrossRef]
- Sadoff, J.; Gray, G.; Vandebosch, A.; Cárdenas, V.; Shukarev, G.; Grinsztejn, B.; Goepfert, P.A.; Truyers, C.; Fennema, H.; Spiessens, B.; et al. Safety and Efficacy of Single-Dose Ad26.COV2.S Vaccine against COVID-19. N. Engl. J. Med. 2021, 384, 2187–2201. [Google Scholar] [CrossRef]
- Logunov, D.Y.; Dolzhikova, I.V.; Shcheblyakov, D.V.; Tukhvatulin, A.I.; Zubkova, O.V.; Dzharullaeva, A.S.; Kovyrshina, A.V.; Lubenets, N.L.; Grousova, D.M.; Erokhova, A.S.; et al. Safety and efficacy of an rAd26 and rAd5 vector-based heterologous prime-boost COVID-19 vaccine: An interim analysis of a randomised controlled phase 3 trial in Russia. Lancet 2021, 397, 671–681. [Google Scholar] [CrossRef]
- Vellinga, J.; Smith, J.P.; Lipiec, A.; Majhen, D.; Lemckert, A.; Van Ooij, M.; Ives, P.; Yallop, C.; Custers, J.; Havenga, M. Challenges in Manufacturing Adenoviral Vectors for Global Vaccine Product Deployment. Hum. Gene Ther. 2014, 25, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Luitjens, A.; Lewis, J.A. Method for the Production of Adenoviral Vectors. U.S. Patent 10,041,049 B2, 7 August 2018. [Google Scholar]
- Altaras, N.E.; Aunins, J.G.; Evans, R.K.; Kamen, A.; Konz, J.O.; Wolf, J.J. Production and Formulation of Adenovirus Vectors; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2005; Volume 99, pp. 193–260. [Google Scholar]
- Lurie, N.; Saville, M.; Hatchett, R.; Halton, J. Developing COVID-19 Vaccines at Pandemic Speed. N. Engl. J. Med. 2020, 382, 1969–1973. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Ball, P. The lightning-fast quest for COVID vaccines—And what it means for other diseases. Nat. Cell Biol. 2021, 589, 16–18. [Google Scholar] [CrossRef]
- Kim, J.; Eygeris, Y.; Gupta, M.; Sahay, G. Self-assembled mRNA vaccines. Adv. Drug Deliv. Rev. 2021, 170, 83–112. [Google Scholar] [CrossRef]
- Bloom, K.; Berg, F.V.D.; Arbuthnot, P. Self-amplifying RNA vaccines for infectious diseases. Gene Ther. 2021, 28, 117–129. [Google Scholar] [CrossRef]
- Hodgson, J. The pandemic pipeline. Nat. Biotechnol. 2020, 38, 523–532. [Google Scholar] [CrossRef]
- Kis, Z.; Kontoravdi, C.; Shattock, R.; Shah, N. Resources, Production Scales and Time Required for Producing RNA Vaccines for the Global Pandemic Demand. Vaccines 2020, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Konz, J.O.J.; Lee, A.L.; Goerke, A.R.; To, C.S.B. Methods of Adenovirus Purification. E.P. Patent 1,506,287 B1, 25 April 2007. [Google Scholar]
- Xie, L.; Pilbrough, W.; Metallo, C.; Zhong, T.; Pikus, L.; Leung, J.; Auniņš, J.G.; Zhou, W. Serum-free suspension cultivation of PER.C6® cells and recombinant adenovirus production under different pH conditions. Biotechnol. Bioeng. 2002, 80, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Luitjens, A.; Van Herk, H. Method for the Production of Ad26 Adenoviral Vectors. W.O. Patent 2011/098592 A1, 18 August 2011. [Google Scholar]
- Kramberger, P.; Urbas, L.; Štrancar, A. Downstream processing and chromatography based analytical methods for production of vaccines, gene therapy vectors, and bacteriophages. Hum. Vaccines Immunother. 2015, 11, 1010–1021. [Google Scholar] [CrossRef]
- Nestola, P.; Peixoto, C.; Silva, R.R.J.S.; Alves, P.; Mota, J.; Carrondo, M. Improved virus purification processes for vaccines and gene therapy. Biotechnol. Bioeng. 2015, 112, 843–857. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.W.; Reichl, U. Downstream processing of cell culture-derived virus particles. Expert Rev. Vaccines 2011, 10, 1451–1475. [Google Scholar] [CrossRef]
- Goerke, A.R.; To, B.C.; Lee, A.L.; Sagar, S.L.; Konz, J.O. Development of a novel adenovirus purification process utilizing selective precipitation of cellular DNA. Biotechnol. Bioeng. 2005, 91, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Berdichevsky, M.; Gentile, M.-P.; Hughes, B.; Meis, P.; Peltier, J.; Blumentals, I.; Aunins, J.; Altaras, N. Establishment of Higher Passage PER.C6 Cells for Adenovirus Manufacture. Biotechnol. Prog. 2008, 24, 158–165. [Google Scholar] [CrossRef]
- Xie, L.; Metallo, C.; Warren, J.; Pilbrough, W.; Peltier, J.; Zhong, T.; Pikus, L.; Yancy, A.; Leung, J.; Auniņs, J.G.; et al. Large-scale propagation of a replication-defective adenovirus vector in stirred-tank bioreactor PER.C6? Cell culture under sparging conditions. Biotechnol. Bioeng. 2003, 83, 45–52. [Google Scholar] [CrossRef]
- Gorfien, S.; Fike, R.; Godwin, G.; Dzimian, J.; Epstein, D.A.; Gruber, D.; McClure, D.; Price, P. Serum-Free Mammalian Cell Culture Medium, And Uses Thereof. U.S. Patent 8,785,194 B2, 22 July 2014. [Google Scholar]
- Bionumbers—Empirical Elemental Formula for Biomass. Available online: https://bionumbers.hms.harvard.edu/bionumber.aspx?id=101801 (accessed on 1 June 2021).
- BioNumbers—Diameter of HEK-293 Cell. Available online: https://bionumbers.hms.harvard.edu/bionumber.aspx?id=108893 (accessed on 1 July 2021).
- Bryan, A.K.; Hecht, V.C.; Shen, W.; Payer, K.R.; Grover, W.; Manalis, S.R. Measuring single cell mass, volume, and density with dual suspended microchannel resonators. Lab Chip 2014, 14, 569–576. [Google Scholar] [CrossRef]
- Palsson, B.Ø. Cellular composition and ultra-structure. In Systems Biology: Simulation of Dynamic Network States, 1st ed.; Cambridge University Press: Cambridge, UK, 2011; pp. 111–115. [Google Scholar]
- Burova, E.; Ioffe, E. Chromatographic purification of recombinant adenoviral and adeno-associated viral vectors: Methods and implications. Gene Ther. 2005, 12, S5–S17. [Google Scholar] [CrossRef]
- Ahi, Y.S.; Mittal, S.K. Components of Adenovirus Genome Packaging. Front. Microbiol. 2016, 7, 1503. [Google Scholar] [CrossRef] [PubMed]
- Fedosyuk, S.; Merritt, T.; Peralta-Alvarez, M.P.; Morris, S.J.; Lam, A.; Laroudie, N.; Kangokar, A.; Wright, D.; Warimwe, G.M.; Angell-Manning, P.; et al. Simian adenovirus vector production for early-phase clinical trials: A simple method applicable to multiple serotypes and using entirely disposable product-contact components. Vaccine 2019, 37, 6951–6961. [Google Scholar] [CrossRef] [PubMed]
- Merck. Clarification of Mammalian Cell Cultures by Depth Filtration. Available online: https://www.merckmillipore.com/Web-GB-Site/en_US/-/GBP/ShowDocument-Pronet?id=201706.004 (accessed on 16 August 2021).
- Weggeman, M. Virus Purification Using Ultrafiltration. E.P. Patent 1,869,171 B2, 14 October 2015. [Google Scholar]
- Moleirinho, M.; Rosa, S.; Carrondo, M.; Silva, R.; Hagner-McWhirter, Å.; Ahlén, G.; Lundgren, M.; Alves, P.; Peixoto, C. Clinical-Grade Oncolytic Adenovirus Purification Using Polysorbate 20 as an Alternative for Cell Lysis. Curr. Gene Ther. 2018, 18, 366–374. [Google Scholar] [CrossRef]
- Moleirinho, M.; Silva, R.; Alves, P.; Carrondo, M.J.T.; Peixoto, C. Current challenges in biotherapeutic particles manufacturing. Expert Opin. Biol. Ther. 2019, 20, 451–465. [Google Scholar] [CrossRef] [PubMed]
- Vicente, T.; Mota, J.P.; Peixoto, C.; Alves, P.M.; Carrondo, M.J. Rational design and optimization of downstream processes of virus particles for biopharmaceutical applications: Current advances. Biotechnol. Adv. 2011, 29, 869–878. [Google Scholar] [CrossRef]
- BIA Separations. Product Sheet & Instruction Manual—CIMmultus QA 8000 mL cGMP Compliant Monolithic Column. Available online: https://www.biaseparations.com/en/download/5472 (accessed on 16 August 2021).
- Peterka, M.; BIA Separations. CIM Monolith Technology: Enabling Economic Vaccines Production 2010. Available online: https://dc.engconfintl.org/cgi/viewcontent.cgi?article=1021&context=vaccine_iii (accessed on 13 May 2021).
- Clendinen, C.; Zhang, Y.; Warburton, R.N.; Light, D.W. Manufacturing costs of HPV vaccines for developing countries. Vaccine 2016, 34, 5984–5989. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Heinzle, E.; Biwer, A.P.; Cooney, C.L. Capital-Cost Estimation. In Development of Sustainable Bioprocesses—Modeling and Assessment, 1st ed.; John Wiley & Sons, Inc.: Chichester, UK, 2007; pp. 83–86. [Google Scholar]
- U.S. Energy Information Administration. Electricity Explained. Available online: https://www.eia.gov/energyexplained/electricity/prices-and-factors-affecting-prices.php (accessed on 13 May 2021).
- Merck/Sigma-Aldrich. Sigma-Aldrich (USA). Available online: https://www.sigmaaldrich.com/united-states.html (accessed on 13 May 2021).
- Cytiva Life Sciences. Cytiva Life Sciences (USA). Available online: https://www.cytivalifesciences.com/en/us/shop (accessed on 13 May 2021).
- US Bureau of Labor Statistics. Hourly mean wage for Chemical Equipment Operators and Tenders in Pharmaceutical and Medicine Manufacturing in the United States. 2020. Available online: https://beta.bls.gov/dataViewer/view/timeseries/OEUN000000032540051901103 (accessed on 13 May 2021).
- Plotkin, S.; Robinson, J.M.; Cunningham, G.; Iqbal, R.; Larsen, S. The complexity and cost of vaccine manufacturing—An overview. Vaccine 2017, 35, 4064–4071. [Google Scholar] [CrossRef] [PubMed]
- Munira, S.L.; Hendriks, J.T.; Atmosukarto, I.I.; Friede, M.H.; Carter, L.M.; Butler, J.R.; Clements, A.C. A cost analysis of producing vaccines in developing countries. Vaccine 2019, 37, 1245–1251. [Google Scholar] [CrossRef]
- UNICEF. COVID-19 Vaccine Market Dashboard. Available online: https://www.unicef.org/supply/covid-19-vaccine-market-dashboard (accessed on 12 June 2021).
- From Pfizer to Moderna: Who’s Making Billions from COVID-19 Vaccines? Available online: https://www.theguardian.com/business/2021/mar/06/from-pfizer-to-moderna-whos-making-billions-from-covid-vaccines (accessed on 14 June 2021).
- Hummel, J.; Pagkaliwangan, M.; Gjoka, X.; Davidovits, T.; Stock, R.; Ransohoff, T.; Gantier, R.; Schofield, M. Modeling the Downstream Processing of Monoclonal Antibodies Reveals Cost Advantages for Continuous Methods for a Broad Range of Manufacturing Scales. Biotechnol. J. 2019, 14, e1700665. [Google Scholar] [CrossRef]
- Pollard, D.; Brower, M.; Abe, Y.; Lopes, A.G.; Sinclair, A. Standardized Economic Cost Modeling for Next-Generation MAb Production. Bioprocess Int. 2016. Available online: https://bioprocessintl.com/business/economics/standardized-economic-cost-modeling-next-generation-mab-production/ (accessed on 8 July 2021).
- Jagschies, G. 55.4.1 Facility Cost Case Study—Drug Substance Bioprocessing Facility. In Biopharmaceutical Processing, 1st ed.; Jagschies, G., Lindskog, E., Łącki, K., Galliher, P., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 1203–1206. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Product Name | Developer | Platform | Status |
|---|---|---|---|
| mRNA-1273 | Moderna | mRNA | Approved in 1, Switzerland EUA in 46 countries |
| Comirnaty | Pfizer (BioNTech) | mRNA | Approved in 5 countries EUA in 21 countries |
| CVnCoV | CureVac | mRNA | Phase 3 |
| Ad26.COV2.S | Janssen | Adenovirus Vector (Ad26) | EUA in 17 countries; stopped use in 2 |
| Vaxzevria | Oxford-Astra Zeneca | Adenovirus Vector(ChAdOx1) | Approved in 1, Brazil EUA in 74 countries; stopped use in 2 |
| NVX-CoV2373 | Novavax | Protein Sub-unit | Phase 3 |
| Sputnik V | Gamaleya Research Institute | Adenovirus Vector (Ad26, Ad5) | EUA in 69 countries |
| Convidecia | CanSino | Adenovirus Vector (Ad5) | Approved in China EUA in 5 countries |
| EpiVacCorona | Vector Institute | Protein Sub-unit | Approved in Turkmenistan Early use in Russia |
| BBIBP-CorV | Sinopharm | Inactivated Virus | Approved in 3 countries EUA in 27 countries |
| CoronaVac | Sinovac | Inactivated Virus | Approved in China EUA in 23 countries |
| Covaxin | Bharat Biotech | Inactivated Virus | EUA in 12 countries |
| Platform | Development Speed | Supply | Cost of Goods | Comments |
|---|---|---|---|---|
| Inactivated Virus | - | + | + |
|
| Protein Subunit | - | +/− | +/− |
|
| mRNA | ++ | + | +/− |
|
| Adenovirus Vector | + | + | + |
|
| Component | Centrifugation | Depth Filtration | Membrane Filtration |
|---|---|---|---|
| Cells | 95% | 100% | 100% |
| Cell Debris | 90% | 95% | 100% |
| Nucleic Acids (s) * | 95% | 100% | 100% |
| Viral Particles | 10% | 10% | 1% |
| Component | Depth Filtration #1 | Depth Filtration #2 | Membrane Filtration |
|---|---|---|---|
| Cells | 95% | 100% | 100% |
| Cell Debris | 90% | 95% | 100% |
| Nucleic Acids (s) * | 95% | 100% | 100% |
| Viral Particles | 10% | 10% | 1% |
| Component | Concentration | Diafiltration |
|---|---|---|
| Nucleic Acids | 0.10 | 0.10 |
| Proteins | 0.10 | 0.10 |
| Viral Particles | 1.00 | 0.98 |
| Step | Buffer | Volume (BV *) | Flow Rate (BV */min) |
|---|---|---|---|
| Equilibration | Loading Buffer | 10 | 0.5 |
| Loading | Loading Buffer | Maximum allowed by the column binding capacity | 0.5 |
| Wash | Loading Buffer | 10 | 0.5 |
| Elution | Elution Buffer | 2 | 0.5 |
| Regeneration | Regen Buffer | 10 | 0.5 |
| Component | Retention (Loading) | Release (Elution) |
|---|---|---|
| Viral Particles | 80% | 100% |
| Nucleic Acids | 30% | 100% |
| Proteins | 5% | 100% |
| Processing Step | Yield |
|---|---|
| DNA Precipitation | 99% |
| Clarification | 80% |
| UF-DF #1 | 90% |
| AEX | 80% |
| UF-DF #2 | 90% |
| Sterile Filtration | 95% |
| Overall | 49% |
| B1 | B2 | P1 | P2 | |
|---|---|---|---|---|
| Virus Production Titer (1011 VP/mL) | 0.5 | 1.0 | 10 | 20 |
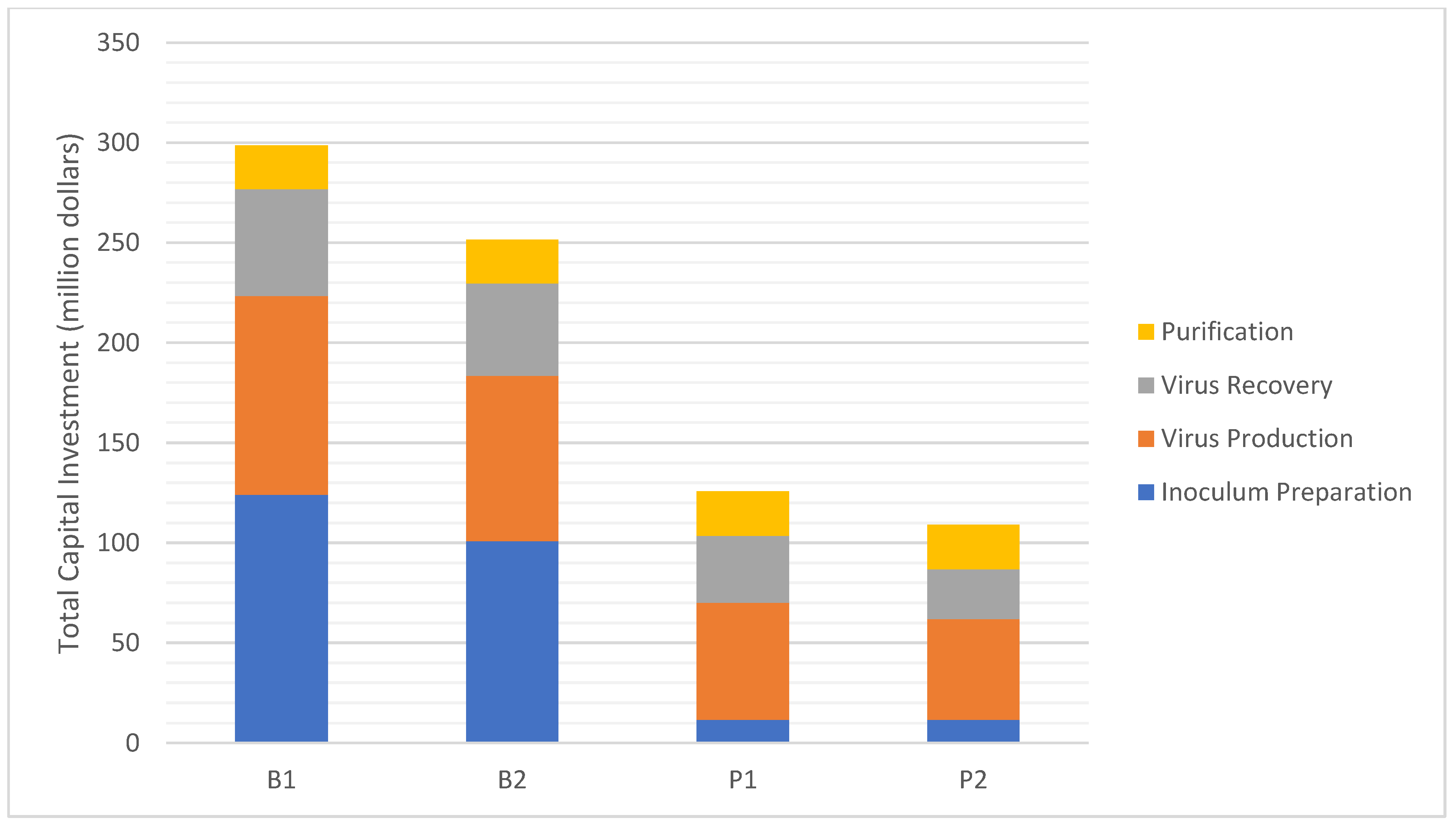
| Total Capital Investment (million $) | 299 | 251 | 126 | 109 |
| Annual Operating Cost (million $) | 93 | 68 | 59 | 43 |
| Batch Yield (g of VP) | 124 | 124 | 124 | 124 |
| Annual Number of Batches | 89 | 89 | 89 | 89 |
| Cost Basis Annual Rate (g of VP/year) | 11,000 | 11,000 | 11,000 | 11,000 |
| Cost Basis Annual Rate (million doses/year) | 400 | 400 | 400 | 400 |
| Unit Production Cost ($/mg VP) | 8.42 | 6.16 | 5.34 | 3.91 |
| Cost per Dose * ($/dose) | 0.23 | 0.17 | 0.15 | 0.11 |
| Vaccine Developer | Vaccine Name | Manufacturer | Price per Dose ($) | Vaccine Dose (VP) |
|---|---|---|---|---|
| AstraZeneca | Vaxzevria | AstraZeneca (Europe/USA/…?) | 2.19–6.50 | 5 × 1010 [14] |
| Vaxzevria | Fiocruz (Brazil) | 3.16 | ||
| Covishield | Serum Institute of India | 1.20–5.25 | ||
| Vaxzevria | Siam Bioscience (Thailand) | 3.25 | ||
| Gamaleya Research Institute | Sputnik V | Gamaleya Research Institute (Russia) | 10.00–19.90 | 1 × 1011 [16] |
| Sputnik V | Shilpa Biologicals (India) | 13.58 | ||
| Sputnik V | Uniao Quimica Farmaceutica (Brazil) | 3.00 | ||
| Janssen | Ad26.COV2.S | Janssen (Europe/USA) | 8.50–10.00 | 5 × 1010 [15] |
| B1 | B2 | P1 | P2 | |
|---|---|---|---|---|
| Single-Use Filters | 1519 | 955 | 917 | 591 |
| TFF (Multi-Use) Membranes | 95 | 65 | 99 | 77 |
| Single-Use Bags | 1977 | 1546 | 5664 | 4567 |
| AEX Chromatography Columns * | 1713 | 1713 | 1713 | 1713 |
| TOTAL | 5304 | 4280 | 8393 | 6948 |
| B2 | B2.2 | B2.4 | P2 | P2.2 | P2.4 | |
|---|---|---|---|---|---|---|
| Working Volume of the Viral Production Vessel (L) | 5053 | 10,104 | 20,205 | 252 | 505 | 1008 |
| Nominal Volume of the Viral Production Vessel (L) | 6579 | 13,154 | 2 × 13,151 * | 700 (500 L bag) | 1300 (1000 L bag) | 1300 (1000 L bag) |
| Production Rate (million doses/year **) | 400 | 800 | 1600 | 400 | 800 | 1600 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira, R.G.; Gordon, N.F.; Stock, R.; Petrides, D. Adenoviral Vector COVID-19 Vaccines: Process and Cost Analysis. Processes 2021, 9, 1430. https://doi.org/10.3390/pr9081430
Ferreira RG, Gordon NF, Stock R, Petrides D. Adenoviral Vector COVID-19 Vaccines: Process and Cost Analysis. Processes. 2021; 9(8):1430. https://doi.org/10.3390/pr9081430
Chicago/Turabian StyleFerreira, Rafael G., Neal F. Gordon, Rick Stock, and Demetri Petrides. 2021. "Adenoviral Vector COVID-19 Vaccines: Process and Cost Analysis" Processes 9, no. 8: 1430. https://doi.org/10.3390/pr9081430
APA StyleFerreira, R. G., Gordon, N. F., Stock, R., & Petrides, D. (2021). Adenoviral Vector COVID-19 Vaccines: Process and Cost Analysis. Processes, 9(8), 1430. https://doi.org/10.3390/pr9081430