Abstract
Despite the significant progress in anticancer drug development over recent years, there is a vital need for newer agents with unique, but still effective, mechanisms of action in order to treat the disease, particularly the highly aggressive and drug-resistant types. Alkylating agents, in particular nitrogen-based alkylators, are commonly used to treat hematological and solid malignancies; they exert their antineoplastic effects at all phases of the cell cycle and prevent reproduction of tumor cells. Certain alkylating agents have been designed to be more lipophilic, enabling the compound to penetrate the cell and enhance its alkylating activity against tumors. This review details the evolution of currently available alkylating agents and their profiles, with a focus on nitrogen-based alkylating agents, as important anticancer therapy strategies.
1. Introduction
It is a well-known fact that cancer is a major worldwide health issue, threatening human health with increasing incidence and mortality. According to the World Health Organization (WHO), cancer is the second leading cause of deaths globally, after cardiovascular disease [1]. There has been significant progress in the development of anticancer drugs in recent years; however, newer agents require unique, yet effective, mechanisms of action in order to treat cancers, including those cancers that are highly aggressive and drug-resistant [2].
Alkylating agents, which prevent cell reproduction through direct deoxyribonucleic acid (DNA) damage, are widely used for the treatment of hematological and solid malignancies, often in combination with other chemotherapy medicines. For example, combining camptothecin (or a camptothecin derivative) with an alkylating agent, such as melphalan or cyclophosphamide, has been shown to significantly reduce tumor development compared with single-agent use [3]. Alkylating agents are electrophilic and covalently bind to electron-rich functional groups of various targeted molecules. Alkylating agents exert their antineoplastic effects at all phases of the cell cycle and occur through the addition of an alkyl group, most often at the N7 position of guanine residues. This alkyl group addition causes DNA fragmentation, intra- and inter-strand DNA cross-linking, or DNA mutations due to nucleotide mispairing. These mechanisms disrupt DNA replication and eventually result in cell death (or apoptosis) [4]. Nitrogen mustard agents (or nitrogen-based alkylators), and their derivatives, were the first anticancer chemotherapy medicines. They were initially used for malignant lymphoma in the 1940s and are becoming increasingly attractive as a proven antineoplastic agent [2,4]. Over the past eight decades, a large number of compounds, particularly nitrogen mustard-based hybrid molecules, have been developed.
However, overcoming intrinsic and acquired resistance to alkylating agents and other chemotherapeutic drugs continues to be a major challenge, especially when chemoresistance may lead to disease recurrence, metastasis, or death [5,6]. Therefore, it is key to understand the molecular mechanisms at play to improve the overall clinical outcomes for cancer patients and minimize the potential of treatment failure. There are various mechanisms that may trigger alkylating drug resistance, including the action of O6-methylguanine methyltransferase (MGMT), which can reverse DNA methylation damage by removing alkylating lesions [5,7]. Studies have shown that the inhibition of MGMT with O6-benzylguanine promotes the antitumor activity of methylating agents (such as temozolomide) both in vitro and in vivo [7], highlighting this major mechanism of resistance to the cytotoxic effects of alkylating agents. Another major obstacle of anticancer treatment is the lack of site specificity, which may lead to adverse events (AEs) on non-tumor cells. Hence, the development of new alkylating agents has focused on reducing the toxicity observed with the early agents to improve selectivity and cytotoxicity [2,8].
Alkylating agents are broadly classified into nitrogen mustards, nitrosoureas, alkyl sulfonates, triazines, ethylenimines, hydrazines, benzoquinone-containing agents, illudins and platinum-containing agents. This article will review the evolution of nitrogen-based alkylating agents, including the first discovery of their anticancer potential and as a developing cancer therapy strategy.
2. Nitrogen-Based Alkylating Agents
Despite its name, mustard gas is not produced from the mustard plant, but from its chemical substances. Mustard gas in its impure form is yellow-brown in color, with an odor similar to mustard plants. When exposed, mustard gas can cause serious injury to the eyes, skin, and respiratory tract [4]. Sulfur mustards (bis [2-chloroethyl]-sulfide; 1,5-dichloro-3-thiapentane), which are related to the mustard gas group, may cause large skin blisters and, due to their high toxicity, are not suitable for medical use.
Mustard gas was initially developed as a devastating chemical warfare agent in World War I and was subsequently banned as part of the 1925 Geneva Protocol [9]. However, during World War II, a ship explosion resulted in a vast number of military personnel being accidentally exposed to 2000 mustard gas bombs [4,8]. To prevent any further harm at the time, research to discover more efficient agents and protective measures was conducted, and it was found that nitrogen mustard agents were more stable (compared with sulfur mustards) [9]. In 1942, Gilman, Goodman, and co-workers at Yale discovered that bis(2-chloromethyl) methylamine (or chlormethine) possessed anticancer effects in animal models [10], and subsequent clinical trials showed encouraging results. Further clinical trials in patients with terminal malignant diseases resulted in successful outcomes. It is interesting to note that these findings were kept confidential due to the war, and were not disclosed for almost two decades when they were revealed by one of the discoverers [11].
Nitrogen-based alkylators are the most frequently used alkylating agents in cancer therapy [4,12]. Following uptake, the nitrogen-based alkylator is metabolized to a highly reactive aziridinium derivative that alkylates DNA and inhibits DNA reduplication [4]. There are several generations of nitrogen-based alkylating drugs (Table 1, Figure 1).

Table 1.
Examples of nitrogen-based alkylators.
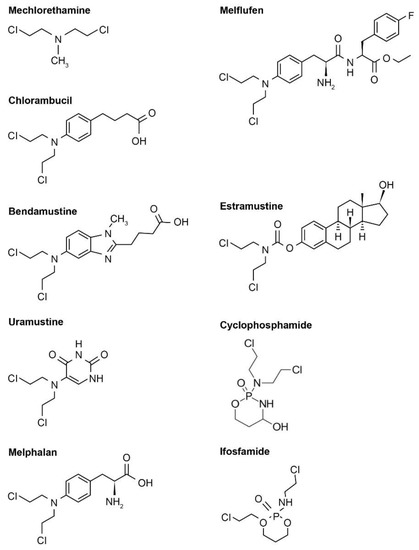
Figure 1.
Examples of various nitrogen-based alkylator chemical structures. Note that these chemical structures are publicly available and adapted from online resources (such as ChemDraw).
Among these highly reactive agents, the bis(2-chloroethyl) group is the main chemical constituent, and they all share a common mechanism of action, an in-situ generation of highly electrophilic cyclopropyl through an intramolecular nucleophilic substitution reaction [12,13]. The resulting aziridinium cation serves as the direct alkylating agent and, once formed, it undergoes nucleophilic attack by DNA bases, most often N-7 of guanine, to form a covalent bond (Figure 2). The remaining molecule is important in determining the physical properties of the agent, including transport, distribution, lipophilicity, and reactivity [12]. It is worth noting that a number of agents are prodrugs that have been designed to be selectively activated inside tumor cells. Overall, the underlying evolution of the nitrogen-based alkylator class is the design and development of molecules that are increasingly lipophilic and distributed effectively.

Figure 2.
Nitrogen mustard alkylation reaction process. Reprinted (adapted) with permission from [9], Copyright American Chemical Society, 2020.
2.1. First-Generation Nitrogen-Based Alkylators
For first-generation agents, aliphatic groups were linked to the mustard pharmacophore, –N(CH2CH2Cl)2. This group includes mechlorethamine hydrochloride (2-chloro-N-[2-chloroethyl]-N-methyl-ethanamine), a salt of a synthetic nitrogen-containing sulfur mustard derivative that has antineoplastic and lympholytic properties. Mechlorethamine hydrochloride, which was first approved in 1949 by the US Food and Drug Administration (FDA) [13], induces a rapid response and is indicated for the treatment of lymphosarcoma, Hodgkin’s disease (stages III and IV), chronic myelocytic or chronic lymphocytic leukemia, polycythemia vera, and bronchogenic carcinoma. Due to its highly reactive profile, when mechlorethamine is combined with water in the body, it no longer exists in an active form. As a result, mechlorethamine cannot be taken orally, and is administered as an intravenous infusion over 20 min. However, dosage may vary depending on the clinical situation. Potential adverse events (AEs) include fatigue, nausea, and vomiting, which can be minimized by administering at bedtime, followed by a barbiturate and an anti-emetic. Serious AEs may include anaphylactic reactions or bleeding. Contraindications to mechlorethamine include hypersensitivity and known infectious diseases. Additionally, due to bone marrow suppression, administering vaccinations around mechlorethamine treatment should be avoided [4].
2.2. Second-Generation Nitrogen-Based Alkylators
Chlormethine is too reactive for oral use and so must be administered intravenously. However, in second-generation agents, an electron-withdrawing aromatic ring is placed adjacent to the nitrogen atom, where the interaction between lone-pair electrons and the aromatic ring electrons makes the agent less prone to displacing the chloride ion that forms a transient aziridinium ring [9]. Such modifications result in more stable molecules that are less reactive with serum and cellular constituents compared with the first-generation nitrogen mustards, allowing oral administration in tablet form. This group includes chlorambucil, melphalan, and bendamustine.
Chlorambucil (4-[bis(2-chloroethyl)amino] benzenebutanoic acid), approved by the FDA in 1957 [13], is indicated for the palliative treatment of chronic lymphocytic leukemia, malignant lymphoma, lymphosarcoma, giant follicular lymphoma, and Hodgkin’s disease [4]. Chlorambucil alkylates and cross-links DNA at all cell cycle phases, and the electron withdrawing properties of its aromatic aryl ring means that reactions with cellular components are slower compared with the first-generation nitrogen-based alkylator agents [4]. Chlorambucil appears to be well tolerated by most patients [4] and less toxic than mechlorethamine [13]. Typically administered at 0.1–0.2 mg/kg/day for 3–6 weeks, the daily dose can be given in a single administration [4]. Severe neutropenia seems to only occur in patients receiving a total dosage of 6.5 mg/kg or more as part of a continuous course of therapy. Once discontinued, this effect may be reversible following approximately 10 days from the last dose; bone marrow failure has been known to occur in rare cases. Other less frequent AEs include diarrhea, oral ulcerations, nausea, vomiting, central nervous system (CNS) damage (such as tremors, hallucinations, muscular twitching, increased risk of seizures in children with nephrotic syndrome), skin reactions, and infertility [2,4].
Melphalan (2-amino-3-[4-(bis[2-chloroethyl]amino) phenyl]propanoic acid) is a phenylalanine derivative of nitrogen mustard and was approved by the FDA in 1964 [4,13]. It is indicated for the palliative treatment of multiple myeloma and for the palliation of non-respectable epithelial ovarian carcinoma. Melphalan is also indicated to treat breast cancer, neuroblastoma, and rhabdomyosarcoma, and occasionally used for malignant melanoma due to lack of melanocytes [2,13]. Similar to other nitrogen-based alkylators, the cytotoxicity of melphalan is harnessed through DNA cross-linking, and melphalan is transported to tumor cells using L-leucine-type transporters [13]. Melphalan can be given orally or intravenously, and dosing varies depending on route of administration, purpose, and the patient’s weight. Like with chlorambucil, daily dosage can be given at one time in an oral form. Melphalan is typically administered 6–10 mg daily for 2 to 3 weeks, followed by a rest interval of several weeks before starting the subsequent cycle. In combination with prednisone, melphalan has been used to enhance treatment effectiveness for multiple myeloma. Melphalan treatment has been linked with myelosuppression and lowered white blood cell count, increased risk of infection, and reduced platelet count; however, bone marrow suppression is generally reversible if melphalan is withdrawn early enough. Commonly reported AEs include nausea and vomiting, with possible allergic reactions following multiple rounds of treatment [4].
Bendamustine hydrochloride (4-[5-[bis(2-chloroethyl)amino]-1-methylbenzimidazol-2-yl]butanoic acid) was designed in the 1960s, first marketed in the early 1970s in East Germany, and approved by the FDA almost 40 years later in 2008 [4]. Bendamustine is indicated for the treatment of chronic lymphocytic leukemia, as well as multiple myeloma and indolent B-cell non-Hodgkin lymphoma [4]. While both bendamustine and chlorambucil are water-soluble, and share metabolic pathways and mechanism of action, bendamustine, with its benzimidazole moiety, possesses a more favorable safety profile [4,13]. Bendamustine is distributed freely in the blood and hydrolyzed to metabolites with low cytotoxic activity. The standard dose (100–120 mg/m2 body surface area) for bendamustine is administered intravenously over 30 to 60 min, with varying cycles for each indicated disease. The most common hematologic complications include lymphopenia, leukopenia, anemia, neutropenia, and thrombocytopenia. The most common non-hematologic AEs included pyrexia, nausea, diarrhea or constipation, headache, fatigue, anorexia, cough, rash, dyspnea, and stomatitis. In order to address any Grade 3/4 AEs, dose reduction or delayed administration could be implemented [4]. There is some caution around the use of bendamustine as an anticancer strategy, mainly due to its potentially genotoxic and neurotoxic characteristics, with a case report describing a patient developing ascending paralysis following treatment [13].
2.3. Third-Generation Nitrogen Mustards
In third-generation nitrogen mustards, the pyrimidine nucleus was selected as the carrier for the pharmacophore, as an approach to support oral administration [4]. This group includes uracil mustard (uramustine; 5-[bis(2-chloroethyl)amino]-1H-pyrimidine-2,4-dione), a bifunctional alkylating agent that acts on the cell cycle non-specifically. The activity of uramustine is derived from the formation of an unstable ethylenimmonium ion. Uramustine is a current treatment option for chronic lymphocytic leukemia, chronic myelogenous leukemia, Hodgkin lymphoma, non-Hodgkin lymphomata of the histiocytic or lymphocytic type, lymphosarcoma, ovarian cancer, and breast cancer. Uramustine is administered orally as capsules and taken once weekly for at least 4 weeks; the typical adult dose is 150 μg/kg. Uramustine is considered to be well tolerated and does not cause alopecia. Reported AEs include nausea, vomiting, diarrhea, dermatitis, nervousness, irritability, and depression. Additionally, due to the bone suppression effects, there is the potential of increased risk of microbial infections, bleeding and delayed healing [4].
2.4. Phosphoramide Mustards
Phosphoramide mustards (1,3,2-oxazaphosphinane) are formed by the conversion of the base nitrogen mustard into non-toxic prodrugs, and then actively transported into cancer cells where they are enzymatically converted into their active, cytotoxic forms [4]. Phosphoramide mustards include cyclophosphamide (2-[bis(2-chloroethyl)amino]-1,3,2λ5-oxazaphosphinan-2-one) and ifosfamide (3-(2-chloroethyl)-2-[(2-chloroethyl)amino]-1,3,2λ5-oxazaphosphinan-2-one), and current phosphoramide prodrugs share a common 2H-oxazaphosphorine scaffold.
As a synthetic alkylating agent approved by the FDA in 1959 [13], cyclophosphamide (N, N-bise[2-chloroethyl]-2-oxo-1-oxa-3-aza-2u{5}-phosphacyclohexan-2-amine) can be administered orally or intravenously. It was initially assumed that cyclophosphamide would be activated by phosphoramidase enzymes present in target tumor cells; however, this turned out not to be the case [13]. Instead, activation occurs via cytochrome P450 (CYP450)-mediated hydroxylation in the liver [14]. Upon cellular uptake, cyclophosphamide is metabolized by the CYP450 oxidase enzyme to form an active phosphoramide mustard product and acrolein, an inactive yet toxic metabolite that affects the bladder epithelium (Figure 3) [3,13]. The active phosphoramide mustard is subsequently metabolized into an active nornitrogen mustard (normeclorethamine) [13]. Cyclophosphamide targets various cancers, including lymphomata (e.g., multiple myeloma, Stage 3–4 malignant lymphomata, Hodgkin’s disease, Burkitt lymphoma, nodular or diffuse lymphocytic lymphoma) and some leukemias (chronic lymphocytic leukemia, acute myelogenous and monocytic leukemia) [2,4]. Depending on the clinical situation, doses vary and can be adjusted to account for antitumor activity and tolerable myelosuppression. Nausea, vomiting, and leukopenia are commonly reported AEs associated with cyclophosphamide treatment, and there is an increased risk of infections. Hematologic effects, such as thrombocytopenia or anemia, are known to occur occasionally, which can usually be reversed using dose reductions or interrupting treatment [4]. Cyclophosphamide is often administered in combination with chemotherapy in order to address drug resistance in tumor cells and minimize AEs. For example, the standard combination treatment for advanced non-Hodgkin lymphoma is cyclophosphamide, doxorubicin, vincristine and prednisone (also known as CHOP) [13].
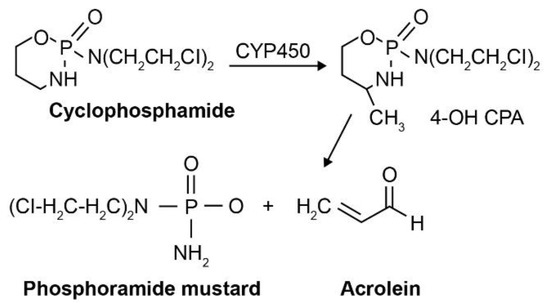
Figure 3.
Enzymatic conversion of non-toxic prodrug to active cytotoxic form, specifically the activation of cyclophosphamide by CYP450 oxidase. Reprinted (adapted) with permission from [3], Copyright Taylor & Francis, 2007.
Ifosfamide (3-[2-chloroethyl]-2-[(2-chloroethyl)amino] tetrahydro-2H-1,3,2-oxazaphosphorine 2-oxide) is an isomeric derivative of cyclophosphamide and was approved by the FDA in 1987 [4,13]. Ifosfamide produces 7-atom DNA cross-links (compared to 5-atom cross-links with cyclophosphamide). As with cyclophosphamide, ifosfamide requires hepatic activation by CYP450; however, due to its distinct metabolic pathway, generation of the cytotoxic metabolite occurs at a much slower rate than for cyclophosphamide [13,15]. Ifosfamide also displays dose-dependent pharmacokinetics, which may be seen at high concentrations in relevant metabolic pathways [4]. Ifosfamide is used across a range of cancers, including germ-cell testicular cancer, breast cancer, non-Hodgkin lymphoma, soft tissue sarcoma, ovarian cancer, cervical cancer and lung cancer. Administered intravenously, the daily dose for ifosfamide is 1.2 g/m2 body surface area for five consecutive days. Ifosfamide induces less myelotoxicity (mainly leukopenia) compared with cyclophosphamide, as well as exhibiting little cross-resistance. Commonly reported AEs include alopecia, nausea, vomiting, and transient nail ridging. There have been reports of neurotoxicity and urotoxicity following ifosfamide treatment, potentially connected to the formation of chloroacetaldehyde via N-dechloroethylation [4].
2.5. Steroid-Coupled Nitrogen Mustards
Recent modifications highlight steroid-coupled nitrogen mustards as a promising approach that, for bifunctionality, bring together the alkylating agent effects and a steroid for selective uptake, or combined anticancer action. By acting as a biological carrier, the steroid allows the nitrogen mustard moiety to be easily transported across the cellular barrier [8]. This group includes estramustine phosphate sodium (estradiol 3-[bis(2-chloroethyl)carbamate] 17-(dihydrogen phosphate), an oral, synthetic drug combining estradiol and mechlorethamine with a carbamate link. It is phosphorylated for improved water solubility and displays anti-androgenic effects. Estramustine phosphate sodium is primarily used for the palliative treatment of metastatic or progressive prostate cancer. It has been shown to be effective as an anticancer treatment option in combination with a 24-hydroxylase inhibitor (such as N-[4-(4-chlorophenyl)benzoyl]-2-(1H-imidazol-1-yl)-2-phenyl-1-aminoethane) [3]. It is taken orally, either before or after food, and the recommended daily dose is 14 mg/kg body weight, administered across three or four doses. Once absorbed, estramustine phosphate is dephosphorylated into estramustine, its estrone analog and estradiol [4]. Reported AEs include difficulty breathing, allergic reactions, and blood clots, where hypertension should be regularly assessed. Contraindications for estramustine include hypersensitivity to estradiol or nitrogen mustard [4].
2.6. Peptide Conjugation
Several aminopeptidases have been known to be overexpressed in malignancies, implicating their role as potential antitumor targets. The multifunctional protein aminopeptidase N (APN) has received the most attention due to its association with the varying phenotypes of human malignancies, particularly the fast-growing, aggressive types [16]. As research into this area develops, peptides are emerging as a promising therapeutic approach, exhibiting traits that make them ideal carriers of anticancer drugs to primary tumor sites (such as ease of synthesis, size, biocompatibility, and ability to target tumor cells).
Melphalan flufenamide (L-melphalanyl-p-L-fluorophenylalanine ethyl ester hydrochloride), also known as melflufen and previously denoted J1, is the ethyl ester of a dipeptide consisting of melphalan and para-fluoro-L-phenylalanine. Melflufen is highly lipophilic, and therefore is rapidly taken up by myeloma cells [16]. Inside the cell, cleavage of melflufen by aminopeptidases leads to enrichment of hydrophilic alkylating metabolites (such as melphalan) (Figure 4). In essence, by employing this simple peptide bond directly to APN-expressing cells, the agent provides a peptidase-potentiated effect, enriching its anti-myeloma activity [16,17,18].
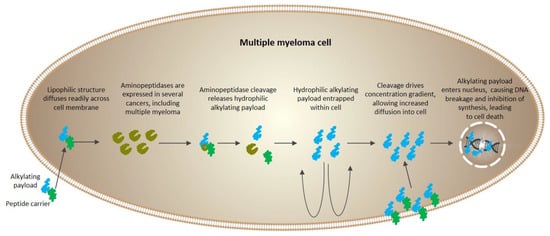
Figure 4.
Schematic of the mechanism of action for melflufen. Reprinted (adapted) with permission from [9], Copyright American Chemical Society, 2020.
Melflufen has been shown from in vitro studies to be more cytotoxic to myeloma cells than to peripheral blood mononuclear cells, as well as exhibiting activity in melphalan-resistant myeloma cells [19,20]. Studies using myeloma tumor samples have shown melphalan exposure to be 50 times higher with melflufen compared with direct administration of melphalan in culture. Melflufen has also shown to be substantially more cytotoxic than melphalan (approximately 50-fold) [19,21].
Melflufen is currently in late-stage clinical development, and US marketing approval was submitted in June 2020 [22]. Priority review was granted in August 2020 [23].
3. Alkylating Anticancer Agents: An Evolving Landscape
Alkylating agents, in particular nitrogen mustards, have been important anticancer treatments for many decades, and continue to evolve as researchers seek to overcome drug resistance and lack of selectivity [4,13]. By the late 1950s, approximately 500 nitrogen-based alkylators had been produced, where around 30 had progressed to the clinical trial stage [24]. As a group, alkylating agents are often used in combination with other anticancer drugs. Cyclophosphamide in combination with docetaxel has been demonstrated as an adjuvant therapy in breast cancer patients, with a favorable safety profile and mild AEs reported in postoperative patients [3]. The rationale of conjugating antitumor drugs with nitrogen-based alkylators may present a novel strategy for antitumor agents with enhanced antitumor effect, selectivity, and lowered toxicity (such as brefeldin A, a macrolide antibiotic with an antitumor, antiviral, and antifungal profile) [2].
Lipophilicity is an important physicochemical property in medicinal chemistry and has a major influence on the pharmacokinetics and pharmacodynamics of a drug substance. Some studies have shown a significant increase of lipophilic nature for the N-mustard construct compared with the parent compound [25]. To reduce the toxicity associated with nitrogen mustards, steroids have been tested as a vehicle to deliver the mustard drugs to a specific target tissue via interaction with steroid receptors, and over time, further developed conjugated alkylating agents have shown improved lipophilicity and solubility [2]. By improving lipophilicity, the active metabolite can sufficiently diffuse from its site of formation [3] and enhance certain agent characteristics, including rapid cellular uptake, cytotoxicity, and selectivity. This shift presents these modified agents as potentially novel and exciting alkylators that may improve clinical outcomes, possibly transforming the treatment landscape for patients with malignancies. However, it must be noted that lipophilicity can limit the dissolution of an antitumor agent. For orally administered agents, which need to be readily absorbed, their pharmacokinetic profile is dependent on possessing a hydrophilic–lipophilic balance to ensure that the agent is adequately distributed.
While this article covers currently approved drugs, preclinical and clinical progress has been made in the targeted delivery of nitrogen-based alkylator agents, including DNA-directed nitrogen mustards, antibody-directed enzyme prodrug therapy (ADEPT), gene-directed enzyme prodrug therapy (GDEPT), and CNS-targeted nitrogen mustards. DNA-targeted alkylating agents help to overcome the sequence-specific challenges with the majority of alkylating anticancer agents by using DNA intercalating carriers that improve the binding component of the DNA interaction. ADEPT involves linking an enzyme to an antibody that catalyzes prodrug conversion into the active form. This approach allows the alkylating agent to be cell cycle-independent and less likely to induce acquired resistance. For the GDEPT method, specific genes encoding an enzyme are introduced into cancer cells capable of catalyzing and activating the non-toxic prodrug into cytotoxic forms. Compared to ADEPT, which occurs extracellularly, prodrug activation occurs intracellularly in GDEPT [8]. Strategies that focus on stimuli-responsive (such as high levels of reactive oxygen species [ROS]) nitrogen mustard prodrugs are also gaining traction as anticancer therapies. For example, a ROS-activated nitrogen mustard prodrug encapsulated in liposomes has shown to specifically target and kill hematoma tumor cells [26]. In some cases, DNA carrier molecules have been used to address the loss of drug by reaction limitation associated with mustard alkylators and have been shown to bind intrinsically to the DNA core with both region and sequence specificity [27].
Author Contributions
F.L. and J.W. contributed equally to all sections of this article, including review and finalization. All authors have read and agreed to the published version of the manuscript.
Funding
This research and APC were funded by Oncopeptides AB, Sweden. F.L. is an employee of Oncopeptides AB; J.W. is an employee of Red Glead Discovery AB. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing not applicable.
Acknowledgments
Medical writing assistance and support was provided by Jaya Shumoogam of Aura, a division of Spirit Medical Communications Group Limited, and funded by Oncopeptides AB, Sweden.
Conflicts of Interest
The authors declare no conflict of interest.
References
- World Health Organization. Cancer. 2020. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 22 December 2020).
- Chen, Y.; Jia, Y.; Song, W.; Zhang, L. Therapeutic Potential of Nitrogen Mustard Based Hybrid Molecules. Front. Pharmacol. 2018, 9, 1453. [Google Scholar] [CrossRef]
- Ralhan, R.; Kaur, J. Alkylating agents and cancer therapy. Expert Opin. Ther. Patents 2007, 17, 1061–1075. [Google Scholar] [CrossRef]
- Weber, G.F. DNA Damaging Drugs. Mol. Ther. Cancer 2015, 9–112. [Google Scholar] [CrossRef]
- Wickström, M.; Dyberg, C.; Milosevic, J.; Einvik, C.; Calero, R.; Sveinbjörnsson, B.; Sandén, E.; Darabi, A.; Siesjö, P.; Kool, M.; et al. Wnt/β-catenin pathway regulates MGMT gene expression in cancer and inhibition of Wnt signalling prevents chemoresistance. Nat. Commun. 2015, 6, 8904. [Google Scholar] [CrossRef]
- Zheng, H.-C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Kitange, G.J.; James, C.D.; Plummer, R.; Calvert, H.; Weller, M.; Wick, W. Mechanisms of Chemoresistance to Alkylating Agents in Malignant Glioma. Clin. Cancer Res. 2008, 14, 2900–2908. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Kumar, S.; Prasad, D.N.; Bhardwaj, T.R. Therapeutic journery of nitrogen mustard as alkylating anticancer agents: Historic to future perspectives. Eur. J. Med. Chem. 2018, 151, 401–433. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, F.; Wennerberg, J. Melflufen: A journey from discovery to multi-kilogram production. In Complete Accounts of Integrated Drug Discovery and Development: Recent Examples from the Pharmaceutical Industry Volume 3; Pesti, J.A., Abdel-Magid, A.F., Vaidyanathan, R., Eds.; ACS Symposium Series; American Chemical Society: Washington DC, USA, 2020; Volume 1369, Chapter 5; pp. 157–177. [Google Scholar] [CrossRef]
- Gilman, A.; Philips, F.S. The Biological Actions and Therapeutic Applications of the B-Chloroethyl Amines and Sulfides. Science 1946, 103, 409–436. [Google Scholar] [CrossRef]
- Gilman, A. The initial clinical trial of nitrogen mustard. Am. J. Surg. 1963, 105, 574–578. [Google Scholar] [CrossRef]
- Colvin, M. Alkylating Agents. In Holland-Frei Cancer Medicine, 6th ed.; Kufe, D.W., Pollock, R.E., Weichselbaum, R.R., Bast, R.C., Gansler, T.S., Holland, J.F., Frei, E., Eds.; BC Decker: Hamilton, ON, Canada; Toronto, ON, Canada, 2003. Available online: https://www.ncbi.nlm.nih.gov/books/NBK12772/ (accessed on 22 December 2020).
- Diethelm-Varela, B.; Ai, Y.; Liang, D.; Xue, F. Nitrogen Mustards as Anticancer Chemotherapies: Historic Perspective, Current Developments and Future Trends. Curr. Top. Med. Chem. 2019, 19, 691–712. [Google Scholar] [CrossRef]
- Pass, G.J.; Carrie, D.; Lorimore, S.; Wright, E.; Houston, B.; Henderson, C.J.; Boylan, M.; Wolf, C.R. Role of Hepatic Cytochrome P450s in the Pharmacokinetics and Toxicity of Cyclophosphamide: Studies with the Hepatic Cytochrome P450 Reductase Null Mouse. Cancer Res. 2005, 65, 4211–4217. [Google Scholar] [CrossRef] [PubMed]
- A Fleming, R. An overview of cyclophosphamide and ifosfamide pharmacology. Pharmacother. J. Hum. Pharmacol. Drug Ther. 1997, 17, 146–154. [Google Scholar]
- Wickström, M.; Nygren, P.; Larsson, R.; Harmenberg, J.; Lindberg, J.; Sjöberg, P.; Jerling, M.; Lehmann, F.; Richardson, P.; Anderson, K.; et al. Melflufen - a peptidase-potentiated alkylating agent in clinical trials. Oncotarget 2017, 8, 66641–66655. [Google Scholar] [CrossRef] [PubMed]
- Gullbo, J.; Tullberg, M.; Våbenø, J.; Ehrsson, H.; Lewensohn, R.; Nygren, P.; Larsson, R.; Luthman, K. Structure–Activity Relationship for Alkylating Dipeptide Nitrogen Mustard Derivatives. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2003, 14, 113–132. [Google Scholar] [CrossRef]
- Wickström, M.; Viktorsson, K.; Lundholm, L.; Aesoy, R.; Nygren, H.; Sooman, L.; Fryknäs, M.; Vogel, L.K.; Lewensohn, R.; Larsson, R.; et al. The alkylating prodrug J1 can be activated by aminopeptidase N, leading to a possible target directed release of melphalan. Biochem. Pharmacol. 2010, 79, 1281–1290. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Ray, A.; Viktorsson, K.; Spira, J.; Paba-Prada, C.; Munshi, N.; Richardson, P.; Lewensohn, R.; Anderson, K.C. In Vitro and In Vivo Antitumor Activity of a Novel Alkylating Agent, Melphalan-Flufenamide, against Multiple Myeloma Cells. Clin. Cancer Res. 2013, 19, 3019–3031. [Google Scholar] [CrossRef]
- Ray, A.; Ravillah, D.; Das, D.S.; Song, Y.; Nordström, E.; Gullbo, J.; Richardson, P.G.; Chauhan, D.; Anderson, K.C. A novel alkylating agent Melflufen induces irreversible DNA damage and cytotoxicity in multiple myeloma cells. Br. J. Haematol. 2016, 174, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Wickström, M.; Haglund, C.; Lindman, H.; Nygren, P.; Larsson, R.; Gullbo, J. The novel alkylating prodrug J1: Diagnosis directed activity profile ex vivo and combination analyses in vitro. Investig. New Drugs 2007, 26, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Oncopeptides Submits a New Drug Application to the FDA for Accelerated Approval of Melflufen in Triple-Class Refractory Multiple Myeloma Patients. Available online: https://www.prnewswire.com/news-releases/oncopeptides-submits-a-new-drug-application-to-the-fda-for-accelerated-approval-of-melflufen-in-triple-class-refractory-multiple-myeloma-patients-301085607.html (accessed on 22 December 2020).
- FDA Grants Priority Review of Melflufen for Patients with Triple-Class Refractory Multiple Myeloma. Available online: https://www.prnewswire.com/news-releases/fda-grants-priority-review-of-melflufen-for-patients-with-triple-class-refractory-multiple-myeloma-301120645.html (accessed on 22 December 2020).
- Gram, H.F.; Mosher, C.W.; Baker, B.R. Potential Anticancer Agents.1XVII. Alkylating Agents to Phenylalanine Mustard. I. J. Am. Chem. Soc. 1959, 81, 3103–3108. [Google Scholar] [CrossRef]
- Bartzatt, R.L. Synthesis and Alkylation Activity of a Nitrogen Mustard Agent to Penetrate the Blood-Brain Barrier. Drug Deliv. 2004, 11, 19–26. [Google Scholar] [CrossRef][Green Version]
- Lin, M.; Guo, W.; Zhang, Z.; Zhou, Y.; Chen, J.; Wang, T.; Zhong, X.; Lu, Y.; Yang, Q.; Wei, Q.; et al. Reduced Toxicity of Liposomal Nitrogen Mustard Prodrug Formulation Activated by an Intracellular ROS Feedback Mechanism in Hematological Neoplasm Models. Mol. Pharm. 2019, 17, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Denny, W.A. DNA minor groove alkylating agents. Curr. Med. Chem. 2001, 8, 533–544. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).