
New Perspectives of CYP1B1 Inhibitors in the Light of Molecular Studies



Abstract
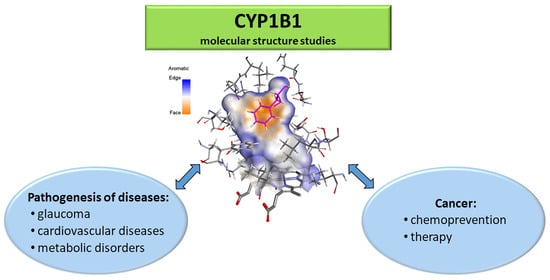
1. Introduction
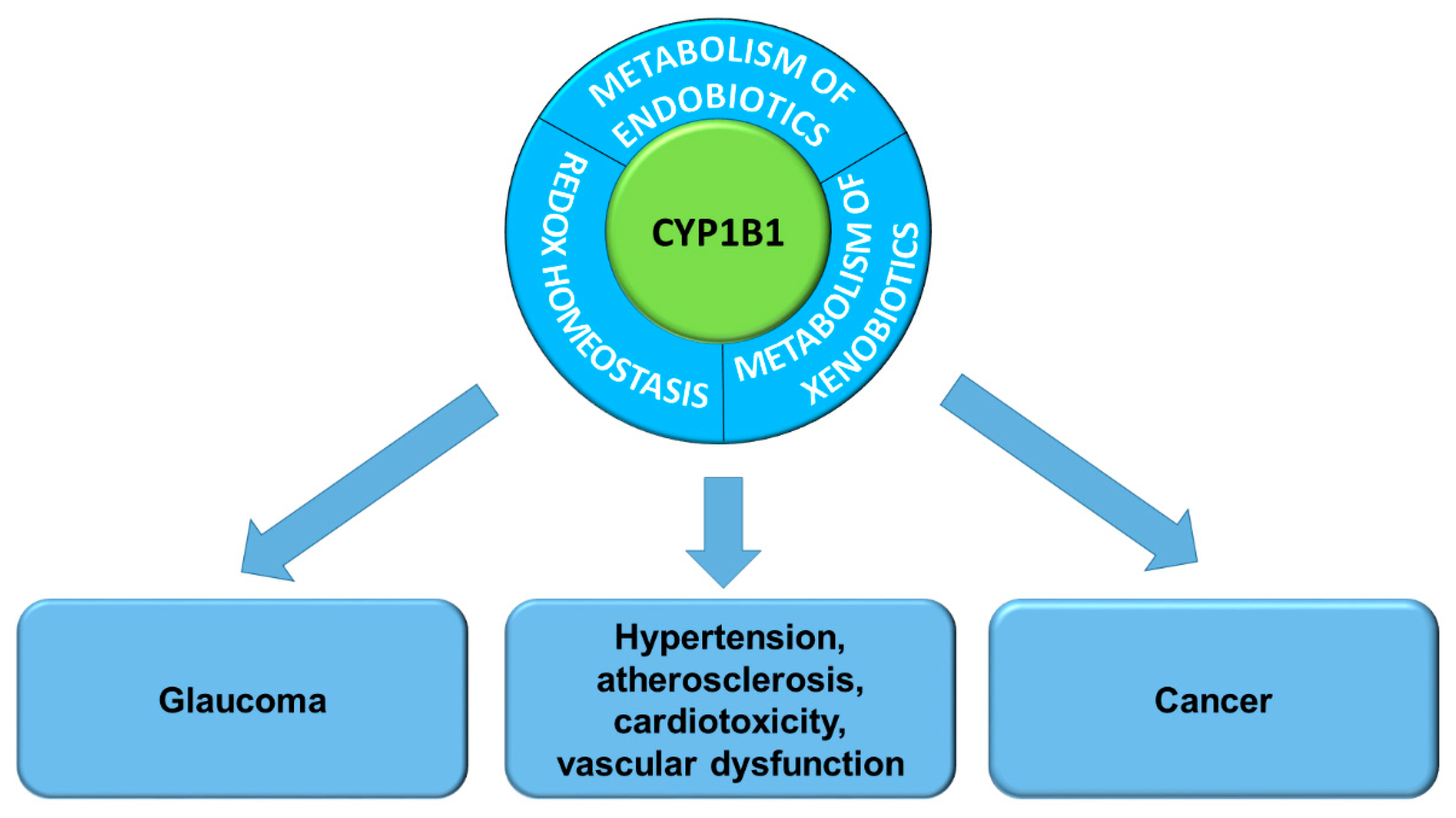
2. The Role of CYP1B1 in the Pathogenesis of Diseases
2.1. CYP1B1 Contribution in Pathogenesis of Eye Diseases
2.2. Redox Homeostasis
2.3. CYP1B1 in Cardiovascular Diseases
2.4. Metabolic Diseases
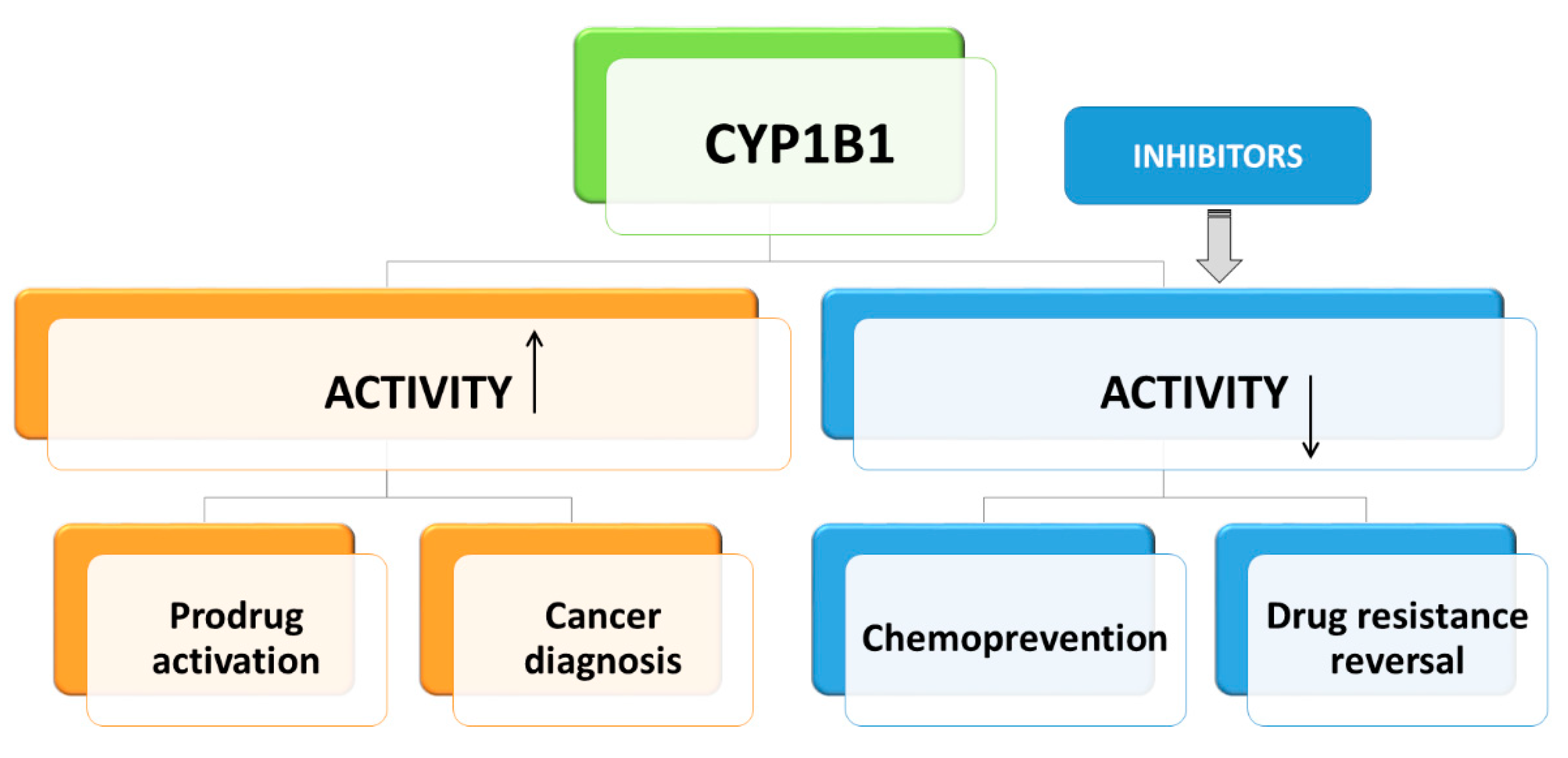
2.5. The Role of CYP1B1 in Cancer Chemoprevention and Therapy
3. CYP1B1 Molecular Structure Studies
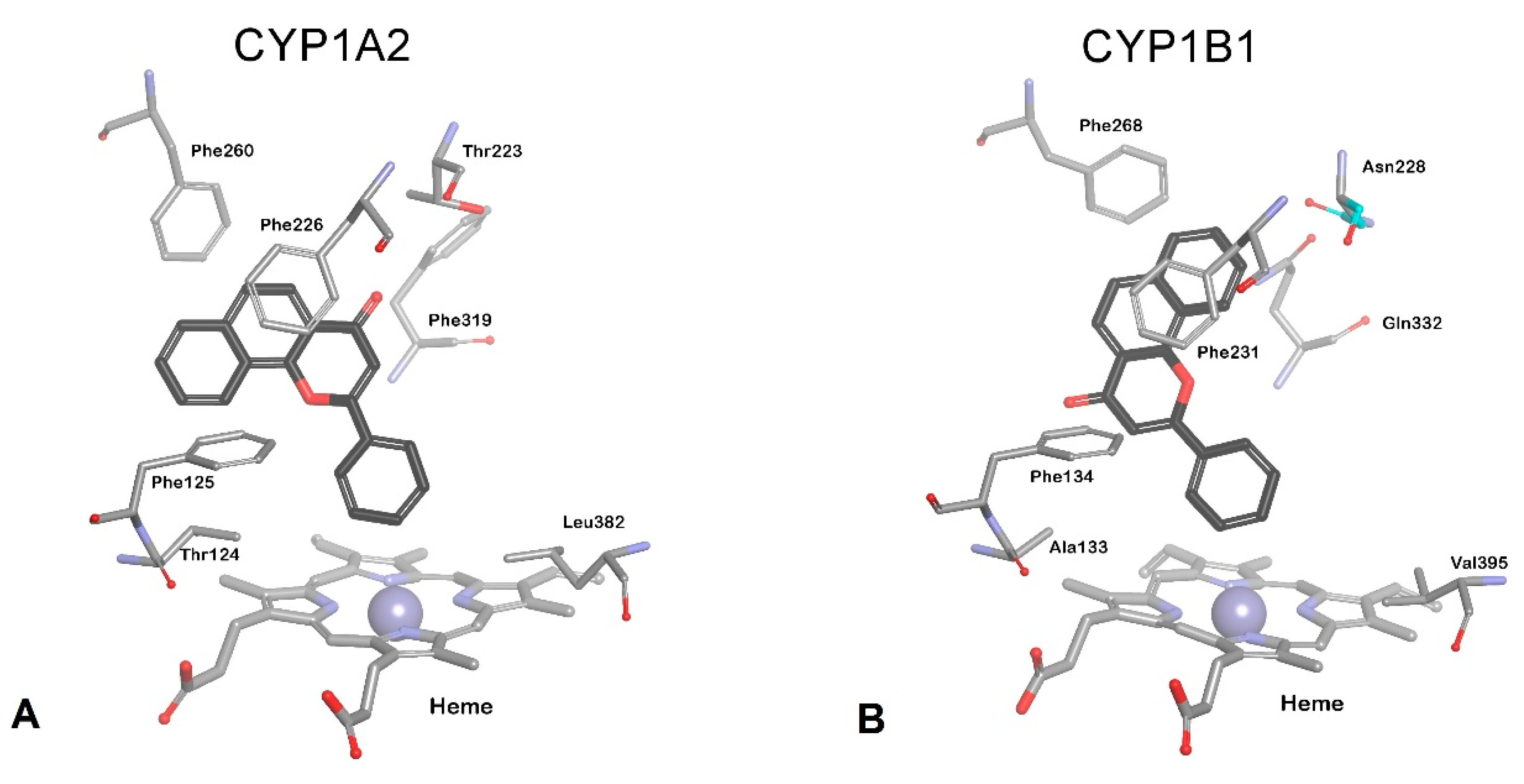
3.1. Family CYP1 Crystal Structures
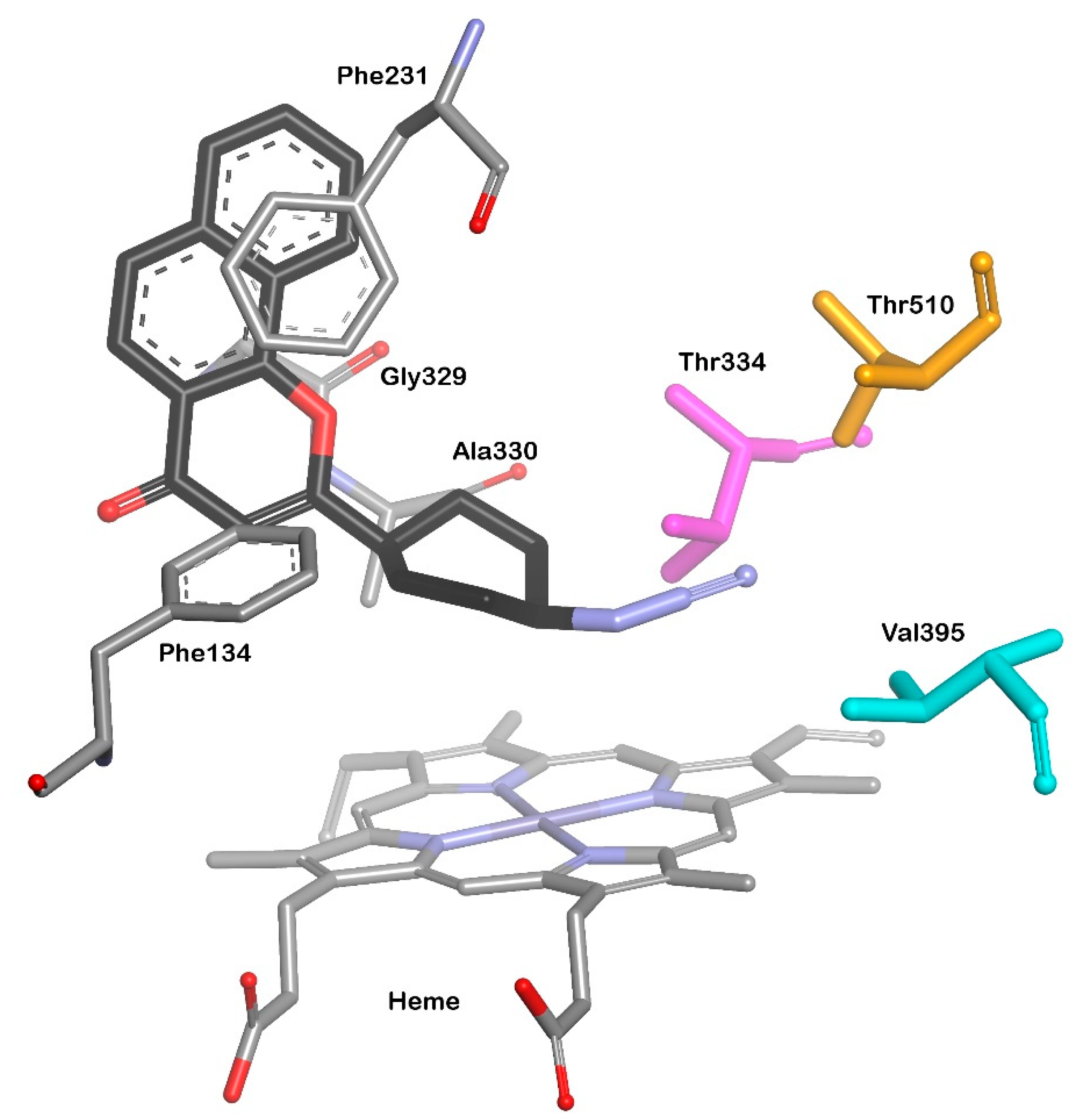
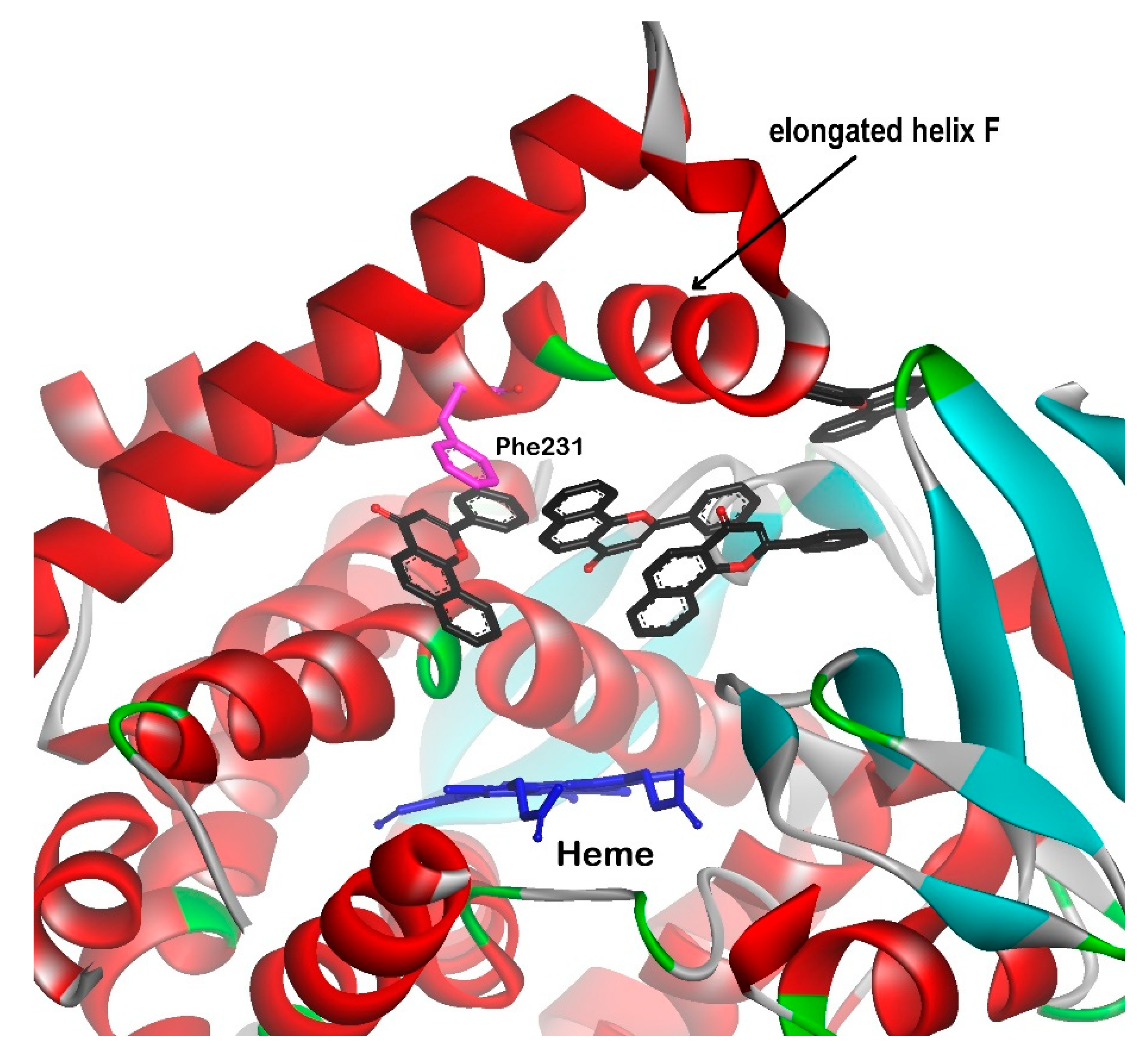
3.2. CYP1B1 Crystal Structure Studies
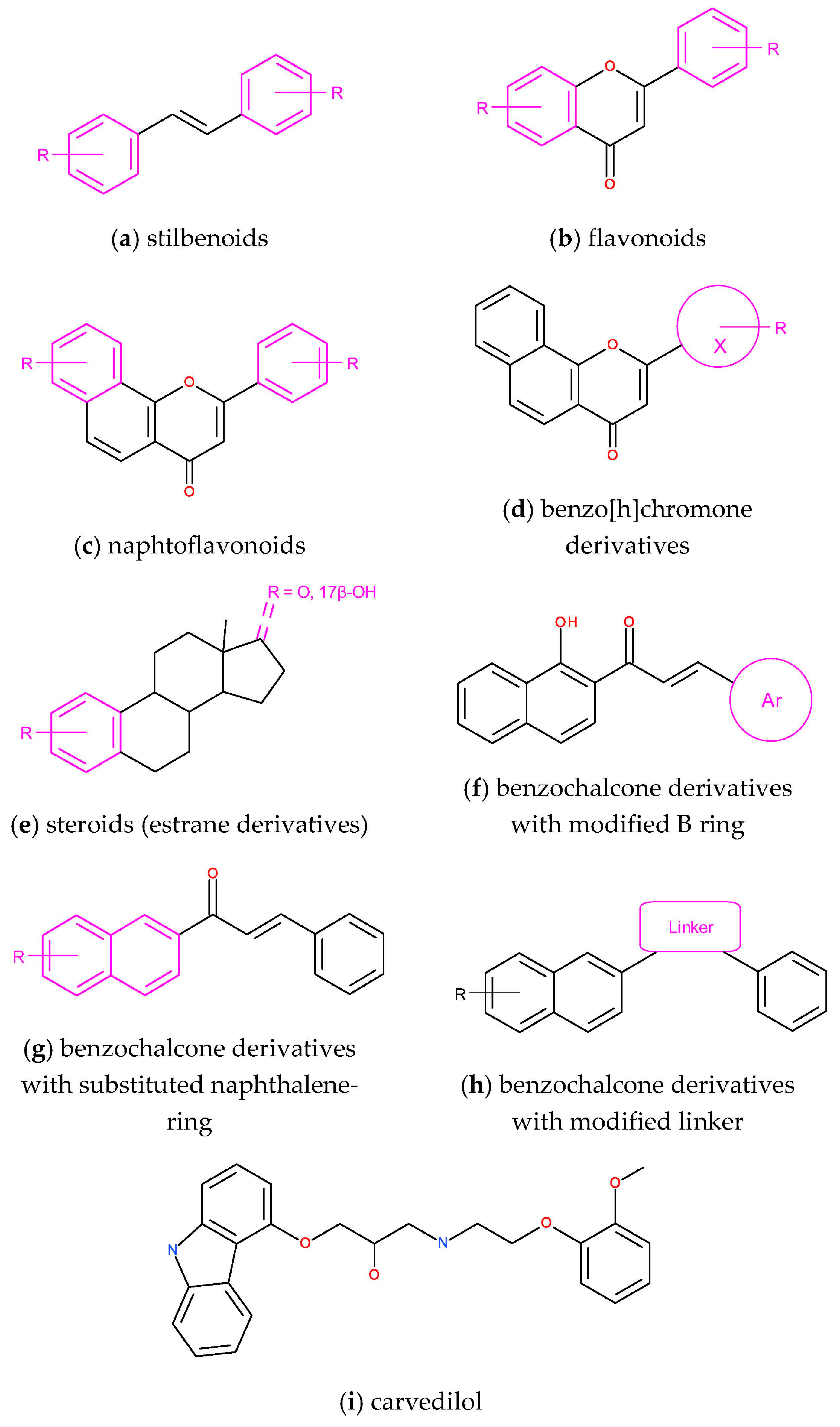
3.3. Design of Selective CYP1B1 Inhibitors
3.4. Genetic Variability
3.5. Characteristics of in Silico Methods
4. Summary: Perspectives of CYP1B1 Studies in Drug Design
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bièche, I.; Narjoz, C.; Asselah, T.; Vacher, S.; Marcellin, P.; Lidereau, R.; Beaune, P.; de Waziers, I. Reverse transcriptase-PCR quantification of mRNA levels from cytochrome (CYP)1, CYP2 and CYP3 families in 22 different human tissues. Pharm. Genom. 2007, 17, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T. Inhibition of carcinogen-activating cytochrome P450 Enzymes by xenobiotic chemicals in relation to antimutagenicity and anticarcinogenicity. Toxicol. Res. 2017, 33, 79–96. [Google Scholar] [CrossRef]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef]
- Go, R.-E.; Hwang, K.-A.; Choi, K.-C. Cytochrome P450 1 family and cancers. J. Steroid Biochem. Mol. Biol. 2015, 147, 24–30. [Google Scholar] [CrossRef]
- McFadyen, M.C.E.; McLeod, H.L.; Jackson, F.C.; Melvin, W.T.; Doehmer, J.; Murray, G.I. Cytochrome P450 CYP1B1 protein expression: A novel mechanism of anticancer drug resistance. Biochem. Pharmacol. 2001, 62, 207–212. [Google Scholar] [CrossRef]
- Rochat, B.; Morsman, J.M.; Figg, W.D.; Murray, G.I.; McLeod, H.L. Human CYP1B1 and anticancer agent metabolism: Mechanism for tumor-specific drug inactivation? J. Pharmacol. Exp. Ther. 2001, 296, 537–541. [Google Scholar]
- Zhu, Z.; Mu, Y.; Qi, C.; Wang, J.; Xi, G.; Guo, J.; Mi, R.; Zhao, F. CYP1B1 enhances the resistance of epithelial ovarian cancer cells to paclitaxel in vivo and in vitro. Int. J. Mol. Med. 2015, 35, 340–348. [Google Scholar] [CrossRef]
- Murray, G.I.; Taylor, M.C.; McFadyen, M.C.E.; McKay, J.A.; Greenlee, W.F.; Burke, M.D.; Melvin, W.T. Tumor-specific expression of cytochrome P450 CYP1B1. Cancer Res. 1997, 57, 3026–3031. [Google Scholar] [PubMed]
- Stoilov, I.; Akarsu, A.N.; Sarfarazi, M. Identification of three different truncating mutations in cytochrome P4501B1 (CYP1B1) as the principal cause of primary congenital glaucoma (Buphthalmos) in families linked to the GLC3A locus on chromosome 2p21. Hum. Mol. Genet. 1997, 6, 641–647. [Google Scholar] [CrossRef]
- Alsubait, A.; Aldossary, W.; Rashid, M.; Algamdi, A.; Alrfaei, B.M. CYP1B1 gene: Implications in glaucoma and cancer. J. Cancer 2020, 11, 4652–4661. [Google Scholar] [CrossRef] [PubMed]
- Carrera, A.N.; Grant, M.K.O.; Zordoky, B.N. CYP1B1 as a therapeutic target in cardio-oncology. Clin. Sci. 2020, 134, 2897–2927. [Google Scholar] [CrossRef]
- Nebert, D.W.; Dalton, T.P.; Okey, A.B.; Gonzalez, F.J. Role of Aryl Hydrocarbon Receptor-mediated Induction of the CYP1 Enzymes in Environmental Toxicity and Cancer. J. Biol. Chem. 2004, 279, 23847–23850. [Google Scholar] [CrossRef]
- Yang, X.; Solomon, S.; Fraser, L.R.; Trombino, A.F.; Liu, D.; Sonenshein, G.E.; Hestermann, E.V.; Sherr, D.H. Constitutive regulation ofCYP1B1 by the aryl hydrocarbon receptor (AhR) in pre-malignant and malignant mammary tissue. J. Cell. Biochem. 2008, 104, 402–417. [Google Scholar] [CrossRef]
- Kawajiri, K.; Fujii-Kuriyama, Y. Cytochrome P450 gene regulation and physiological functions mediated by the aryl hydrocarbon receptor. Arch. Biochem. Biophys. 2007, 464, 207–212. [Google Scholar] [CrossRef]
- Avilla, M.N.; Malecki, K.M.C.; Hahn, M.E.; Wilson, R.H.; Bradfield, C.A. The Ah receptor: Adaptive metabolism, ligand diversity, and the Xenokine model. Chem. Res. Toxicol. 2020, 33, 860–879. [Google Scholar] [CrossRef]
- Ingelman-Sundberg, M.; Zhong, X.B.; Hankinson, O.; Beedanagari, S.; Yu, A.M.; Peng, L.; Osawa, Y. Potential role of epigenetic mechanisms in the regulation of drug metabolism and transport. Drug Metab. Dispos. 2013, 41, 1725–1731. [Google Scholar] [CrossRef] [PubMed]
- Beedanagari, S.R.; Taylor, R.T.; Hankinson, O. Differential regulation of the dioxin-induced Cyp1a1 and Cyp1b1 genes in mouse hepatoma and fibroblast cell lines. Toxicol. Lett. 2010, 194, 26–33. [Google Scholar] [CrossRef]
- Beedanagari, S.R.; Taylor, R.T.; Bui, P.; Wang, F.; Nickerson, D.W.; Hankinson, O. Role of epigenetic mechanisms in differential regulation of the dioxin-inducible human CYP1A1 and CYP1B1 genes. Mol. Pharmacol. 2010, 78, 608–616. [Google Scholar] [CrossRef] [PubMed]
- Kang, G.H.; Lee, S.; Cho, N.Y.; Gandamihardja, T.; Long, T.I.; Weisenberger, D.J.; Campan, M.; Laird, P.W. DNA methylation profiles of gastric carcinoma characterized by quantitative DNA methylation analysis. Lab. Investig. 2008, 88, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Habano, W.; Gamo, T.; Sugai, T.; Otsuka, K.; Wakabayashi, G.; Ozawa, S. CYP1B1, but not CYP1A1, is downregulated by promoter methylation in colorectal cancers. Int. J. Oncol. 2009, 34, 1085–1091. [Google Scholar] [CrossRef] [PubMed]
- Shah, B.R.; Xu, W.; Mraz, J. Cytochrome P450 1B1: Role in health and disease and effect of nutrition on its expression. RSC Adv. 2019, 9, 21050–21062. [Google Scholar] [CrossRef]
- Achary, M.S.; Reddy, A.B.M.; Chakrabarti, S.; Panicker, S.G.; Mandal, A.K.; Ahmed, N.; Balasubramanian, D.; Hasnain, S.E.; Nagarajaram, H.A. Disease-causing mutations in proteins: Structural analysis of the CYP1b1 mutations causing primary congenital glaucoma in humans. Biophys. J. 2006, 91, 4329–4339. [Google Scholar] [CrossRef]
- Banerjee, A.; Chakraborty, S.; Chakraborty, A.; Chakrabarti, S.; Ray, K. Functional and Structural Analyses of CYP1B1 Variants Linked to Congenital and Adult-Onset Glaucoma to Investigate the Molecular Basis of These Diseases. PLoS ONE 2016, 11, e0156252. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.; Yousaf, S.; Sheikh, S.A.; Sajid, Z.; Shabbir, A.S.; Kausar, T.; Tariq, N.; Usman, M.; Shaikh, R.S.; Ali, M.; et al. Identities and frequencies of variants in CYP1B1 causing primary congenital glaucoma in Pakistan. Mol. Vis. 2019, 25, 144–154. [Google Scholar] [PubMed]
- Ou, Z.; Liu, G.; Liu, W.; Deng, Y.; Zheng, L.; Zhang, S.; Feng, G. Bioinformatics analysis of CYP1B1 mutation hotspots in Chinese primary congenital glaucoma patients. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef]
- Falero-Perez, J.; Song, Y.-S.; Zhao, Y.; Teixeira, L.; Sorenson, C.M.; Sheibani, N. Cyp1b1 expression impacts the angiogenic and inflammatory properties of liver sinusoidal endothelial cells. PLoS ONE 2018, 13, e0206756. [Google Scholar] [CrossRef] [PubMed]
- Falero-Perez, J.; Song, Y.-S.; Sorenson, C.M.; Sheibani, N. CYP1B1: A key regulator of redox homeostasis. Trends Cell Mol. Biol. 2018, 13, 27–45. [Google Scholar] [PubMed]
- Maguire, M.; Larsen, M.C.; Foong, Y.H.; Tanumihardjo, S.; Jefcoate, C.R. Cyp1b1 deletion and retinol deficiency coordinately suppress mouse liver lipogenic genes and hepcidin expression during post-natal development. Mol. Cell. Endocrinol. 2017, 454, 50–68. [Google Scholar] [CrossRef] [PubMed]
- Andriopoulos, B.; Corradini, E.; Xia, Y.; Faasse, S.A.; Chen, S.; Grgurevic, L.; Knutson, M.D.; Pietrangelo, A.; Vukicevic, S.; Lin, H.Y.; et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat. Genet. 2009, 41, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Meynard, D.; Kautz, L.; Darnaud, V.; Canonne-Hergaux, F.; Coppin, H.; Roth, M.P. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat. Genet. 2009, 41, 478–481. [Google Scholar] [CrossRef] [PubMed]
- Dinu, D.; Chu, C.; Veith, A.; Lingappan, K.; Couroucli, X.; Jefcoate, C.R.; Sheibani, N.; Moorthy, B. Mechanistic role of cytochrome P450 (CYP)1B1 in oxygen-mediated toxicity in pulmonary cells: A novel target for prevention of hyperoxic lung injury. Biochem. Biophys. Res. Commun. 2016, 476, 346–351. [Google Scholar] [CrossRef]
- Veith, A.C.; Aram, B.B.; Jiang, W.; Wang, L.; Zhou, G.; Jefcoate, C.R.; Couroucli, X.I.; Lingappan, K.; Moorthy, B. Mice Lacking the Cytochrome P450 1B1 Gene Are Less Susceptible to Hyperoxic Lung Injury Than Wild Type. Toxicol. Sci. 2018, 165, 462–474. [Google Scholar] [CrossRef]
- Kubli, S.P.; Bassi, C.; Roux, C.; Wakeham, A.; Göbl, C.; Zhou, W.; Jafari, S.M.; Snow, B.; Jones, L.; Palomero, L.; et al. AhR controls redox homeostasis and shapes the tumor microenvironment in BRCA1-associated breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 3604–3613. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhu, W.; Gonzalez, F.J. Potential role of CYP1B1 in the development and treatment of metabolic diseases. Pharmacol. Ther. 2017, 178, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.; Mazzoccoli, G. Left Ventricular Hypertrophy: Roles of Mitochondria CYP1B1 and Melatonergic Pathways in Co-Ordinating Wider Pathophysiology. Int. J. Mol. Sci. 2019, 20, 4068. [Google Scholar] [CrossRef]
- Elkhatali, S.; Maayah, Z.H.; El-Sherbeni, A.A.; Elshenawy, O.H.; Abdelhamid, G.; Shoieb, S.M.; El-Kadi, A.O.S. Inhibition of mid-chain HETEs protects against angiotensin II-induced cardiac hypertrophy. J. Cardiovasc. Pharmacol. 2017, 70, 16–24. [Google Scholar] [CrossRef]
- Maayah, Z.H.; Althurwi, H.N.; Abdelhamid, G.; Lesyk, G.; Jurasz, P.; El-Kadi, A.O.S. CYP1B1 inhibition attenuates doxorubicin-induced cardiotoxicity through a mid-chain HETEs-dependent mechanism. Pharmacol. Res. 2016, 105, 28–43. [Google Scholar] [CrossRef]
- Maayah, Z.H.; Althurwi, H.N.; El-Sherbeni, A.A.; Abdelhamid, G.; Siraki, A.G.; El-Kadi, A.O.S. The role of cytochrome P450 1B1 and its associated mid-chain hydroxyeicosatetraenoic acid metabolites in the development of cardiac hypertrophy induced by isoproterenol. Mol. Cell. Biochem. 2017, 429, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Sahan-Firat, S.; Jennings, B.L.; Yaghini, F.A.; Song, C.Y.; Estes, A.M.; Fang, X.R.; Farjana, N.; Khan, A.I.; Malik, K.U. 2,3′,4,5′-Tetramethoxystilbene prevents deoxycorticosterone—Salt-induced hypertension: Contribution of cytochrome P-450 1B1. Am. J. Physiol. Hear. Circ. Physiol. 2010, 299. [Google Scholar] [CrossRef] [PubMed]
- Jennings, B.L.; Anderson, L.J.; Estes, A.M.; Yaghini, F.A.; Fang, X.R.; Porter, J.; Gonzalez, F.J.; Campbell, W.B.; Malik, K.U. Cytochrome P450 1B1 contributes to renal dysfunction and damage caused by angiotensin II in mice. Hypertension 2012, 59, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Jennings, B.L.; Montanez, D.E.; May, M.E.; Estes, A.M.; Fang, X.R.; Yaghini, F.A.; Kanu, A.; Malik, K.U. Cytochrome P450 1B1 contributes to increased blood pressure and cardiovascular and renal dysfunction in spontaneously hypertensive rats. Cardiovasc. Drugs Ther. 2014, 28, 145–161. [Google Scholar] [CrossRef] [PubMed]
- Pingili, A.K.; Davidge, K.N.; Thirunavukkarasu, S.; Khan, N.S.; Katsurada, A.; Majid, D.S.A.; Gonzalez, F.J.; Navar, L.G.; Malik, K.U. 2-Methoxyestradiol reduces angiotensin II-induced hypertension and renal dysfunction in ovariectomized female and intact male mice. Hypertension 2017, 69, 1104–1112. [Google Scholar] [CrossRef]
- Malik, K.U.; Jennings, B.L.; Yaghini, F.A.; Sahan-Firat, S.; Song, C.Y.; Estes, A.M.; Fang, X.R. Contribution of cytochrome P450 1B1 to hypertension and associated pathophysiology: A novel target for antihypertensive agents. Prostaglandins Other Lipid Mediat. 2012, 98, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Song, C.Y.; Ghafoor, K.; Ghafoor, H.U.; Khan, N.S.; Thirunavukkarasu, S.; Jennings, B.L.; Estes, A.M.; Zaidi, S.; Bridges, D.; Tso, P.; et al. Cytochrome P450 1B1 contributes to the development of atherosclerosis and hypertension in apolipoprotein E-deficient mice. Hypertension 2016, 67, 206–213. [Google Scholar] [CrossRef]
- Pingili, A.K.; Jennings, B.L.; Mukherjee, K.; Akroush, W.; Gonzalez, F.J.; Malik, K.U. 6β-Hydroxytestosterone, a metabolite of testosterone generated by CYP1B1, contributes to vascular changes in angiotensin II-induced hypertension in male mice. Biol. Sex Differ. 2020, 11, 4. [Google Scholar] [CrossRef]
- Liu, X.; Huang, T.; Li, L.; Tang, Y.; Tian, Y.; Wang, S.; Fan, C. CYP1B1 deficiency ameliorates obesity and glucose intolerance induced by high fat diet in adult C57BL/6J mice. Am. J. Transl. Res. 2015, 7, 761–771. [Google Scholar]
- Larsen, M.C.; Bushkofsky, J.R.; Gorman, T.; Adhami, V.; Mukhtar, H.; Wang, S.; Reeder, S.B.; Sheibani, N.; Jefcoate, C.R. Cytochrome P450 1B1: An unexpected modulator of liver fatty acid homeostasis. Arch. Biochem. Biophys. 2015, 571, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Conway, D.E.; Sakurai, Y.; Weiss, D.; Vega, J.D.; Taylor, W.R.; Jo, H.; Eskin, S.G.; Marcus, C.B.; McIntire, L.V. Expression of CYP1A1 and CYP1B1 in human endothelial cells: Regulation by fluid shear stress. Cardiovasc. Res. 2009, 81, 669–677. [Google Scholar] [CrossRef]
- Yaghini, F.A.; Song, C.Y.; Lavrentyev, E.N.; Ghafoor, H.U.B.; Fang, X.R.; Estes, A.M.; Campbell, W.B.; Malik, K.U. Angiotensin II-induced vascular smooth muscle cell migration and growth are mediated by cytochrome p450 1b1-dependent superoxide generation. Hypertension 2010, 55, 1461–1467. [Google Scholar] [CrossRef]
- Thompson, P.A.; Khatami, M.; Baglole, C.J.; Sun, J.; Harris, S.A.; Moon, E.-Y.; Al-Mulla, F.; Al-Temaimi, R.; Brown, D.G.; Colacci, A.M.; et al. Environmental immune disruptors, inflammation and cancer risk. Carcinogenesis 2015, 36, S232–S253. [Google Scholar] [CrossRef]
- Wang, Z.; Dabrosin, C.; Yin, X.; Fuster, M.M.; Arreola, A.; Rathmell, W.K.; Generali, D.; Nagaraju, G.P.; El-Rayes, B.; Ribatti, D.; et al. Broad targeting of angiogenesis for cancer prevention and therapy. Semin. Cancer Biol. 2015, 35, S224–S243. [Google Scholar] [CrossRef] [PubMed]
- Ramjiawan, R.R.; Griffioen, A.W.; Duda, D.G. Anti-angiogenesis for cancer revisited: Is there a role for combinations with immunotherapy? Angiogenesis 2017, 20, 185–204. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Poveda, B.; Torres-Vargas, J.A.; del Ocaña, M.C.; García-Caballero, M.; Medina, M.Á.; Quesada, A.R. The mediterranean diet, a rich source of angiopreventive compounds in cancer. Nutrients 2019, 11, 2036. [Google Scholar] [CrossRef]
- Kanda, Y.; Osaki, M.; Okada, F. Chemopreventive strategies for inflammation-related carcinogenesis: Current status and future direction. Int. J. Mol. Sci. 2017, 18, 867. [Google Scholar] [CrossRef]
- D’Uva, G.; Baci, D.; Albini, A.; Noonan, D.M. Cancer chemoprevention revisited: Cytochrome P450 family 1B1 as a target in the tumor and the microenvironment. Cancer Treat. Rev. 2018, 63, 1–18. [Google Scholar] [CrossRef]
- Dutour, R.; Poirier, D. Inhibitors of cytochrome P450 (CYP) 1B1. Eur. J. Med. Chem. 2017, 135, 296–306. [Google Scholar] [CrossRef]
- Cui, J.; Li, S. Inhibitors and prodrugs targeting CYP1: A novel approach in cancer prevention and therapy. Curr. Med. Chem. 2014, 21, 519–552. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Zhang, S.; Gong, Z.; Li, X.; Cao, K.; Deng, H.; He, Y.; et al. The role of microenvironment in tumor angiogenesis. J. Exp. Clin. Cancer Res. 2020, 39, 204. [Google Scholar] [CrossRef]
- Ware, W.R. Natural cancer therapy and prevention targeted on cancer cells and cancer stem cells based on the cytochrome P45O enzyme CYP1B1: A commentary. Altern Ther Heal. Med. 2017, 23, 50–58. [Google Scholar]
- Taylor, W.F.; Jabbarzadeh, E. The use of natural products to target cancer stem cells. Am. J. Cancer Res. 2017, 7, 1588–1605. [Google Scholar] [PubMed]
- Dutkiewicz, Z.; Mikstacka, R. Structure-based drug design for cytochrome P450 family 1 inhibitors. Bioinorg. Chem. Appl. 2018, 2018. [Google Scholar] [CrossRef]
- Sirerol, J.A.; Rodríguez, M.L.; Mena, S.; Asensi, M.A.; Estrela, J.M.; Ortega, A.L. Role of natural stilbenes in the prevention of cancer. Oxid. Med. Cell. Longev. 2016, 2016, 1–15. [Google Scholar] [CrossRef]
- Mikstacka, R.; Wierzchowski, M.; Dutkiewicz, Z.; Gielara-Korzańska, A.; Korzański, A.; Teubert, A.; Sobiak, S.; Baer-Dubowska, W. 3,4,2′-Trimethoxy-trans-stilbene—A potent CYP1B1 inhibitor. MedChemComm 2014, 5, 496–501. [Google Scholar] [CrossRef]
- Gao, X.; Xie, C.; Wang, Y.; Luo, Y.; Yagai, T.; Sun, D.; Qin, X.; Krausz, K.W.; Gonzalez, F.J. The antiandrogen flutamide is a novel aryl hydrocarbon receptor ligand that disrupts bile acid homeostasis in mice through induction of Abcc4. Biochem. Pharmacol. 2016, 119, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, Y.; Chang, I.; Fukuhara, S.; Hiraki, M.; Arichi, N.; Yasumoto, H.; Hirata, H.; Yamamura, S.; Shahryari, V.; Deng, G.; et al. CYP1B1 promotes tumorigenesis via altered expression of CDC20 and DAPK1 genes in renal cell carcinoma. BMC Cancer 2015, 15, 942. [Google Scholar] [CrossRef]
- Cui, J.; Meng, Q.; Zhang, X.; Cui, Q.; Zhou, W.; Li, S. Design and synthesis of new α-naphthoflavones as cytochrome P450 (CYP) 1B1 inhibitors to overcome docetaxel-resistance associated with CYP1B1 overexpression. J. Med. Chem. 2015, 58, 3534–3547. [Google Scholar] [CrossRef]
- Kwon, Y.-J.; Baek, H.-S.; Ye, D.-J.; Shin, S.; Kim, D.; Chun, Y.-J. CYP1B1 enhances cell proliferation and metastasis through induction of EMT and activation of Wnt/β-catenin signaling via Sp1 upregulation. PLoS ONE 2016, 11, e0151598. [Google Scholar] [CrossRef]
- Chang, I.; Mitsui, Y.; Kim, S.K.; Sun, J.S.; Jeon, H.S.; Kang, J.Y.; Kang, N.J.; Fukuhara, S.; Gill, A.; Shahryari, V.; et al. Cytochrome P450 1B1 inhibition suppresses tumorigenicity of prostate cancer via caspase-1 activation. Oncotarget 2017, 8, 39087–39100. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, Y.; Drbohlav, L.M.; Wu, J.Q.; Wang, M.Z. development of an in vitro model to screen CYP1B1-targeted anticancer prodrugs. J. Biomol. Screen. 2016, 21, 1090–1099. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Zhang, X.; Huang, G.; Zhang, Q.; Dong, J.; Sun, G.; Meng, Q.; Li, S. DMAKO-20 as a new multitarget anticancer prodrug activated by the tumor specific CYP1B1 enzyme. Mol. Pharm. 2019, 16, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Wang, Z.; Cui, J.; Cui, Q.; Dong, J.; Zhang, Q.; Li, S. Design, synthesis, and biological evaluation of cytochrome P450 1B1 targeted molecular imaging probes for colorectal tumor detection. J. Med. Chem. 2018, 61, 10901–10909. [Google Scholar] [CrossRef] [PubMed]
- Itoh, T.; Takemura, H.; Shimoi, K.; Yamamoto, K. A 3D model of CYP1B1 explains the dominant 4-hydroxylation of estradiol. J. Chem. Inf. Model. 2010, 50, 1173–1178. [Google Scholar] [CrossRef]
- Wang, A.; Savas, U.; Stout, C.D.; Johnson, E.F. Structural characterization of the complex between α-naphthoflavone and human cytochrome P450 1B1. J. Biol. Chem. 2011, 286, 5736–5743. [Google Scholar] [CrossRef] [PubMed]
- Walsh, A.A.; Szklarz, G.D.; Scott, E.E. Human cytochrome P450 1A1 structure and utility in understanding drug and xenobiotic metabolism. J. Biol. Chem. 2013, 288, 12932–12943. [Google Scholar] [CrossRef] [PubMed]
- Bart, A.G.; Scott, E.E. Structures of human cytochrome P450 1A1 with bergamottin and erlotinib reveal active-site modifications for binding of diverse ligands. J. Biol. Chem. 2018, 293, 19201–19210. [Google Scholar] [CrossRef]
- Bart, A.G.; Takahashi, R.H.; Wang, X.; Scott, E.E. Human cytochrome P450 1A1 adapts active site for atypical nonplanar substrate. Drug Metab. Dispos. 2020, 48, 86–92. [Google Scholar] [CrossRef]
- Sansen, S.; Yano, J.K.; Reynald, R.L.; Schoch, G.A.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. Adaptations for the oxidation of polycyclic aromatic hydrocarbons exhibited by the structure of human P450 1A2. J. Biol. Chem. 2007, 282, 14348–14355. [Google Scholar] [CrossRef]
- Kubo, M.; Yamamoto, K.; Itoh, T. Design and synthesis of selective CYP1B1 inhibitor via dearomatization of α-naphthoflavone. Bioorganic Med. Chem. 2019, 27, 285–304. [Google Scholar] [CrossRef] [PubMed]
- Bart, A.G.; Harris, K.L.; Gillam, E.M.J.; Scott, E.E. Structure of an ancestral mammalian family 1B1 cytochrome P450 with increased thermostability. J. Biol. Chem. 2020, 295, 5640–5653. [Google Scholar] [CrossRef]
- Liu, J.; Taylor, S.F.; Dupart, P.S.; Arnold, C.L.; Sridhar, J.; Jiang, Q.; Wang, Y.; Skripnikova, E.V.; Zhao, M.; Foroozesh, M. Pyranoflavones: A group of small-molecule probes for exploring the active site cavities of cytochrome P450 enzymes 1A1, 1A2, and 1B1. J. Med. Chem. 2013, 56, 4082–4092. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, J.; Goyal, N.; Liu, J.; Foroozesh, M. Review of Ligand Specificity Factors for CYP1A Subfamily Enzymes from Molecular Modeling Studies Reported to-Date. Molecules 2017, 22, 1143. [Google Scholar] [CrossRef]
- Yu, X.; Cojocaru, V.; Wade, R.C. Conformational diversity and ligand tunnels of mammalian cytochrome P450s. Biotechnol. Appl. Biochem. 2013, 60, 134–145. [Google Scholar] [CrossRef]
- Dutkiewicz, Z. Computational methods for calculation of protein-ligand binding affinities in structure-based drug design. Phys. Sci. Rev. 2020. [Google Scholar] [CrossRef]
- Zhan, P.; Itoh, Y.; Suzuki, T.; Liu, X. Strategies for the discovery of target-specific or isoform-selective modulators. J. Med. Chem. 2015, 58, 7611–7633. [Google Scholar] [CrossRef]
- Lee, J.Y.; Cho, H.; Thangapandian, S.; Lim, C.; Chun, Y.J.; Lee, Y.; Choi, S.; Kim, S. Adaptable small ligand of CYP1 enzymes for use in understanding the structural features determining isoform selectivity. ACS Med. Chem. Lett. 2018, 9, 1247–1252. [Google Scholar] [CrossRef]
- Huggins, D.J.; Sherman, W.; Tidor, B. Rational approaches to improving selectivity in drug design. J. Med. Chem. 2012, 55, 1424–1444. [Google Scholar] [CrossRef] [PubMed]
- Chun, Y.-J.; Kim, S.; Kim, D.; Lee, S.-K.; Guengerich, F.P. A new selective and potent inhibitor of human cytochrome P450 1B1 and its application to antimutagenesis. Cancer Res. 2001, 61, 8164–8170. [Google Scholar] [PubMed]
- Chun, Y.J.; Oh, Y.K.; Kim, B.J.; Kim, D.; Kim, S.S.; Choi, H.K.; Kim, M.Y. Potent inhibition of human cytochrome P450 1B1 by tetramethoxystilbene. Toxicol. Lett. 2009, 189, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Chun, Y.J.; Lim, C.; Ohk, S.O.; Lee, J.M.; Lee, J.H.; Choi, S.; Kim, S. trans-Stilbenoids: Potent and selective inhibitors for human cytochrome P450 1B1. MedChemComm 2011, 2, 402–405. [Google Scholar] [CrossRef]
- Wierzchowski, M.; Dutkiewicz, Z.; Gielara-Korzańska, A.; Korzański, A.; Teubert, A.; Teżyk, A.; Stefański, T.; Baer-Dubowska, W.; Mikstacka, R. Synthesis, biological evaluation and docking studies of trans-stilbene methylthio derivatives as cytochromes P450 family 1 inhibitors. Chem. Biol. Drug Des. 2017, 90, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Sridhar, J.; Foroozesh, M. Cytochrome P450 family 1 inhibitors and structure-activity relationships. Molecules 2013, 18, 14470–14495. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Zhang, Q.; Cui, Q.; Huang, G.; Pan, X.; Li, S. Flavonoids and naphthoflavonoids: Wider roles in the modulation of cytochrome P450 family 1 enzymes. ChemMedChem 2016, 11, 2102–2118. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Tanaka, K.; Takenaka, S.; Murayama, N.; Martin, M.V.; Foroozesh, M.K.; Yamazaki, H.; Guengerich, F.P.; Komori, M. Structure–function relationships of inhibition of human cytochromes P450 1A1, 1A2, 1B1, 2C9, and 3A4 by 33 flavonoid derivatives. Chem. Res. Toxicol. 2010, 23, 1921–1935. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Huang, G.; Cui, Q.; Meng, Q.; Li, S.; Cui, J. Discovery of heterocycle-containing α-naphthoflavone derivatives as water-soluble, highly potent and selective CYP1B1 inhibitors. Eur. J. Med. Chem. 2021, 209, 112895. [Google Scholar] [CrossRef]
- Dong, J.; Wang, Z.; Cui, J.; Meng, Q.; Li, S. Synthesis and structure-activity relationship studies of α-naphthoflavone derivatives as CYP1B1 inhibitors. Eur. J. Med. Chem. 2020, 187, 111938. [Google Scholar] [CrossRef]
- Dong, J.; Huang, G.; Zhang, Q.; Wang, Z.; Cui, J.; Wu, Y.; Meng, Q.; Li, S. Development of benzochalcone derivatives as selective CYP1B1 inhibitors and anticancer agents. MedChemComm 2019, 10, 1606–1614. [Google Scholar] [CrossRef]
- Dutour, R.; Roy, J.; Cortés-Benítez, F.; Maltais, R.; Poirier, D. Targeting cytochrome P450 (CYP) 1B1 enzyme with four series of a-ring substituted estrane derivatives: Design, synthesis, inhibitory activity, and selectivity. J. Med. Chem. 2018, 61, 9229–9245. [Google Scholar] [CrossRef]
- Wang, Y.; He, X.; Li, C.; Ma, Y.; Xue, W.; Hu, B.; Wang, J.; Zhang, T.; Zhang, F. Carvedilol serves as a novel CYP1B1 inhibitor, a systematic drug repurposing approach through structure-based virtual screening and experimental verification. Eur. J. Med. Chem. 2020, 193, 112235. [Google Scholar] [CrossRef] [PubMed]
- Gajjar, K.; Martin-Hirsch, P.L.; Martin, F.L. CYP1B1 and hormone-induced cancer. Cancer Lett. 2012, 324, 13–30. [Google Scholar] [CrossRef]
- Chun, Y.J.; Kim, D. Cancer activation and polymorphisms of human cytochrome P450 1B1. Toxicol. Res. 2016, 32, 89–93. [Google Scholar] [CrossRef]
- Stoilov, I.; Akarsu, A.N.; Alozie, I.; Child, A.; Barsoum-Homsy, M.; Turacli, M.E.; Or, M.; Lewis, R.A.; Ozdemir, N.; Brice, G.; et al. Sequence analysis and homology modeling suggest that primary congenital glaucoma on 2p21 results from mutations disrupting either the hinge region or the conserved core structures of cytochrome P4501B1. Am. J. Hum. Genet. 1998, 62, 573–584. [Google Scholar] [CrossRef]
- McLellan, R.A.; Oscarson, M.; Hidestrand, M.; Leidvik, B.; Jonsson, E.; Otter, C.; Ingelman-Sundberg, M. Characterization and functional analysis of two common human cytochrome P450 1B1 variants. Arch. Biochem. Biophys. 2000, 378, 175–181. [Google Scholar] [CrossRef]
- Aklillu, E.; Oscarson, M.; Hidestrand, M.; Leidvik, B.; Otter, C.; Ingelman-Sundberg, M. Functional analysis of six different polymorphic CYP1B1 enzyme variants found in an ethiopian population. Mol. Pharmacol. 2002, 61, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Vasiliou, V.; Gonzalez, F.J. Role of CYP1B1 in Glaucoma. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 333–358. [Google Scholar] [CrossRef]
- Prokudin, I.; Simons, C.; Grigg, J.R.; Storen, R.; Kumar, V.; Phua, Z.Y.; Smith, J.; Flaherty, M.; Davila, S.; Jamieson, R.V. Exome sequencing in developmental eye disease leads to identification of causal variants in GJA8, CRYGC, PAX6 and CYP1B1. Eur. J. Hum. Genet. 2014, 22, 907–915. [Google Scholar] [CrossRef]
- Nishida, C.R.; Everett, S.; de Montellano, P.R.O. Specificity determinants of CYP1B1 estradiol hydroxylation. Mol. Pharmacol. 2013, 84, 451–458. [Google Scholar] [CrossRef]
- Shimada, T.; Murayama, N.; Kakimoto, K.; Takenaka, S.; Lim, Y.R.; Yeom, S.; Kim, D.; Yamazaki, H.; Guengerich, F.P.; Komori, M. Oxidation of 1-chloropyrene by human CYP1 family and CYP2A subfamily cytochrome P450 enzymes: Catalytic roles of two CYP1B1 and five CYP2A13 allelic variants. Xenobiotica 2018, 48, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Kim, D.; Murayama, N.; Tanaka, K.; Takenaka, S.; Nagy, L.D.; Folkman, L.M.; Foroozesh, M.K.; Komori, M.; Yamazaki, H.; et al. Binding of diverse environmental chemicals with human cytochromes P450 2A13, 2A6, and 1B1 and enzyme inhibition. Chem. Res. Toxicol. 2013, 26, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Du, Z.; Liu, J.; Zhou, Y.; Liang, F.; Lü, Q. Association between polymorphisms in estrogen metabolism genes and breast cancer development in Chinese women A prospective case-control study. Medicine 2018, 97, e13337. [Google Scholar] [CrossRef]
- Yang, J.; Xu, D.L.; Lu, Q.; Han, Z.J.; Tao, J.; Lu, P.; Wang, C.; Di, X.K.; Gu, M. Prostate cancer risk and aggressiveness associated with the CYP1B1 4326C/G (Leu432Val) polymorphism: A meta-analysis of 2788 cases and 2968 controls. Asian J. Androl. 2012, 14, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Dillehay, K.; Chen, W.; Shen, D.; Dong, Z.; Li, W. Association of the CYP1B1 Leu432Val polymorphism with the risk of prostate cancer: A meta-analysis. Mol. Biol. Rep. 2012, 39, 7465–7471. [Google Scholar] [CrossRef]
- Xie, Y.; Liu, G.Q.; Miao, X.Y.; Liu, Y.; Zhou, W.; Zhong, D.W. CYP1B1 Leu432Val polymorphism and colorectal cancer risk among Caucasians: A meta-analysis. Tumor Biol. 2012, 33, 809–816. [Google Scholar] [CrossRef]
- Liu, Y.; Lin, C.S.; Zhang, A.M.; Song, H.; Fan, C.C. The CYP1B1 Leu432Val polymorphism and risk of urinary system cancers. Tumor Biol. 2014, 35, 4719–4725. [Google Scholar] [CrossRef]
- Jain, A.N. Scoring functions for protein-ligand docking. Curr. Protein Pept. Sci. 2006, 7, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Moitessier, N.; Englebienne, P.; Lee, D.; Lawandi, J.; Corbeil, C.R. Towards the development of universal, fast and highly accurate docking/scoring methods: A long way to go. Br. J. Pharmacol. 2008, 153, S7–S26. [Google Scholar] [CrossRef]
- Spyrakis, F.; Cozzini, P.; Kellogg, G.E. Docking and Scoring in Drug Discovery. In Burger’s Medicinal Chemistry and Drug Discovery; Wiley: Hoboken, NJ, USA, 2010; pp. 601–684. [Google Scholar]
- Shen, C.; Ding, J.; Wang, Z.; Cao, D.; Ding, X.; Hou, T. From machine learning to deep learning: Advances in scoring functions for protein–ligand docking. WIREs Comput. Mol. Sci. 2020, 10, e1429. [Google Scholar] [CrossRef]
- Raha, K.; Merz, K.M. A quantum mechanics-based scoring function: Study of zinc ion-mediated ligand binding. J. Am. Chem. Soc. 2004, 126, 1020–1021. [Google Scholar] [CrossRef] [PubMed]
- Fanfrlík, J.; Bronowska, A.K.; Řezáč, J.; Přenosil, O.; Konvalinka, J.; Hobza, P. A reliable docking/scoring scheme based on the semiempirical quantum mechanical PM6-DH2 method accurately covering dispersion and H-bonding: HIV-1 protease with 22 ligands. J. Phys. Chem. B 2010, 114, 12666–12678. [Google Scholar] [CrossRef] [PubMed]
- Lepšík, M.; Řezáč, J.; Kolář, M.; Pecina, A.; Hobza, P.; Fanfrlík, J. The semiempirical quantum mechanical scoring function for in silico drug design. ChemPlusChem 2013, 78, 921–931. [Google Scholar] [CrossRef]
- Ajani, H.; Pecina, A.; Eyrilmez, S.M.; Fanfrlík, J.; Haldar, S.; Řezáč, J.; Hobza, P.; Lepšík, M. Superior performance of the SQM/COSMO scoring functions in native pose recognition of diverse protein-ligand complexes in cognate docking. ACS Omega 2017, 2, 4022–4029. [Google Scholar] [CrossRef]
- Cavasotto, C.N.; Aucar, M.G. High-throughput docking using quantum mechanical scoring. Front. Chem. 2020, 8, 246. [Google Scholar] [CrossRef] [PubMed]
- Alonso, H.; Bliznyuk, A.A.; Gready, J.E. Combining docking and molecular dynamic simulations in drug design. Med. Res. Rev. 2006, 26, 531–568. [Google Scholar] [CrossRef]
- Rao, B.C.; Subramanian, J.; Sharma, S.D. Managing protein flexibility in docking and its applications. Drug Discov. Today 2009, 14, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Kokh, D.B.; Wade, R.C.; Wenzel, W. Receptor flexibility in small-molecule docking calculations. WIREs Comput. Mol. Sci. 2011, 1, 298–314. [Google Scholar] [CrossRef]
- Lexa, K.W.; Carlson, H.A. Protein flexibility in docking and surface mapping. Q. Rev. Biophys. 2012, 45, 301–343. [Google Scholar] [CrossRef] [PubMed]
- Feixas, F.; Lindert, S.; Sinko, W.; McCammon, J.A. Exploring the role of receptor flexibility in structure-based drug discovery. Biophys. Chem. 2014, 186, 31–45. [Google Scholar] [CrossRef]
- Antunes, D.A.; Devaurs, D.; Kavraki, L.E. Understanding the challenges of protein flexibility in drug design. Expert Opin. Drug Discov. 2015, 10, 1301–1313. [Google Scholar] [CrossRef]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, A.; Coote, M.L.; Barakat, K. Molecular dynamics-driven drug discovery: Leaping forward with confidence. Drug Discov. Today 2017, 22, 249–269. [Google Scholar] [CrossRef]
- Hollingsworth, S.A.; Dror, R.O. Molecular dynamics simulation for all. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef]
- Bernardi, R.C.; Melo, M.C.R.; Schulten, K. Enhanced sampling techniques in molecular dynamics simulations of biological systems. Biochim. Biophys. Acta Gen. Subj. 2015, 1850, 872–877. [Google Scholar] [CrossRef]
- Spiwok, V.; Sucur, Z.; Hosek, P. Enhanced sampling techniques in biomolecular simulations. Biotechnol. Adv. 2015, 33, 1130–1140. [Google Scholar] [CrossRef]
- Yang, Y.I.; Shao, Q.; Zhang, J.; Yang, L.; Gao, Y.Q. Enhanced sampling in molecular dynamics. J. Chem. Phys. 2019, 151, 070902. [Google Scholar] [CrossRef] [PubMed]
- Lazim, R.; Suh, D.; Choi, S. Advances in molecular dynamics simulations and enhanced sampling methods for the study of protein systems. Int. J. Mol. Sci. 2020, 21, 6339. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Dirks, R.M.; Grossman, J.P.; Xu, H.; Shaw, D.E. Biomolecular simulation: A computational microscope for molecular biology. Annu. Rev. Biophys. 2012, 41, 429–452. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CYP | PDB Id | Ligand | Reference |
|---|---|---|---|
| CYP1A1 | 4I8V | α-naphthoflavone | [74] |
| 6DWM | bergamottin | [75] | |
| 6DWN | erlotinib | [75] | |
| 6O5Y | Pim kinase inhibitor GDC-0339 | [76] | |
| 6UDM | Duocarmycin Prodrug (S) ICT-2726 | to be published | |
| 6UDL | Duocarmycin Prodrug (S) ICT-2700 | to be published | |
| CYP1A2 | 2HI4 | α-naphthoflavone | [77] |
| CYP1B1 | 3PM0 | α-naphthoflavone | [73] |
| 6IQ5 | inhibitor having azide group | [78] | |
| Ancestral | 6OYU | α-naphthoflavone | [79] |
| CYP1B1 | 6OYV | estradiol | [79] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikstacka, R.; Dutkiewicz, Z. New Perspectives of CYP1B1 Inhibitors in the Light of Molecular Studies. Processes 2021, 9, 817. https://doi.org/10.3390/pr9050817
Mikstacka R, Dutkiewicz Z. New Perspectives of CYP1B1 Inhibitors in the Light of Molecular Studies. Processes. 2021; 9(5):817. https://doi.org/10.3390/pr9050817
Chicago/Turabian StyleMikstacka, Renata, and Zbigniew Dutkiewicz. 2021. "New Perspectives of CYP1B1 Inhibitors in the Light of Molecular Studies" Processes 9, no. 5: 817. https://doi.org/10.3390/pr9050817
APA StyleMikstacka, R., & Dutkiewicz, Z. (2021). New Perspectives of CYP1B1 Inhibitors in the Light of Molecular Studies. Processes, 9(5), 817. https://doi.org/10.3390/pr9050817