
PBPK Modeling and Simulation and Therapeutic Drug Monitoring: Possible Ways for Antibiotic Dose Adjustment



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
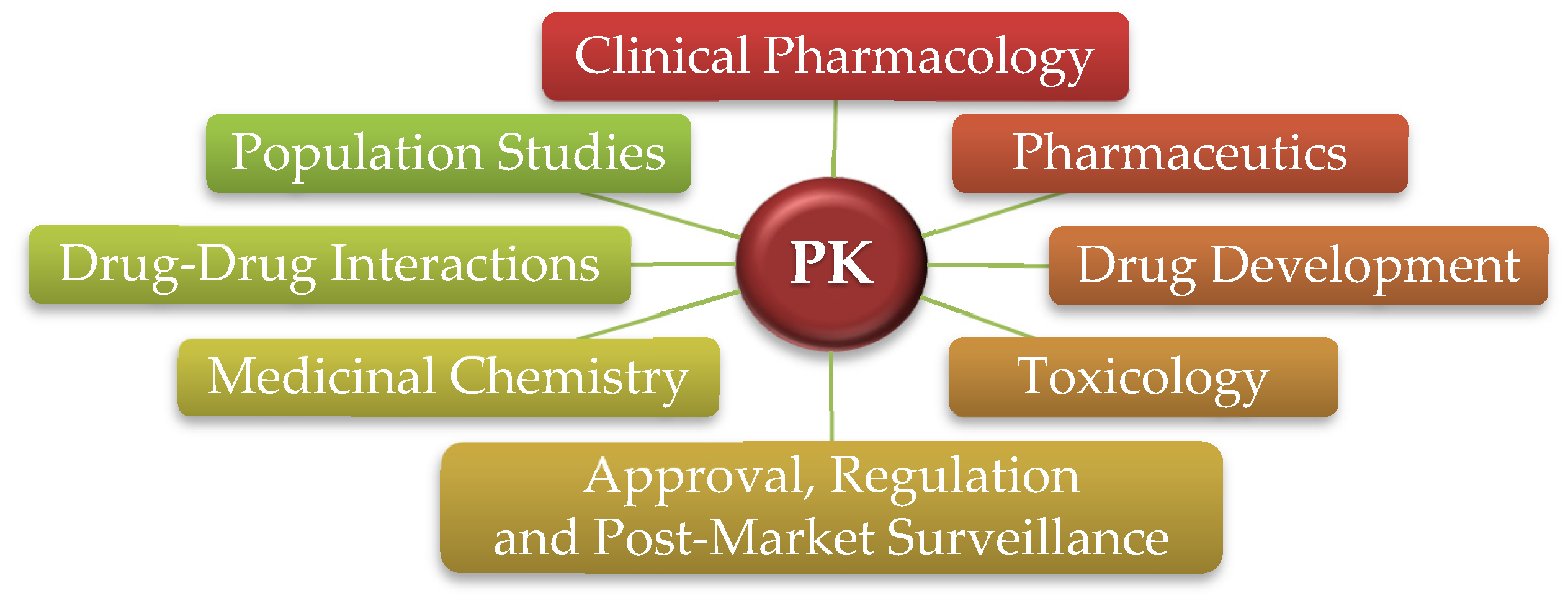
:1. Introduction–Pharmacokinetics
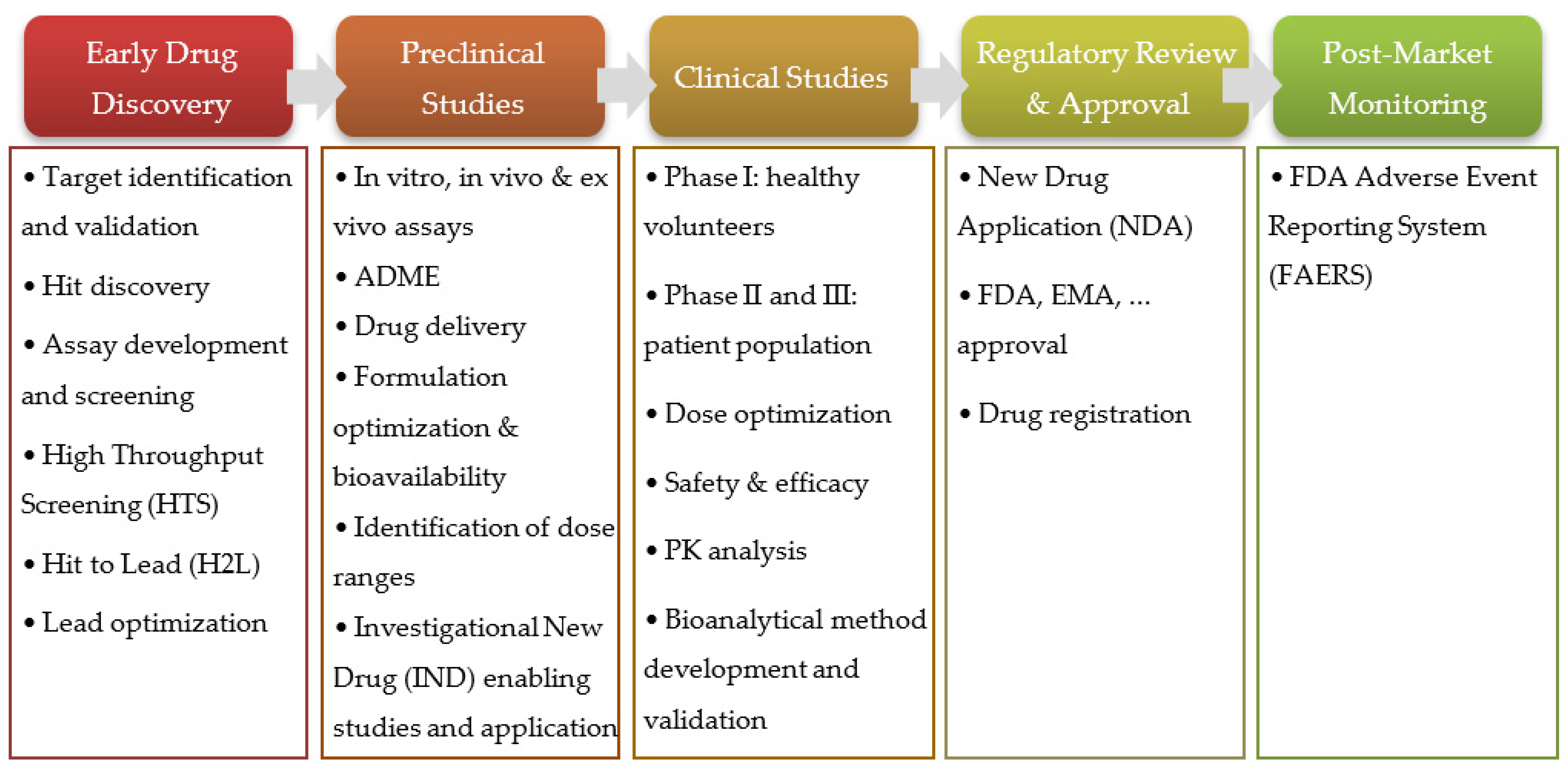
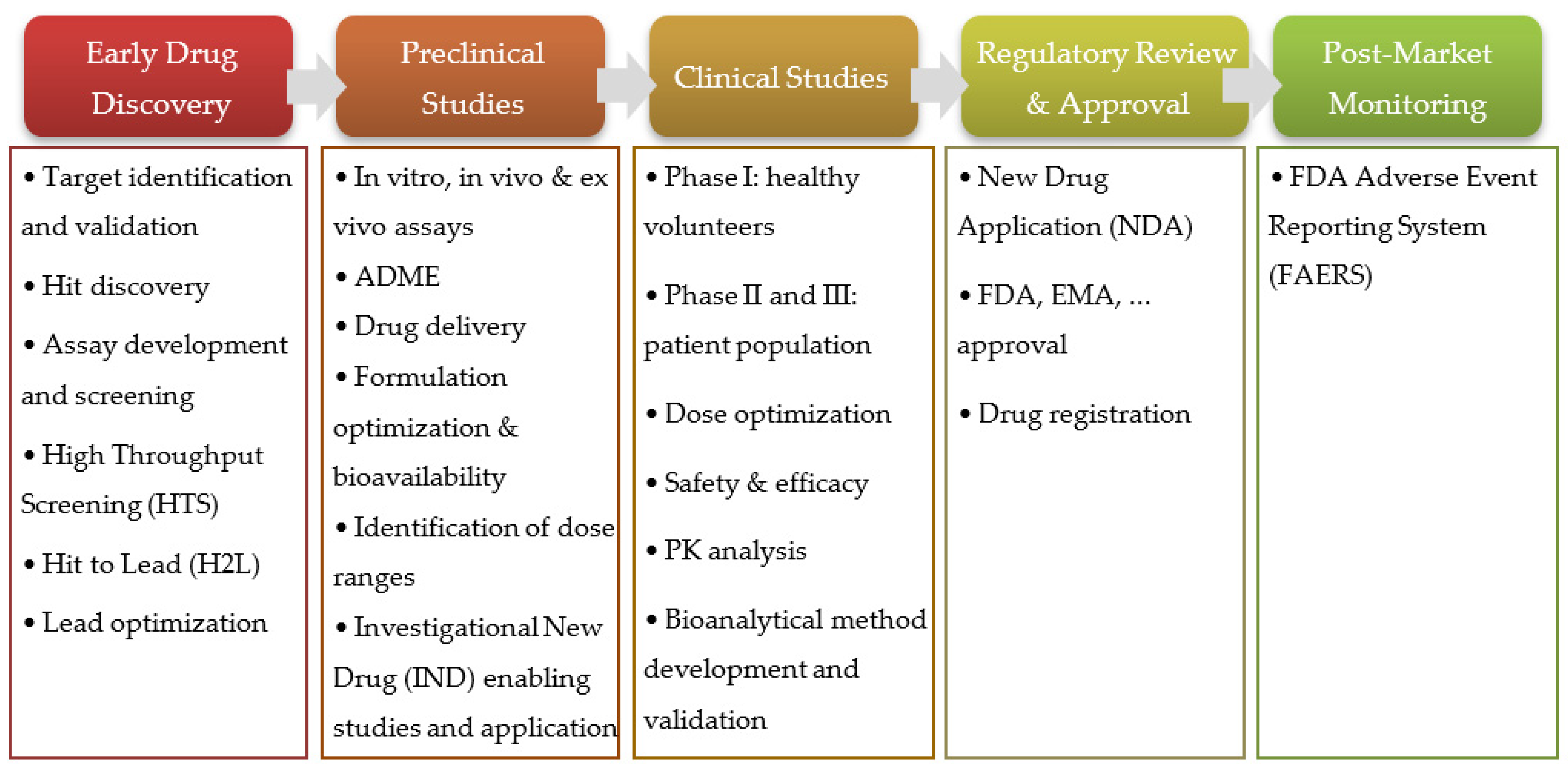
2. Prediction of ADME Properties and PK Modeling and Simulation
2.1. Noncompartmental PK Analysis (NCA)
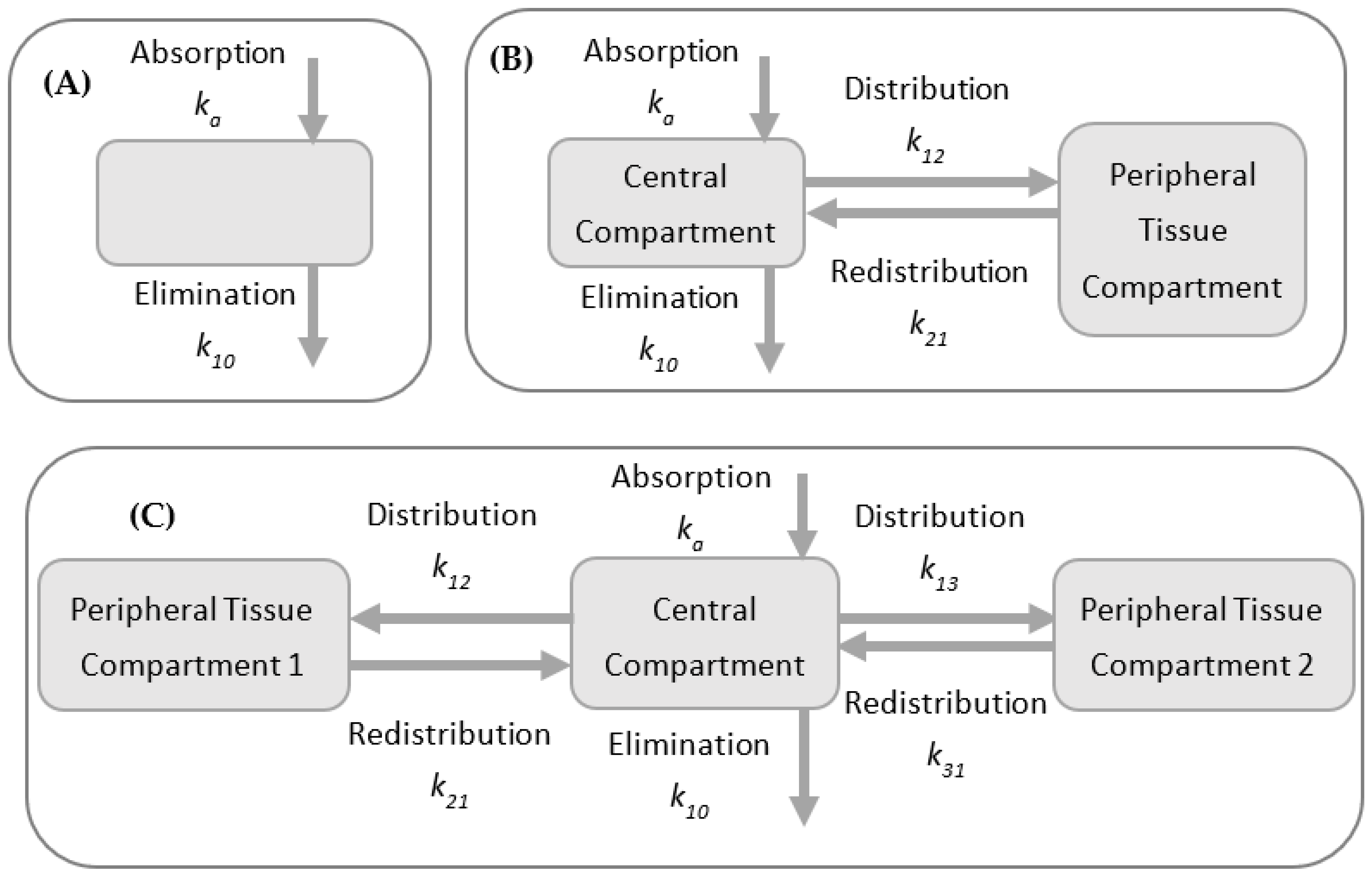
2.2. Compartmental Models
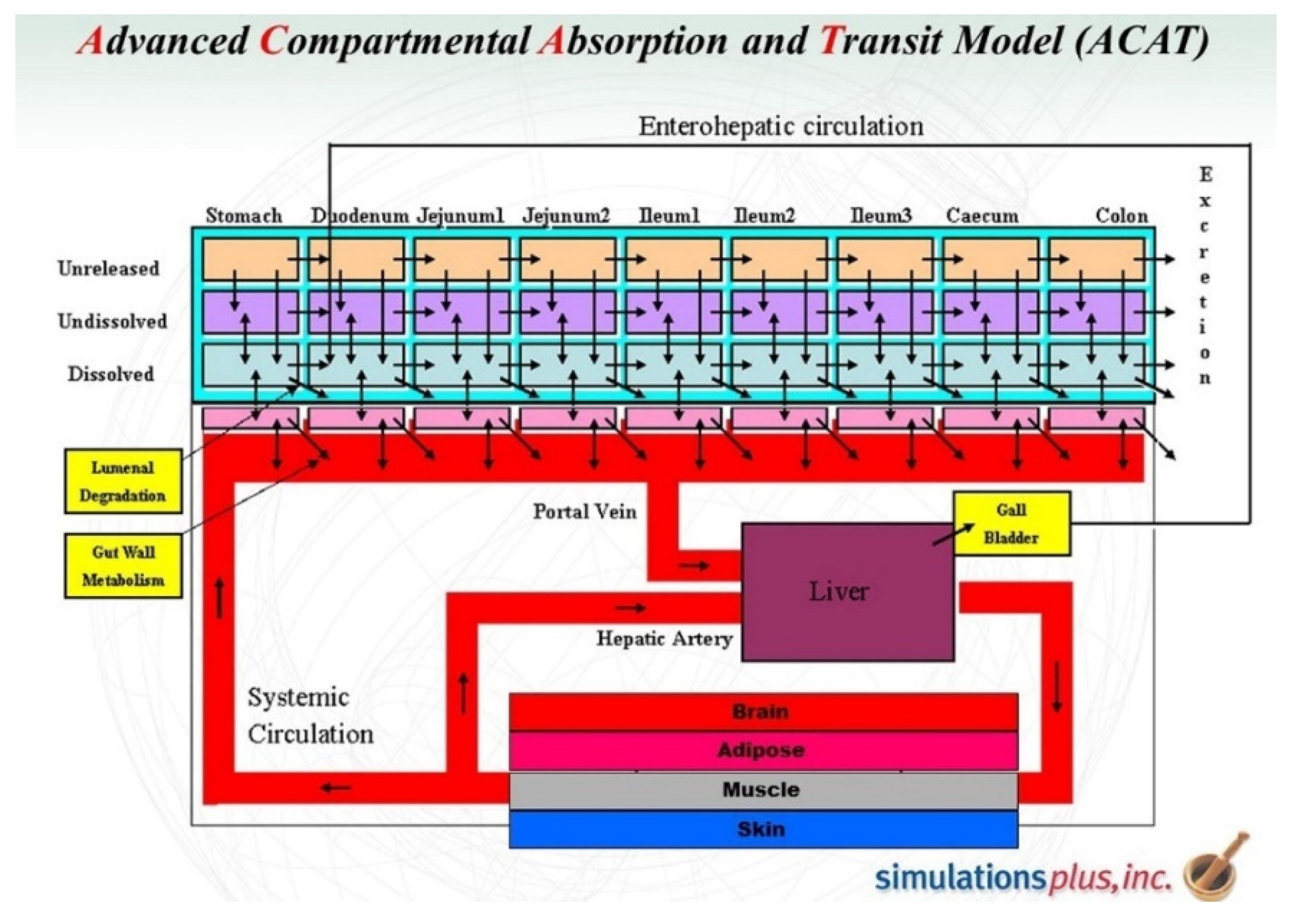
2.3. Physiologically Based Pharmacokinetic Models (PBPK)
2.4. Population PK
3. PK Prediction in Silico Tools
4. Therapeutic Drug Monitoring (TDM)
- Poorly predictable PK and significant interpatient variability, resulting in a wide range of concentration levels between patients after standard dosage administration;
- Narrow therapeutic window that, combined with interpatient variability, poses a high risk of misdoing. The standard dosage could be subtherapeutic for some patients, but the use of very high standard doses in all patients to ensure overall efficacy is forbidden due to the risk of toxicity [44];
- Consistent concentration exposure and response and/or toxicity (PD) relationships; moreover, effects following changes in drug exposure should be reversible, enabling the definition of a range of concentrations associated with optimal efficacy and minimal toxicity;
- Lack of readily assessable PD markers and quick response to dosage changes;
- Acceptable PK stability, considering within-patient PK variability over time (inter-occasion variability) and assay and/or model-related errors [45].
4.1. Target Concentration Intervention (TCI)
4.2. Model-Informed Precision Dosing (MIPD)
5. Infections and Antibiotic Therapy
Antibiotics and the Need for TDM and Dosage Adjustment
6. Final Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tozer, T.N.; Rowland, M. Essentials of Pharmacokinetics and Pharmacodynamics; Wolters Kluwer: Alphen aan den Rijn, The Netherlands, 2015; ISBN 9781451194425. [Google Scholar]
- Fan, J.; de Lannoy, I.A. Pharmacokinetics. Biochem. Pharmacol. 2014, 87, 93–120. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, M.; Duffus, J.; Templeton, D.M. Glossary of terms used in toxicokinetics (IUPAC Recommendations 2003). Pure Appl. Chem. 2004, 76, 1033–1082. [Google Scholar] [CrossRef]
- Research and Development in the Pharmaceutical Industry. Available online: https://www.cbo.gov/publication/57126 (accessed on 1 July 2021).
- Sertkaya, A.; Wong, H.-H.; Jessup, A.; Beleche, T. Key cost drivers of pharmaceutical clinical trials in the United States. Clin. Trials 2016, 13, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. New drugs cost US$2.6 billion to develop. Nat. Rev. Drug Discov. 2014, 13, 877. [Google Scholar] [CrossRef]
- USA Food and Drug Admistration. The Drug Development Process. Available online: https://www.fda.gov/patients/learn-about-drug-and-device-approvals/drug-development-process (accessed on 1 July 2021).
- Alavijeh, M.; Palmer, A. The pivotal role of drug metabolism and pharmacokinetics in the discovery and development of new medicines. IDrugs 2004, 7, 755–763. [Google Scholar]
- Pappalardo, F.; Russo, G.; Tshinanu, F.M.; Viceconti, M. In silico clinical trials: Concepts and early adoptions. Br. Bioinform. 2019, 20, 1699–1708. [Google Scholar] [CrossRef]
- Marsousi, N.; Desmeules, J.A.; Rudaz, S.; Daali, Y. Usefulness of PBPK Modeling in Incorporation of Clinical Conditions in Personalized Medicine. J. Pharm. Sci. 2017, 106, 2380–2391. [Google Scholar] [CrossRef] [Green Version]
- Michelson, S.; Sehgal, A.; Friedrich, C. In silico prediction of clinical efficacy. Curr. Opin. Biotechnol. 2006, 17, 666–670. [Google Scholar] [CrossRef]
- Kimko, H.; Pinheiro, J. Model-based clinical drug development in the past, present and future: A commentary. Br. J. Clin. Pharmacol. 2015, 79, 108–116. [Google Scholar] [CrossRef] [Green Version]
- Bolger, M.B.; Fraczkiewicz, R.; Lukacova, V. Simulations of Absorption, Metabolism, and Bioavailability. In Drug Bioavailability; Mannhold, R., Kubinyi, H., Folkers, G., van de Waterbeemd, H., Testa, B., Eds.; John Wiley Sons: Hoboken, NJ, USA, 2008; pp. 453–495. [Google Scholar] [CrossRef]
- Huang, Q.; Riviere, J.E. The application of allometric scaling principles to predict pharmacokinetic parameters across species. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1241–1253. [Google Scholar] [CrossRef]
- Cho, H.J.; Kim, J.E.; Kim, D.D.; Yoon, I.S. In Vitro-In Vivo extrapolation (IVIVE) for predicting human intestinal absorption and first-pass elimination of drugs: Principles and applications. Drug Dev. Ind. Pharm. 2014, 40, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.T. Predictions of the ADMET properties of candidate drug molecules utilizing different QSAR/QSPR modelling approaches. Curr. Drug Metab. 2010, 11, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Norinder, U.; Bergström, C.A.S. Prediction of ADMET properties. ChemMedChem Chem. Enabling Drug Discov. 2006, 1, 920–937. [Google Scholar]
- Van der Graaf, P.H.; Nilsson, J.; Van Schaick, E.A.; Danhof, M. Multivariate quantitative structure-pharmacokinetic relationships (QSPKR) analysis of adenosine A1 receptor agonists in rat. J. Pharm. Sci. 1999, 88, 306–312. [Google Scholar] [CrossRef]
- Van de Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef]
- Tylutki, Z.; Polak, S.; Wiśniowska, B. Top-down, Bottom-up and Middle-out Strategies for Drug Cardiac Safety Assessment via Modeling and Simulations. Curr. Pharmacol. Rep. 2016, 2, 171–177. [Google Scholar] [CrossRef] [Green Version]
- Tsamandouras, N.; Rostami-Hodjegan, A.; Aarons, L. Combining the “bottom up” and “top down” approaches in pharmacokinetic modelling: Fitting PBPK models to observed clinical data. Br. J. Clin. Pharmacol. 2015, 79, 48–55. [Google Scholar] [CrossRef]
- Gabrielsson, J.; Weiner, D. Pharmacokinetic and Pharmacodynamic Data Analysis: Concepts and Applications, 3rd ed.; Taylor & Francis: Abingdon, UK, 2001; ISBN 9789186274924. [Google Scholar]
- Marchenko, O.V.; Katenka, N.V. Quantitative Methods in Pharmaceutical Research and Development: Concepts and Applications; Springer International Publishing: Cham, Switzerland, 2020; ISBN 9783030485559. [Google Scholar]
- Gabrielsson, J.; Weiner, D. Non-compartmental analysis. Methods Mol. Biol. 2012, 929, 377–389. [Google Scholar] [CrossRef]
- Bulitta, J.B.; Holford, N.H.G. Non-Compartmental Analysis. In Wiley Encyclopedia of Clinical Trials; Wiley: Hoboken, NJ, USA, 2007; pp. 1–21. [Google Scholar]
- Chen, B.; Abuassba, A.O.M. Compartmental Models with Application to Pharmacokinetics. Procedia Comput. Sci. 2021, 187, 60–70. [Google Scholar] [CrossRef]
- Parrott, N.; Lave, T. Applications of physiologically based absorption models in drug discovery and development. Mol. Pharm. 2008, 5, 760–775. [Google Scholar] [CrossRef]
- Jones, H.; Rowland-Yeo, K. Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, e63. [Google Scholar] [CrossRef]
- Sager, J.E.; Yu, J.; Ragueneau-Majlessi, I.; Isoherranen, N. Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulation Approaches: A Systematic Review of Published Models, Applications, and Model Verification. Drug Metab. Dispos. 2015, 43, 1823–1837. [Google Scholar] [CrossRef]
- Upton, R.N.; Foster, D.J.R.; Abuhelwa, A.Y. An introduction to physiologically-based pharmacokinetic models. Pediatr. Anesth. 2016, 26, 1036–1046. [Google Scholar] [CrossRef] [PubMed]
- Nestorov, I. Whole Body Pharmacokinetic Models. Clin. Pharmacokinet. 2003, 42, 883–908. [Google Scholar] [CrossRef] [PubMed]
- Charles, B. Population pharmacokinetics: An overview. Aust. Prescr. 2014, 37, 210–213. [Google Scholar] [CrossRef]
- Ette, E.I.; Williams, P.J. Population Pharmacokinetics I: Background, Concepts, and Models. Ann. Pharmacother. 2004, 38, 1702–1706. [Google Scholar] [CrossRef]
- Di, L.; Feng, B.; Goosen, T.C.; Lai, Y.; Steyn, S.J.; Varma, M.V.; Obach, R.S. A Perspective on the Prediction of Drug Pharmacokinetics and Disposition in Drug Research and Development. Drug Metab. Dispos. 2013, 41, 1975–1993. [Google Scholar] [CrossRef] [Green Version]
- Daga, P.R.; Bolger, M.B.; Haworth, I.S.; Clark, R.D.; Martin, E.J. Physiologically Based Pharmacokinetic Modeling in Lead Optimization. 1. Evaluation and Adaptation of GastroPlus To Predict Bioavailability of Medchem Series. Mol. Pharm. 2018, 15, 821–830. [Google Scholar] [CrossRef]
- Xia, B.; Yang, Z.; Zhou, H.; Lukacova, V.; Zhu, W.; Milewski, M.; Kesisoglou, F. Development of a Novel Oral Cavity Compartmental Absorption and Transit Model for Sublingual Administration: Illustration with Zolpidem. Am. Assoc. Pharm. Sci. J. 2015, 17, 631–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, J.; Lobato, L.; Vale, N. Clinical pharmacokinetic study of latrepirdine via in silico sublingual administration. Silico Pharmacol. 2021, 9, 29. [Google Scholar] [CrossRef]
- Kostewicz, E.S.; Aarons, L.; Bergstrand, M.; Bolger, M.B.; Galetin, A.; Hatley, O.; Jamei, M.; Lloyd, R.; Pepin, X.; Rostami-Hodjegan, A.; et al. PBPK models for the prediction of In Vivo performance of oral dosage forms. Eur. J. Pharm. Sci. 2014, 57, 300–321. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, B.A.; Marshall, J.L.; Hartley, M.; Salem, M.E. A paradigm shift from one-size-fits-all to tailor-made therapy for metastatic colorectal cancer. Clin. Adv. Hematol. Oncol. 2016, 14, 116–128. [Google Scholar] [PubMed]
- Gastmeier, P. From “one size fits all” to personalized infection prevention. J. Hosp. Infect. 2020, 104, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Clarke, W.; Dasgupta, A. Clinical Challenges in Therapeutic Drug Monitoring: Special Populations, Physiological Conditions and Pharmacogenomics; Elsevier Science: Amsterdam, The Netherlands, 2016; ISBN 9780128020524. [Google Scholar]
- Kang, J.S.; Lee, M.H. Overview of therapeutic drug monitoring. Korean J. Intern. Med. 2009, 24, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Buclin, T.; Thoma, Y.; Widmer, N.; André, P.; Guidi, M.; Csajka, C.; Decosterd, L.A. The Steps to Therapeutic Drug Monitoring: A Structured Approach Illustrated with Imatinib. Front. Pharmacol. 2020, 11, 177. [Google Scholar] [CrossRef]
- Holford, N.H.G.; Buclin, T. Safe and Effective Variability—A Criterion for Dose Individualization. Ther. Drug Monit. 2012, 34, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Abrantes, J.A.; Jönsson, S.; Karlsson, M.O.; Nielsen, E.I. Handling interoccasion variability in model-based dose individualization using therapeutic drug monitoring data. Br. J. Clin. Pharmacol. 2019, 85, 1326–1336. [Google Scholar] [CrossRef] [PubMed]
- Widmer, N.; Bardin, C.; Chatelut, E.; Paci, A.; Beijnen, J.; Levêque, D.; Veal, G.; Astier, A. Review of therapeutic drug monitoring of anticancer drugs part two-Targeted therapies. Eur. J. Cancer 2014, 50, 2020–2036. [Google Scholar] [CrossRef]
- Muller, A.E.; Huttner, B.; Huttner, A. Therapeutic Drug Monitoring of Beta-Lactams and Other Antibiotics in the Intensive Care Unit: Which Agents, Which Patients and Which Infections? Drugs 2018, 78, 439–451. [Google Scholar] [CrossRef]
- Maitre, T.; Muret, P.; Blot, M.; Waldner, A.; Duong, M.; Si-Mohammed, A.; Chavanet, P.; Aho, S.; Piroth, L. Benefits and Limits of Antiretroviral Drug Monitoring in Routine Practice. Curr. HIV Res. 2019, 17, 190–197. [Google Scholar] [CrossRef]
- Papamichael, K.; Cheifetz, A.S. Therapeutic drug monitoring in patients on biologics: Lessons from gastroenterology. Curr. Opin. Rheumatol. 2020, 32, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Schoretsanitis, G.; Paulzen, M.; Unterecker, S.; Schwarz, M.; Conca, A.; Zernig, G.; Gründer, G.; Haen, E.; Baumann, P.; Bergemann, N.; et al. TDM in psychiatry and neurology: A comprehensive summary of the consensus guidelines for therapeutic drug monitoring in neuropsychopharmacology, update 2017; a tool for clinicians. World J. Biol. Psychiatry 2018, 19, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Keller, F.; Schröppel, B.; Ludwig, U. Pharmacokinetic and pharmacodynamic considerations of antimicrobial drug therapy in cancer patients with kidney dysfunction. World J. Nephrol. 2015, 4, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Holford, N. Pharmacodynamic principles and target concentration intervention. Transl. Clin. Pharmacol. 2018, 26, 150–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holford, N.; Ma, G.; Metz, D. TDM is dead. Long live TCI! Br. J. Clin. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Keizer, R.J.; ter Heine, R.; Frymoyer, A.; Lesko, L.J.; Mangat, R.; Goswami, S. Model-Informed Precision Dosing at the Bedside: Scientific Challenges and Opportunities. CPT Pharmacomet. Syst. Pharmacol. 2018, 7, 785–787. [Google Scholar] [CrossRef]
- Guidi, M.; Csajka, C.; Buclin, T. Parametric Approaches in Population Pharmacokinetics. J. Clin. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Goutelle, S.; Woillard, J.B.; Neely, M.; Yamada, W.; Bourguignon, L. Nonparametric Methods in Population Pharmacokinetics. J. Clin. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Xu, X.S.; Yuan, M.; Zhu, H.; Yang, Y.; Wang, H.; Zhou, H.; Xu, J.; Zhang, L.; Pinheiro, J. Full covariate modelling approach in population pharmacokinetics: Understanding the underlying hypothesis tests and implications of multiplicity. Br. J. Clin. Pharmacol. 2018, 84, 1525–1534. [Google Scholar] [CrossRef] [Green Version]
- Abbiati, R.A.; Manca, D. A modeling tool for the personalization of pharmacokinetic predictions. Comput. Chem. Eng. 2016, 91, 28–37. [Google Scholar] [CrossRef]
- Hartmanshenn, C.; Scherholz, M.; Androulakis, I.P. Physiologically-based pharmacokinetic models: Approaches for enabling personalized medicine. J. Pharmacokinet. Pharmacodyn. 2016, 43, 481–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, M.; Hirani, R.; Tsiu, M.; Kabani, K.; Chaturvedula, A.; Palasik, B. Utility of Physiologically Based Pharmacokinetic Modeling in Point-of-Care Decisions: An Example Using Digoxin Dosing in Continuous Venovenous Hemodiafiltration. Ther. Drug Monit. 2020, 42, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K. Use of Bayesian statistics in drug development: Advantages and challenges. Int. J. Appl. Basic Med. Res. 2012, 2, 3–6. [Google Scholar] [CrossRef]
- Donagher, J.; Barras, M.A. Therapeutic drug monitoring: Using Bayesian methods to evaluate hospital practice. J. Pharm. Pract. Res. 2018, 48, 522–529. [Google Scholar] [CrossRef]
- Drennan, P.; Doogue, M.; van Hal, S.J.; Chin, P. Bayesian therapeutic drug monitoring software: Past, present and future. Int. J. Pharmacokinet. 2018, 3, 109–114. [Google Scholar] [CrossRef]
- World Health Organization. Antimicrobial Resistance: Global Report on Surveillance; World Health Organization: Geneva, Switzerland, 2014; ISBN 9789241564748. [Google Scholar]
- WHO’s First Global Report on Antibiotic Resistance Reveals Serious, Worldwide Threat to Public Health. Available online: https://www.who.int/southeastasia/news/detail/30-04-2014-who-s-first-global-report-on-antibiotic-resistance-reveals-serious-worldwide-threat-to-public-health (accessed on 1 July 2021).
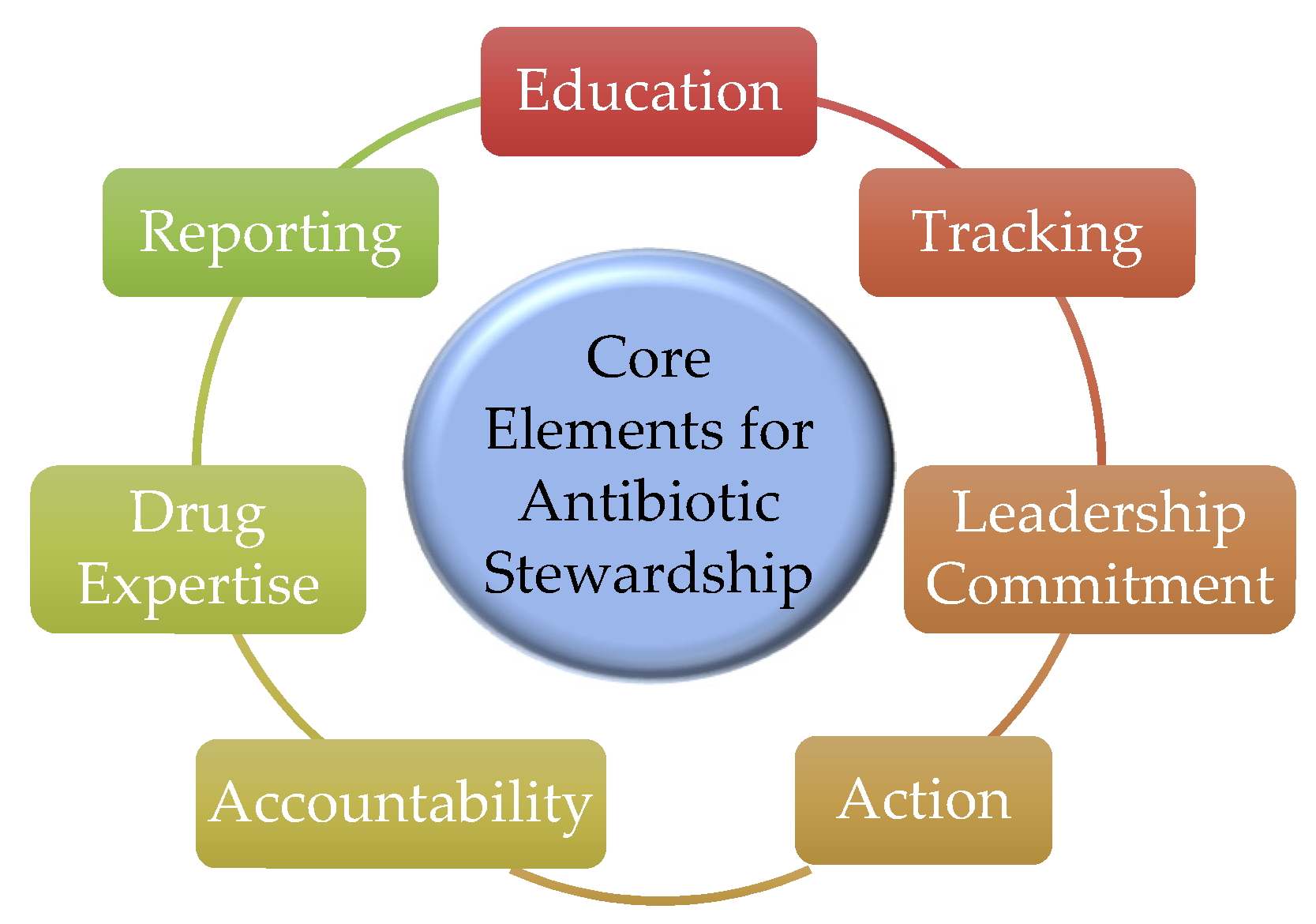
- Pollack, L.A.; Srinivasan, A. Core elements of hospital antibiotic stewardship programs from the Centers for Disease Control and Prevention. Clin. Infect. Dis. 2014, 59 (Suppl. 3), S97–S100. [Google Scholar] [CrossRef] [Green Version]
- Roberts, J.A.; Abdul-Aziz, M.H.; Lipman, J.; Mouton, J.W.; Vinks, A.A.; Felton, T.W.; Hope, W.W.; Farkas, A.; Neely, M.N.; Schentag, J.J.; et al. Individualised antibiotic dosing for patients who are critically ill: Challenges and potential solutions. Lancet Infect. Dis. 2014, 14, 498–509. [Google Scholar] [CrossRef] [Green Version]
- Cunha, C.B.; Opal, S.M. Antibiotic Stewardship: Strategies to Minimize Antibiotic Resistance While Maximizing Antibiotic Effectiveness. Med. Clin. N. Am. 2018, 102, 831–843. [Google Scholar] [CrossRef]
- Leekha, S.; Terrell, C.L.; Edson, R.S. General Principles of Antimicrobial Therapy. Mayo Clin. Proc. 2011, 86, 156–167. [Google Scholar] [CrossRef] [Green Version]
- Abdul-Aziz, M.H.; Alffenaar, J.C.; Bassetti, M.; Bracht, H.; Dimopoulos, G.; Marriott, D.; Neely, M.N.; Paiva, J.A.; Pea, F.; Sjovall, F.; et al. Antimicrobial therapeutic drug monitoring in critically ill adult patients: A Position Paper. Intensive Care Med. 2020, 46, 1127–1153. [Google Scholar] [CrossRef]
- Mabilat, C.; Gros, M.F.; Nicolau, D.; Mouton, J.W.; Textoris, J.; Roberts, J.A.; Cotta, M.O.; van Belkum, A.; Caniaux, I. Diagnostic and medical needs for therapeutic drug monitoring of antibiotics. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 791–797. [Google Scholar] [CrossRef] [Green Version]
- Wicha, S.G.; Märtson, A.G.; Nielsen, E.I.; Koch, B.C.P.; Friberg, L.E.; Alffenaar, J.W.; Minichmayr, I.K. From Therapeutic Drug Monitoring to Model-Informed Precision Dosing for Antibiotics. Clin. Pharmacol. Ther. 2021, 109, 928–941. [Google Scholar] [CrossRef]
- Choi, R.; Woo, H.I.; Park, H.D.; Lee, S.Y. A nationwide utilization survey of therapeutic drug monitoring for five antibiotics in South Korea. Infect. Drug Resist. 2019, 12, 2163–2173. [Google Scholar] [CrossRef]
- Begg, E.J.; Barclay, M.L.; Kirkpatrick, C.M. The therapeutic monitoring of antimicrobial agents. Br. J. Clin. Pharmacol. 2001, 52 (Suppl. 1), 35s–43s. [Google Scholar] [CrossRef]
- Reeves, D.; Lovering, A.; Thomson, A. Therapeutic drug monitoring in the past 40 years of the Journal of Antimicrobial Chemotherapy. J. Antimicrob. Chemother. 2016, 71, 3330–3332. [Google Scholar] [CrossRef] [Green Version]
- Cunha, B.A. Antibiotic side effects. Med. Clin. N. Am. 2001, 85, 149–185. [Google Scholar] [CrossRef]
- Yılmaz, Ç.; Özcengiz, G. Antibiotics: Pharmacokinetics, toxicity, resistance and multidrug efflux pumps. Biochem. Pharmacol. 2017, 133, 43–62. [Google Scholar] [CrossRef] [PubMed]
- Coulthard, K.P.; Peckham, D.G.; Conway, S.P.; Smith, C.A.; Bell, J.; Turnidge, J. Therapeutic drug monitoring of once daily tobramycin in cystic fibrosis--caution with trough concentrations. J. Cyst. Fibros. 2007, 6, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.K.; Tang, H.L.; Zhai, S.D. Benefits of therapeutic drug monitoring of vancomycin: A systematic review and meta-analysis. PLoS ONE 2013, 8, e77169. [Google Scholar] [CrossRef] [PubMed]
- Rybak, M.; Lomaestro, B.; Rotschafer, J.C.; Moellering Jr., R.; Craig, W.; Billeter, M.; Dalovisio, J.R.; Levine, D.P. Therapeutic monitoring of vancomycin in adult patients: A consensus review of the American Society of Health-System Pharmacists, the Infectious Diseases Society of America, and the Society of Infectious Diseases Pharmacists. Am. J. Health-Syst. Pharm. 2009, 66, 82–98. [Google Scholar] [CrossRef]
- Jenkins, A.; Thomson, A.H.; Brown, N.M.; Semple, Y.; Sluman, C.; MacGowan, A.; Lovering, A.M.; Wiffen, P.J. Amikacin use and therapeutic drug monitoring in adults: Do dose regimens and drug exposures affect either outcome or adverse events? A systematic review. J. Antimicrob. Chemother. 2016, 71, 2754–2759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovačević, T.; Avram, S.; Milaković, D.; Špirić, N.; Kovačević, P. Therapeutic monitoring of amikacin and gentamicin in critically and noncritically ill patients. J. Basic. Clin. Pharm. 2016, 7, 65–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Velde, F.; Mouton, J.W.; de Winter, B.C.M.; van Gelder, T.; Koch, B.C.P. Clinical applications of population pharmacokinetic models of antibiotics: Challenges and perspectives. Pharmacol. Res. 2018, 134, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Hodiamont, C.J.; Janssen, J.M.; de Jong, M.D.; Mathôt, R.A.; Juffermans, N.P.; van Hest, R.M. Therapeutic Drug Monitoring of Gentamicin Peak Concentrations in Critically Ill Patients. Ther. Drug Monit. 2017, 39, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Drennan, P.G.; Thoma, Y.; Barry, L.; Matthey, J.; Sivam, S.; van Hal, S.J. Bayesian Forecasting for Intravenous Tobramycin Dosing in Adults with Cystic Fibrosis Using One Versus Two Serum Concentrations in a Dosing Interval. Ther. Drug Monit. 2021, 43, 505–511. [Google Scholar] [CrossRef]
- Möhlmann, J.E.; van Luin, M.; Mascini, E.M.; van Leeuwen, H.J.; de Maat, M.R. Monitoring of tobramycin serum concentrations in selected critically ill patients receiving selective decontamination of the digestive tract: A retrospective evaluation. Eur. J. Clin. Pharmacol. 2019, 75, 831–836. [Google Scholar] [CrossRef]
- Roberts, J.A.; Lipman, J. Pharmacokinetic issues for antibiotics in the critically ill patient. Crit. Care Med. 2009, 37, 840–851. [Google Scholar] [CrossRef] [Green Version]
- Zapke, S.E.; Willmann, S.; Grebe, S.-O.; Menke, K.; Thürmann, P.A.; Schmiedl, S. Comparing Predictions of a PBPK Model for Cyclosporine with Drug Levels from Therapeutic Drug Monitoring. Front. Pharmacol. 2021, 12, 1134. [Google Scholar] [CrossRef]
- Emoto, C.; McPhail, B.T.; Fukuda, T. Clinical applications of physiologically based pharmacokinetic modeling: Perspectives on the advantages and challenges. Ther. Drug Monit. 2020, 42, 157–158. [Google Scholar] [CrossRef]
- Perry, C.; Davis, G.; Conner, T.M.; Zhang, T. Utilization of physiologically based pharmacokinetic modeling in clinical pharmacology and therapeutics: An overview. Curr. Pharmacol. Rep. 2020, 6, 71–84. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira, A.; Lapa, R.; Vale, N. PBPK Modeling and Simulation and Therapeutic Drug Monitoring: Possible Ways for Antibiotic Dose Adjustment. Processes 2021, 9, 2087. https://doi.org/10.3390/pr9112087
Ferreira A, Lapa R, Vale N. PBPK Modeling and Simulation and Therapeutic Drug Monitoring: Possible Ways for Antibiotic Dose Adjustment. Processes. 2021; 9(11):2087. https://doi.org/10.3390/pr9112087
Chicago/Turabian StyleFerreira, Abigail, Rui Lapa, and Nuno Vale. 2021. "PBPK Modeling and Simulation and Therapeutic Drug Monitoring: Possible Ways for Antibiotic Dose Adjustment" Processes 9, no. 11: 2087. https://doi.org/10.3390/pr9112087
APA StyleFerreira, A., Lapa, R., & Vale, N. (2021). PBPK Modeling and Simulation and Therapeutic Drug Monitoring: Possible Ways for Antibiotic Dose Adjustment. Processes, 9(11), 2087. https://doi.org/10.3390/pr9112087