
Underestimation of Heritability across the Molecular Layers of the Gene Expression Process



Abstract
:1. Introduction
2. Materials and Methods
2.1. Expression and Genotype Data
2.2. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brynedal, B.; Choi, J.; Raj, T.; Bjornson, R.; Stranger, B.E.; Neale, B.M.; Voight, B.F.; Cotsapas, C. Large-scale trans-eQTLs affect hundreds of transcripts and mediate patterns of transcriptional co-regulation. Am. J. Hum. Genet. 2017, 100, 581–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spielman, R.S.; Bastone, L.A.; Burdick, J.T.; Morley, M.; Ewens, W.J.; Cheung, V.G. Common genetic variants account for differences in gene expression among ethnic groups. Nat. Genet. 2007, 39, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Liu, Y.; Jiang, N.; Chen, J.; Leach, L.; Luo, Z.; Wang, M. Genome-wide eQTLs and heritability for gene expression traits in unrelated individuals. BMC Genom. 2014, 15, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Gay, N.R.; Gloudemans, M.; Antonio, M.L.; Abell, N.S.; Balliu, B.; Park, Y.; Martin, A.R.; Musharoff, S.; Rao, A.S.; Aguet, F.; et al. Impact of admixture and ancestry on eQTL analysis and GWAS colocalization in GTEx. Genome Biol. 2020, 21, 1–20. [Google Scholar] [CrossRef]
- Lee, C. Genome-wide expression quantitative trait loci analysis using mixed models. Front. Genet. 2018, 9, 341. [Google Scholar] [CrossRef]
- Shin, J.; Lee, C. A mixed model reduces spurious genetic associations produced by population stratification in genome-wide association studies. Genomics 2015, 105, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Carayol, J.; Chabert, C.; Di Cara, A.; Armenise, C.; Lefebvre, G.; Langin, D.; Viguerie, N.; Metairon, S.; Saris, W.H.M.; Astrup, A.; et al. Protein quantitative trait locus study in obesity during weight-loss identifies a leptin regulator. Nat. Commun. 2017, 8, 2084. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, H.; Lee, C. Underestimation of heritability using a mixed model with a polygenic covariance structure in a genome-wide association study for complex traits. Eur. J. Hum. Genet. 2014, 22, 851–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickrell, J.K.; Pai, A.A.; Gilad, Y.; Pritchard, J.K. Noisy splicing drives mRNA isoform diversity in human cells. PLoS Genet. 2010, 6, e1001236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battle, A.; Khan, Z.; Wang, S.H.; Mitrano, A.; Ford, M.J.; Pritchard, J.K.; Gilad, Y. Genomic variation. Impact of regulatory variation from RNA to protein. Science 2015, 347, 664–667. [Google Scholar] [CrossRef] [Green Version]
- Ryu, J.; Lee, C. Regulatory nucleotide sequence signals for expression of the genes encoding ribosomal proteins. Front. Genet. 2020, 11, 501. [Google Scholar] [CrossRef]
- Jansen, R.; Hottenga, J.J.; Nivard, M.G.; Abdellaoui, A.; Laport, B.; de Geus, E.J.; Wright, F.A.; Penninx, B.W.J.H.; Boomsma, D.I. Conditional eQTL analysis reveals allelic heterogeneity of gene expression. Hum. Mol. Genet. 2017, 26, 1444–1451. [Google Scholar] [CrossRef] [PubMed]
- GTEx Consortium. The GTEx consortium atlas of genetic regulatory effects across human tissues. GT Sci. 2020, 369, 1318–1330. [Google Scholar]
- Kaina, B.; Christmann, M.; Naumann, S.; Roos, W.P. MGMT: Key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair 2007, 6, 1079–1099. [Google Scholar] [CrossRef] [PubMed]
- Liang, R.; Shen, X.L.; Zhang, B.; Li, Y.; Xu, W.; Zhao, C.; Luo, Y.; Huang, K. Apoptosis signal-regulating kinase 1 promotes ochratoxin A-induced renal cytotoxicity. Sci. Rep. 2015, 5, 8078. [Google Scholar] [CrossRef] [Green Version]
- Massart, F.; Saggese, G. Morphogenetic targets and genetics of undescended testis. Sex Dev. 2010, 4, 326–335. [Google Scholar] [CrossRef]
- Yang, L.; Liu, Q.; Zhang, X.; Liu, X.; Zhou, B.; Chen, J.; Huang, D.; Li, J.; Li, H.; Chen, F.; et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature 2020, 583, 133–138. [Google Scholar] [CrossRef]
- Vaquerizas, J.M.; Kummerfeld, S.K.; Teichmann, S.A.; Luscombe, N.M. A census of human transcription factors: Function, expression and evolution. Nat. Rev. Genet. 2009, 10, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Lupo, A.; Cesaro, E.; Montano, G.; Zurlo, D.; Izzo, P.; Costanzo, P. KRAB-zinc finger proteins: A repressor family displaying multiple biological functions. Curr. Genom. 2013, 14, 268–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alders, M.; Ryan, A.; Hodges, M.; Bliek, J.; Feinberg, A.P.; Privitera, O.; Westerveld, A.; Little, P.F.; Mannens, M. Disruption of a novel imprinted zinc-finger gene, ZNF215, in Beckwith–Wiedemann Syndrome. Am. J. Hum. Genet. 2000, 66, 1473–1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
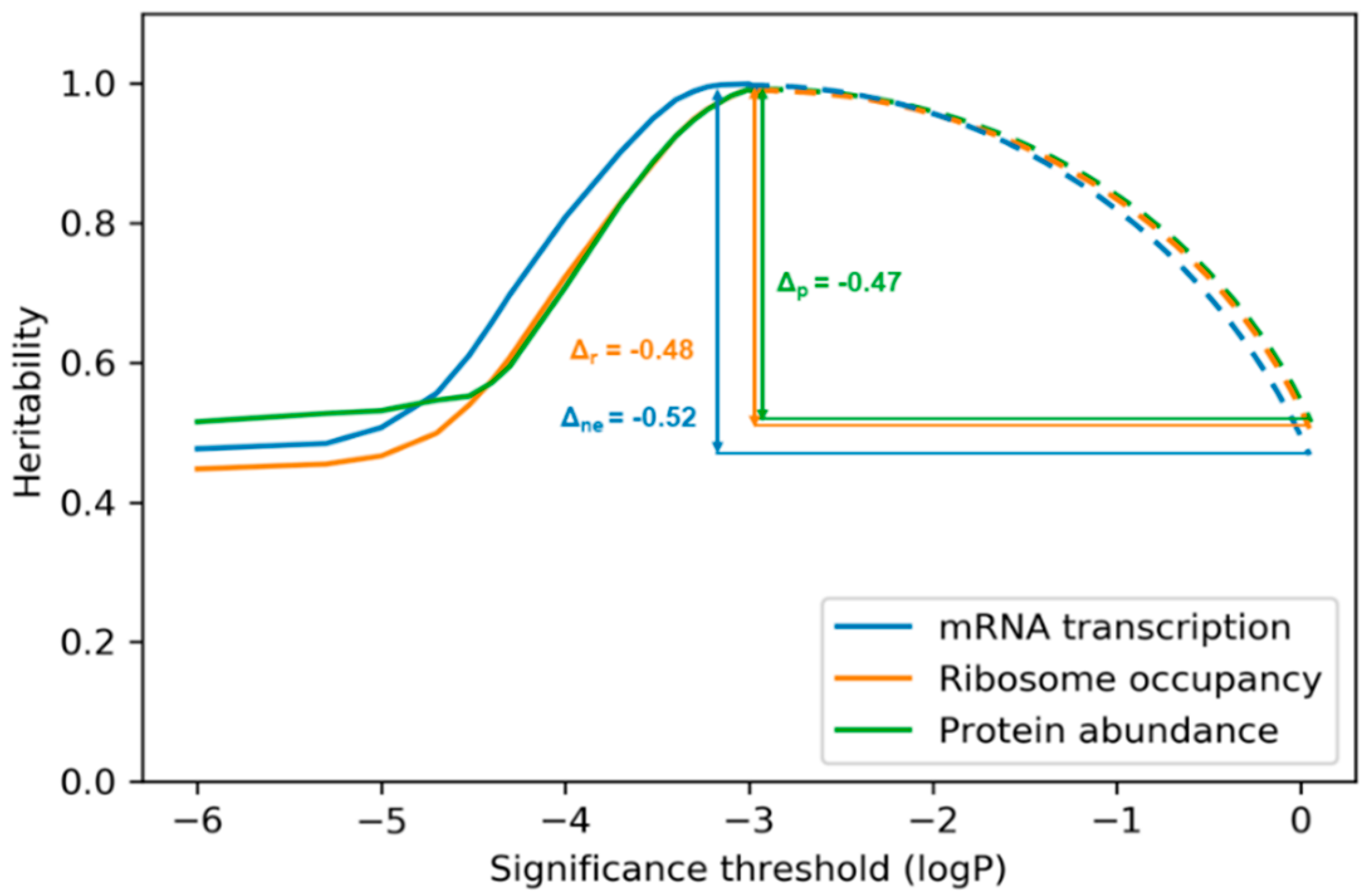

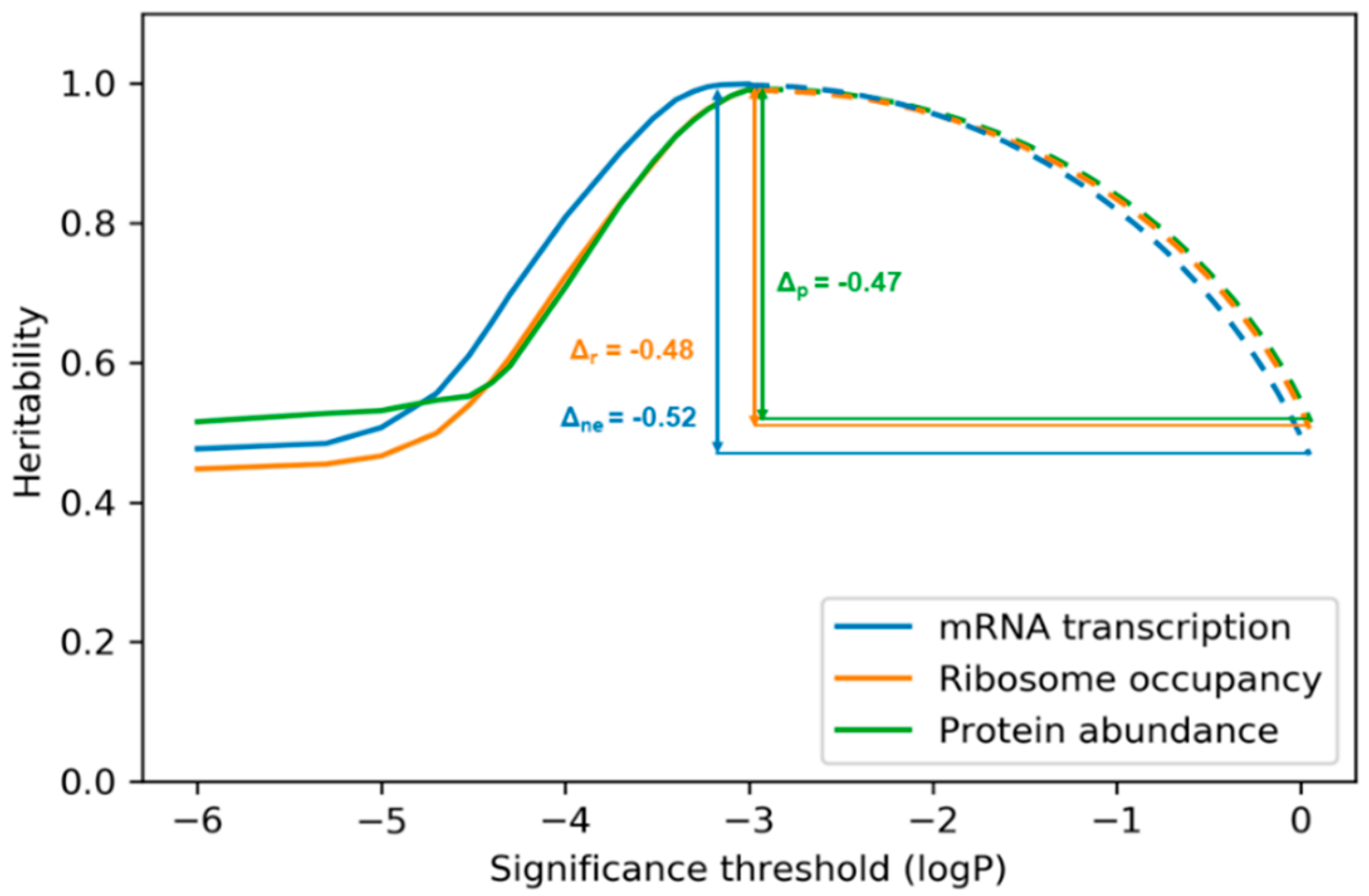{kind=link}
| Features | mRNA Transcription (A) | Protein Abundance (B) | Difference (A-B) | p-Value | |
|---|---|---|---|---|---|
| h2 | 0.477 | 0.525 | −0.048 | 1.06 × 10−6 | |
| No. of eQTLs | 13.9 | 16.9 | −3.0 | 2.82 × 10−1 | |
| h2/No. of eQTLs | 0.221 | 0.281 | −0.060 | 1.48 × 10−5 | |
| h2 | 0.801 | 0.699 | 0.102 | 1.32 × 10−156 | |
| No. of eQTLs | 138.2 | 75.6 | 62.6 | 9.56 × 10−46 | |
| h2/No. of eQTLs | 0.011 | 0.044 | −0.033 | 4.80 × 10−151 |
| eQTL Type | SNP | Position § | Allele ¶ | MAF | eGene (Chr) | cis/trans Regulation | Significance w/ ‡ | |
|---|---|---|---|---|---|---|---|---|
| Conventional GSM | Optimal GSM | |||||||
| neQTL | rs11016815 | 10; 129502351 | G/C | 0.1032 | SCP2 (1) | trans | 2.84 × 10−3 | 2.98 × 10−6 |
| rQTL | rs1041872 | 11; 6979775 | G/A | 0.1048 | CCDC25 (8) | trans | 3.40 × 10−3 | 9.46 × 10−6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryu, J.; Lee, C. Underestimation of Heritability across the Molecular Layers of the Gene Expression Process. Processes 2021, 9, 2144. https://doi.org/10.3390/pr9122144
Ryu J, Lee C. Underestimation of Heritability across the Molecular Layers of the Gene Expression Process. Processes. 2021; 9(12):2144. https://doi.org/10.3390/pr9122144
Chicago/Turabian StyleRyu, Jihye, and Chaeyoung Lee. 2021. "Underestimation of Heritability across the Molecular Layers of the Gene Expression Process" Processes 9, no. 12: 2144. https://doi.org/10.3390/pr9122144
APA StyleRyu, J., & Lee, C. (2021). Underestimation of Heritability across the Molecular Layers of the Gene Expression Process. Processes, 9(12), 2144. https://doi.org/10.3390/pr9122144