High-Throughput Raman Spectroscopy Combined with Innovate Data Analysis Workflow to Enhance Biopharmaceutical Process Development

 ,
,  ,
,
Abstract
1. Introduction
1.1. PAT for Upstream Processing
1.1.1. USP Monitoring and Analytics
1.1.2. PAT within USP Mammalian Cell Cultures
1.1.3. Applications of Raman Spectroscopy in USP
1.2. PAT for Downstream Processing (DSP)
1.2.1. DSP Monitoring and Analytics of Mammalian Therapeutic Products
1.2.2. Applications of PAT in DSP
1.2.3. Applications of Raman Spectroscopy in DSP
1.3. High-Throughput Raman Spectroscopy and Its Advantages for Bioprocess Development
2. Materials and Methods
2.1. Product Materials
2.2. Cell Line and Cell Culture Propagation
2.3. Bioreactor Systems and Cell Culture Process
2.4. Titre Analysis
2.5. CEX Sample Generation
2.6. Protein Concentration and Analytical Size Exclusion Chromatography
2.7. Spectral Data Acquisition
2.8. Spectral Data Pre-Processing and Model Set-Up
2.9. Partial Least Squares Model Generation
3. Results and Discussion
3.1. Demonstration of HT Raman Spectroscopy Microscope to USP

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | PLS Regression Peak (β) Wavenumber [cm−1] [USP Model] | Reference Wavenumber of Analyte [cm−1] | Assignment | Reference | ||
|---|---|---|---|---|---|---|
| start | max | end | ||||
| Glucose * | 442 | 446 | 450 | 443 | - | [62] |
| 450 | δ (C-C-O) | [63] | ||||
| 451 | - | [64] | ||||
| 510 | 517 | 521 | 514 | - | [62] | |
| 516 | - | [65] | ||||
| 518 | δ (C-CO) | [63] | ||||
| 559 | 562 | 567 | - | - | - | |
| 583 | 588 | 590 | 585 | δ (C-C-O) | [63] | |
| 716 | 718 | 721 | - | - | - | |
| 871 | 928 | 988 | 893 | ν (C-C) | [63] | |
| 895 | δ (C-H), δ (C-OH) | [66] | ||||
| 898 | - | [62] | ||||
| 910 | δ (C-H) | [63] | ||||
| 913 | - | [62] | ||||
| 914 | - | [67] | ||||
| 990 | - | [65] | ||||
| 1006 | 1002 | - | [67] | |||
| 1010 | 1019 | 1024 | 1018 | δ (COH) | [68] | |
| 1032 | - | - | - | |||
| 1059 | 1062 | 1066 | 1060 | ν (CO) | [63,66,68] | |
| 1128 | 1120 | δ (COH) | [63] | |||
| 1121 | - | [65] | ||||
| 1122 | - | [67] | ||||
| 1124 | ν (CO) | [68] | ||||
| 1125 | ν (C-C) | [66] | ||||
| 1130 | - | [62] | ||||
| 1358 | 1373 | 1395 | 1360 | w (CH2) | [63] | |
| 1370 | δ (C-H) | [66] | ||||
| 1372 | w (CH2) | [68] | ||||
| 1373 | - | [62] | ||||
| 1374 | - | [65] | ||||
| 1490 | - | - | - | |||
| Lactate ** | 381 | 402 | 417 | - | - | - |
| 535 | 540 | w (CO2-) | [70,71] | |||
| 840 | 855 | 866 | 855 | - | [65] | |
| 860 | ν (C-CO2-) | [70,71] | ||||
| 926 | 930 | r(CH3) | [70,71] | |||
| 1040 | 1045 | ν (C-CH3) | [65,70,71] | |||
| 1084 | 1085 | ν (C-O) | [65,70,71] | |||
| 1419 | 1420 | ν (CO2-) | [70,71] | |||
| 1456 | 1455 | δ (CH3) | [70,71] | |||
| 1456 | - | [65] | ||||
| VCD *** | 538 | 540 | lactate (w (CO2-)) | [70,71] | ||
| 840 | 855 | 866 | 855 | - | [65] | |
| 860 | lactate ν (C-CO2-) | [70,71] | ||||
| 924 | 930 | lactate (r (CH3)) | [70,71] | |||
| 1040 | 1045 | lactate (ν (C-CH3)) | [65,70,71] | |||
| 1084 | 1085 | lactate (ν (C-O)) | [65,70,71] | |||
| 1420 | 1420 | lactate (ν (CO2-)) | [70,71] | |||
| 1456 | 1455 | lactate (δ (CH3)) | [70,71] | |||
| 1456 | - | [65] | ||||
| Antibody **** | 538 | 540 | lactate (w (CO2-)) | [70,71] | ||
| 843 | 855 | 864 | 855 | - | [65] | |
| 860 | lactate ν (C-CO2-) | [70,71] | ||||
| 1041 | 1045 | lactate (ν (C-CH3)) | [65,70,71] | |||
| 1362 | 1368 | 1384 | 1360 | glucose (w (CH2)) | [63] | |
| 1370 | glucose (δ (C-H)) | [66] | ||||
| 1372 | glucose (w (CH2)) | [68] | ||||
| 1373 | - | [62] | ||||
| 1374 | - | [65] | ||||
| 1453 | 1456 | 1466 | 1455 | lactate (δ (CH3)) | [70,71] | |
| 1456 | - | [65] | ||||
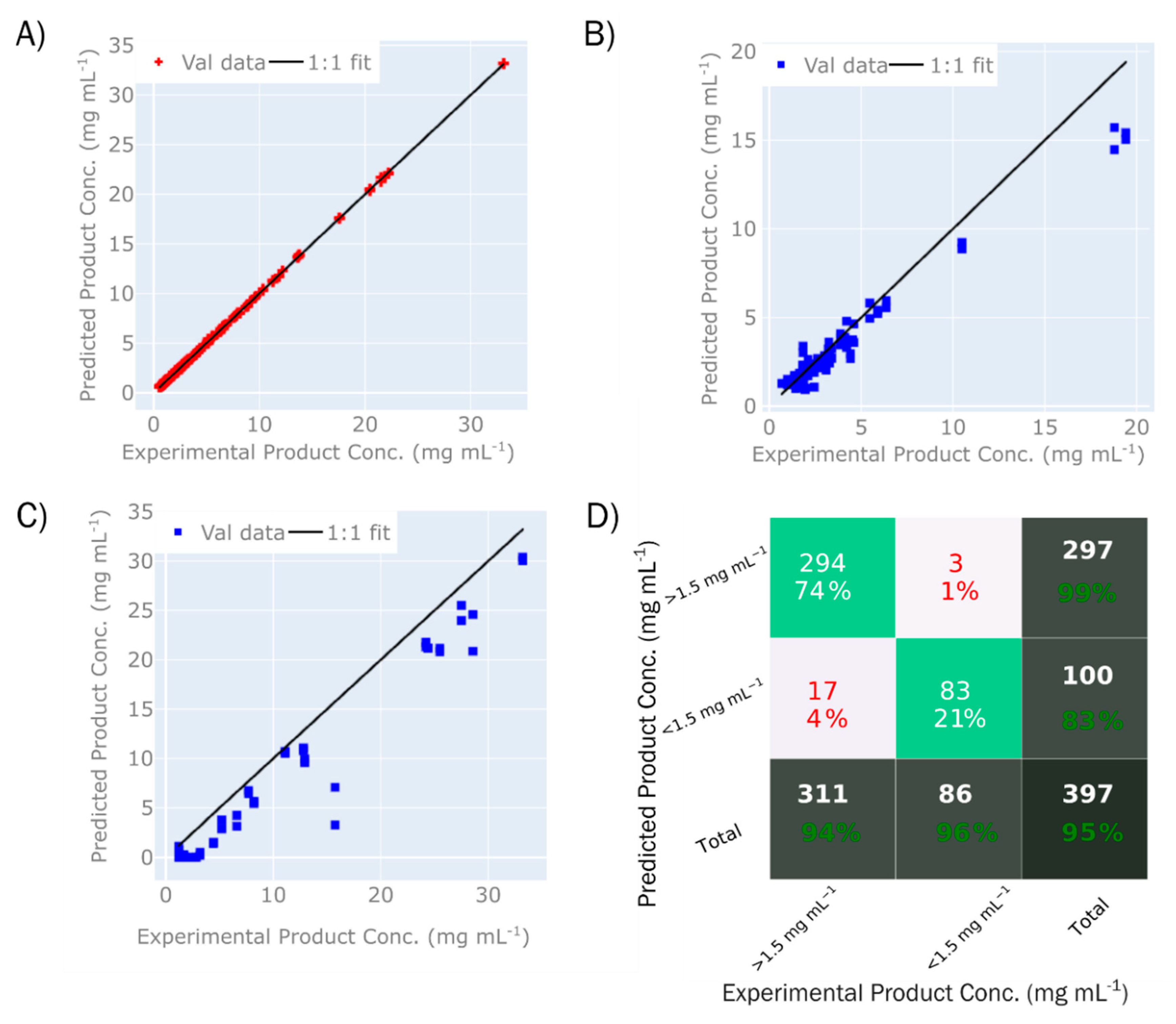
3.2. Application of Raman Spectroscopy to DSP
3.2.1. Strategies for Improving Predictions
3.2.2. Raman Spectroscopy Future Perspective
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AUC | Area under curve |
| CEX | Cation exchange chromatography |
| CHO | Chinese hamster ovary |
| CPP | Critical process parameters |
| CQA | Critical quality attribute |
| DLS | Dynamic light scattering |
| DSP | Downstream processing |
| FDA | Food and drug administration |
| GMP | Good manufacturing practice |
| HCP | Host cell proteins |
| HMW | High molecular weight |
| HPLC | High performance liquid chromatography |
| HT | High throughput |
| IEX | Ion exchange |
| LMW | Low molecular Weight |
| MIR | Mid-infrared |
| MVDA | Multivariate data analysis |
| NIR | Near-infrared |
| PAT | Process analytical technology |
| PLS | Partial least squares |
| PLS-DA | Partial least squares discrimination analysis |
| RMSE | Root mean square error |
| RMSEP | Root mean square error of prediction |
| R2 | Correlation coefficient |
| R&D | Research and development |
| SE | Size exclusion |
| SERS | Surface-enhanced Raman Spectroscopy |
| SNV | Standard normal variate |
| SS | Stainless steel |
| UHPLC | Ultra high-performance liquid chromatography |
| USP | Upstream processing |
| UV | Ultraviolet |
| VCD | Viable cell density |
References
- PAT—A Framework for Innovative Pharmaceutical, Manufacturing and Quality Assurance. Available online: https://www.fda.gov/media/71012/download (accessed on 1 September 2020).
- Brunner, M.; Fricke, J.; Kroll, P.; Herwig, C. Investigation of the interactions of critical scale-up parameters (pH, pO2 and pCO2) on CHO batch performance and critical quality attributes. Bioprocess Biosyst. Eng. 2017, 40, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Rathore, A.S.; Kapoor, G. Process analytical technology: Strategies for biopharmaceuticals. In Encyclopedia of Industrial Biotechnology: Bioprocess, Bioseparation, and Cell Technology; Flickinger, M., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2010; pp. 1543–1565. [Google Scholar]
- Biophorum. In-Line Monitoring/Real-Time Release Testing in Biopharmaceutical Processes—Prioritization and Cost Benefit Analysis. Available online: https://www.biophorum.com/download/in-line-monitoring-real-time-release-testing-in-biopharmaceutical-processes-prioritization-and-cost-benefit-analysis (accessed on 1 September 2020).
- Challa, S.; Potumarthi, R. Chemometrics-based process analytical technology (PAT) tools: Applications and adaptation in pharmaceutical and biopharmaceutical industries. Appl. Biochem. Biotechnol. 2013, 169, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Konakovsky, V.; Yagtu, A.C.; Clemens, C.; Müller, M.M.; Berger, M.; Schlatter, S.; Herwig, C. Universal capacitance model for real-time biomass in cell culture. Sensors 2015, 15, 22128–22150. [Google Scholar] [CrossRef] [PubMed]
- Hakemeyer, C.; Strauss, U.; Werz, S.; Jose, G.E.; Folque, F.; Menezes, J.C. At-line NIR spectroscopy as effective PAT monitoring technique in Mab cultivations during process development and manufacturing. Talanta 2012, 90, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Sandor, M.; Rüdinger, F.; Bienert, R.; Grimm, C.; Solle, D.; Scheper, T. Comparative study of non-invasive monitoring via infrared spectroscopy for mammalian cell cultivations. J. Biotechnol. 2013, 168, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Ude, C.; Schmidt-Hager, J.; Findeis, M.; John, G.T.; Scheper, T.; Beutel, S. Application of an online-biomass sensor in an optical multisensory platform prototype for growth monitoring of biotechnical relevant microorganism and cell lines in single-use shake flasks. Sensors 2014, 14, 17390–17405. [Google Scholar] [CrossRef]
- Ude, C.; Schmidt-Hager, J.; Findeis, M.; John, G.T.; Scheper, T.; Beutel, S. Fluorescence-based soft sensor for at situ monitoring of chinese hamster ovary cell cultures. Biotechnol. Bioeng. 2014, 111, 1577–1586. [Google Scholar]
- Calvet, A.; Li, B.; Ryder, A.G. Rapid quantification of tryptophan and tyrosine in chemically defined cell culture media using fluorescence spectroscopy. J. Pharm. Biomed. Anal. 2012, 71, 89–98. [Google Scholar] [CrossRef]
- Goldrick, S.; Lovett, D.; Montague, G.; Lennox, B. Influence of Incident Wavelength and Detector Material Selection on Fluorescence in the Application of Raman Spectroscopy to a Fungal Fermentation Process. Bioengineering 2018, 5, 79. [Google Scholar] [CrossRef]
- Buckley, K.; Ryder, A.G. Applications of Raman Spectroscopy in Biopharmaceutical Manufacturing: A Short Review. Appl. Spectrosc. 2017, 71, 1085–1116. [Google Scholar] [CrossRef]
- Wu, Q.; Hamilton, T.; Nelson, W.H.; Elliott, S.; Sperry, J.F.; Wu, M. UV Raman Spectral Intensities of E. Coli and Other Bacteria Excited at 228.9, 244.0, and 248.2 nm. Anal. Chem. 2001, 73, 3432–3440. [Google Scholar] [CrossRef] [PubMed]
- Matthews, T.E.; Berry, B.N.; Smelko, J.; Moretto, J.; Moore, B.; Wiltberger, K. Closed loop control of lactate concentration in mammalian cell culture by Raman spectroscopy leads to improved cell density, viability, and biopharmaceutical protein production. Biotechnol. Bioeng. 2016, 113, 2416–2424. [Google Scholar] [CrossRef] [PubMed]
- Matthews, T.E.; Smelko, J.P.; Berry, B.; Romero-Torres, S.; Hill, D.; Kshirsagar, R.; Wiltberger, K. Glucose monitoring and adaptive feeding of mammalian cell culture in the presence of strong autofluorescence by near infrared Raman spectroscopy. Biotechnol. Prog. 2018, 34, 1574–1580. [Google Scholar] [CrossRef] [PubMed]
- Abu-Absi, N.R.; Kenty, B.M.; Cuellar, M.E.; Borys, M.C.; Sakhamuri, S.; Strachan, D.J.; Hausladen, M.C.; Li, Z.J. Real time monitoring of multiple parameters in mammalian cell culture bioreactors using an in-line Raman spectroscopy probe. Biotechnol. Bioeng. 2011, 108, 1215–1221. [Google Scholar] [CrossRef]
- Webster, T.A.; Hadley, B.C.; Hilliard, W.; Jaques, C.; Mason, C. Development of generic raman models for a GS-KOTM CHO platform process. Biotechnol. Prog. 2018, 34, 730–737. [Google Scholar] [CrossRef]
- Craven, S.J.; Whelan, J.; Glennon, B. Glucose concentration control of a fed-batch mammalian cell bioprocess using a nonlinear model predictive controller. J. Process Control 2014, 24, 344–357. [Google Scholar] [CrossRef]
- Rafferty, C.; O’Mahony, J.; Burgoyne, B.; Rea, R.; Balss, K.M.; Latshaw, D.C. Raman spectroscopy as a method to replace off-line pH during mammalian cell culture processes. Biotechnol. Bioeng. 2020, 117, 146–156. [Google Scholar] [CrossRef]
- Li, M.Y.; Ebel, B.; Blanchard, F.; Paris, C.; Guedon, E.; Marc, A. Control of IgG glycosylation by in situ and real-time estimation of specific growth rate of CHO cells cultured in bioreactor. Biotechnol. Bioeng. 2019, 116, 985–993. [Google Scholar] [CrossRef]
- Hong, P.; Koza, S.; Bouvier, E.S.P. A review size exclusion chromatography for the analysis of protein biotherapeutics and their aggregates. J. Liq. Chromatogr. Relat. Technol. 2012, 35, 2923–2950. [Google Scholar] [CrossRef]
- Fekete, S.; Beck, A.; Veuthey, J.L.; Guillarme, D. Theory and practice of size exclusion chromatography for the analysis of protein aggregates. J. Pharm. Biomed. Anal. 2014, 101, 161–173. [Google Scholar] [CrossRef]
- Swartz, M.E. UPLC™: An Introduction and Review. J. Liq. Chromatogr. Relat. Technol. 2005, 28, 1253–1263. [Google Scholar] [CrossRef]
- Bouvier, E.S.P.; Koza, S.M. Advances in size-exclusion separations of proteins and polymers by UHPLC. TrAC Trends Anal. Chem. 2014, 63, 85–94. [Google Scholar] [CrossRef]
- Haberger, M.; Leiss, M.; Heidenreich, A.K.; Pester, O.; Hafenmair, G.; Hook, M.; Bonnington, L.; Wegele, H.; Haindl, M.; Reusch, D.; et al. Rapid characterization of biotherapeutic proteins by size-exclusion chromatography coupled to native mass spectrometry. mAbs 2016, 8, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Madadkar, P.; Umatheva, U.; Hale, G.; Durocher, Y.; Ghosh, R. Ultrafast Separation and Analysis of Monoclonal Antibody Aggregates Using Membrane Chromatography. Anal. Chem. 2017, 89, 4716–4720. [Google Scholar] [CrossRef]
- Tiwari, A.; Kateja, N.; Chanana, S.; Rathore, A.S. Use of HPLC as an Enabler of Process Analytical Technology in Process Chromatography. Anal. Chem. 2018, 90, 7824–7829. [Google Scholar] [CrossRef]
- Patel, B.A.; Pinto, N.D.; Gospodarek, A.; Kilgore, B.; Goswami, K.; Napoli, W.N.; Desai, J.; Heo, J.H.; Panzera, D.; Pollard, D.; et al. On-Line Ion Exchange Liquid Chromatography as a Process Analytical Technology for Monoclonal Antibody Characterization in Continuous Bioprocessing. Anal. Chem. 2017, 89, 11357–11365. [Google Scholar] [CrossRef]
- Heo, J.H.; Heo, J.H.; Mou, X.; Wang, F.; Troisi, J.M.; Christopher, W.S.; Kirby, S.; Driscoll, D.; Mercorelli, S.; Pollard, D.J. A microfluidic approach to high throughput quantification of host cell protein impurities for bioprocess development. Pharm. Bioprocess. 2014, 2, 129–139. [Google Scholar] [CrossRef]
- Kumar, M.A.; Mazlomi, M.A.; Hedström, M.; Mattiasson, B. Versatile automated continuous flow system (VersAFlo) for bioanalysis and bioprocess control. Sens. Actuators B Chem. 2012, 161, 855–861. [Google Scholar] [CrossRef]
- Rüdt, M.; Briskot, T.; Hubbuch, J. Advances in downstream processing of biologics—Spectroscopy: An emerging process analytical technology. J. Chromatogr. A 2017, 1490, 2–9. [Google Scholar] [CrossRef]
- Rolinger, L.; Rüdt, M.; Hubbuch, J. A critical review of recent trends, and a future perspective of optical spectroscopy as PAT in biopharmaceutical downstream processing. Anal. Bioanal. Chem. 2020, 412, 2047–2064. [Google Scholar] [CrossRef]
- Rüdt, M.; Brestrich, N.; Rolinger, L.; Hubbuch, J. Real-time monitoring and control of the load phase of a protein a capture step. Biotechno. Bioeng. 2017, 114, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Brestrich, N.; Ruedt, M.; Buechler, D.; Hubbuch, J. Selective protein quantification for preparative chromatography using variable pathlength UV/Vis spectroscopy and partial least squares regression. Chem. Eng. Sci. 2018, 176, 157–164. [Google Scholar] [CrossRef]
- Rolinger, L.; Ruedt, M.; Diehm, J.; Chow-Hubbertz, J.; Heitmann, M.; Schleper, S.; Hubbuch, J. Multi-attribute PAT for UF/DF of Proteins—Monitoring Concentration, particle sizes, and Buffer Exchange. Anal. Bioanal. Chem. 2020, 412, 2123–2136. [Google Scholar] [CrossRef]
- Thakur, G.; Hebbi, V.; Rathore, A.S. An NIR-based PAT approach for real-time control of loading in Protein a chromatography in continuous manufacturing of monoclonal antibodies. Biotechnol. Bioeng. 2020, 117, 673–686. [Google Scholar] [CrossRef]
- Capito, F.; Skudas, R.; Kolmar, H.; Hunzinger, C. At-line mid infrared spectroscopy for monitoring downstream processing unit operations. Process Biochem. 2015, 50, 997–1005. [Google Scholar] [CrossRef]
- Walch, N.; Scharl, T.; Felföldi, E.; Sauer, D.G.; Melcher, M.; Leisch, F.; Dürauer, A.; Jungbauer, A. Prediction of the Quantity and Purity of an Antibody Capture Process in Real Time. Biotechnol. J. 2019, 14, 1800521. [Google Scholar] [CrossRef]
- André, S.; Cristau, L.S.; Gaillard, S.; Devos, O.; Calvosa, É.; Duponchel, L. In-line and real-time prediction of recombinant antibody titer by in situ Raman spectroscopy. Anal. Chim. Acta 2015, 892, 148–152. [Google Scholar] [CrossRef]
- Feidl, F.; Garbellini, S.; Vogg, S.; Sokolov, M.; Souquet, J.; Broly, H.; Butté, A.; Morbidelli, M. A new flow cell and chemometric protocol for implementing in-line Raman spectroscopy in chromatography. Biotechnol. Prog. 2019, 35, e2847. [Google Scholar] [CrossRef] [PubMed]
- Feidl, F.; Garbellini, S.; Luna, M.F.; Vogg, S.; Souquet, J.; Broly, H.; Morbidelli, M.; Butté, A. Combining Mechanistic Modeling and Raman Spectroscopy for Monitoring Antibody Chromatographic Purification. Processes 2019, 7, 683. [Google Scholar] [CrossRef]
- Yilmaz, D.; Mehdizadeh, H.; Navarro, D.; Shehzad, A.; O’Connor, M.; McCormick, P. Application of Raman spectroscopy in monoclonal antibody producing continuous systems for downstream process intensification. Biotechnol. Prog. 2019, 36, e2947. [Google Scholar] [CrossRef]
- Chmielowski, R.A.; Mathiasson, L.; Blom, H.; Go, D.; Ehring, H.; Khan, H.; Li, H.; Cutler, C.; Lacki, K.; Tugcu, N.; et al. Definition and dynamic control of a continuous chromatography process independent of cell culture titer and impurities. J. Chromatogr. A 2017, 1526, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Qi, W.; Lewis, E.N.; Carpenter, J.F. Concomitant Raman spectroscopy and dynamic light scattering for characterization of therapeutic proteins at high concentrations. Anal. Biochem. 2015, 472, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Gómez de la Cuesta, R.; Goodacre, R.; Ashton, L. Monitoring Antibody Aggregation in Early Drug Development Using Raman Spectroscopy and Perturbation-Correlation Moving Windows. Anal. Chem. 2014, 86, 11133–11140. [Google Scholar] [CrossRef] [PubMed]
- Paidi, S.K.; Siddhanta, S.; Strouse, R.; McGivney, J.B.; Larkin, C.; Barman, I. Rapid Identification of Biotherapeutics with Label-Free Raman Spectroscopy. Anal. Chem. 2016, 88, 4361–4368. [Google Scholar] [CrossRef]
- Zhang, C.; Springall, J.S.; Wang, X.; Barman, I. Rapid, quantitative determination of aggregation and particle formation for antibody drug conjugate therapeutics with label-free Raman spectroscopy. Anal. Chim. Acta 2019, 1081, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Ashton, L.; Brewster, V.L.; Correa, E.; Goodacre, R. Detection of glycosylation and iron-binding protein modifications using Raman spectroscopy. Analyst 2017, 142, 808–814. [Google Scholar] [CrossRef]
- Brewster, V.L.; Ashton, L.; Goodacre, R. Monitoring the glycosylation status of proteins using Raman spectroscopy. Anal. Chem. 2011, 83, 6074–6081. [Google Scholar] [CrossRef]
- Cowcher, D.P.; Deckert-Gaudig, T.; Brewster, V.L.; Ashton, L.; Deckert, V.; Goodacre, R. Detection of protein glycosylation using tip-enhanced Raman scattering. Anal. Chem. 2016, 88, 2105–2112. [Google Scholar] [CrossRef]
- Capito, F.; Skudas, R.; Kolmar, H.; Stanislawski, B. Host cell protein quantification by fourier transform mid infrared spectroscopy (FT-MIR). Biotechnol. Bioeng. 2013, 110, 252–259. [Google Scholar] [CrossRef]
- Virtanen, T.; Reinikainen, S.P.; Kögler, M.; Mänttäri, M.; Viitala, T.; Kallioinen, M. Real-time fouling monitoring with Raman spectroscopy. J. Membr. Sci. 2017, 525, 312–319. [Google Scholar] [CrossRef]
- Kojima, T.; Tsutsumi, S.; Yamamoto, K.; Ikeda, Y.; Moriwaki, T. High-throughput cocrystal slurry screening by use of in situ Raman microscopy and multi-well plate. Int. J. Pharm. 2010, 399, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Medipally, D.K.; Maguire, A.; Bryant, J.; Armstrong, J.; Dunne, M.; Finn, M.; Lyng, F.M.; Meade, A.D. Development of a high throughput (HT) Raman spectroscopy method for rapid screening of liquid blood plasma from prostate cancer patients. Analyst 2017, 142, 1216–1226. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Shih, W.C. Performance of line-scan Raman microscopy for high-throughput chemical imaging of cell population. Appl. Opt. 2014, 53, 2881–2885. [Google Scholar] [CrossRef] [PubMed]
- Schie, I.W.; Rüger, J.; Mondol, A.S.; Ramoji, A.; Neugebauer, U.; Krafft, C.; Popp, J. High-Throughput Screening Raman Spectroscopy Platform for Label-Free Cellomics. Anal. Chem. 2018, 90, 2023–2030. [Google Scholar] [CrossRef] [PubMed]
- Le Losq, C. Rampy: A Python Library for Processing Spectroscopic (IR, Raman, XAS) Data; Zenodo: Geneva, Switzerland, 2018. [Google Scholar]
- Wold, S.; Geladi, P.; Esbensen, K.; Öhman, J. Multi-way principal components-and PLS-analysis. J. Chemom. 1987, 1, 41–56. [Google Scholar] [CrossRef]
- Rowland-Jones, R.C.; Jaques, C. At-line raman spectroscopy and design of experiments for robust monitoring and control of miniature bioreactor cultures. Biotechnol. Prog. 2019, 35, e2740. [Google Scholar] [CrossRef]
- Berry, B.; Moretto, J.; Matthews, T.; Smelko, J.; Wiltberger, K. Cross-scale predictive modeling of CHO cell culture growth and metabolites using Raman spectroscopy and multivariate analysis. Biotechnol. Prog. 2015, 31, 566–577. [Google Scholar] [CrossRef]
- Vasko, P.D.; Blackwell, J.; Koenig, J.L. Infrared and raman spectroscopy of carbohydrates: Part I: Identification of OH and CH-related vibrational modes for D-glucose, maltose, cellobiose, and dextran by deuterium-substitution methods. Carbohydr. Res. 1971, 19, 297–310. [Google Scholar] [CrossRef]
- Mathlouthi, M.; Vinh Luu, D. Laser-raman spectra of d-glucose and sucrose in aqueous solution. Carbohydr. Res. 1980, 81, 203–212. [Google Scholar] [CrossRef]
- Söderholm, S.; Roos, Y.H.; Meinander, N.; Hotokka, M. Raman spectra of fructose and glucose in the amorphous and crystalline states. J. Raman Spectrosc. 1999, 30, 1009–1018. [Google Scholar] [CrossRef]
- Singh, G.P.; Goh, S.; Canzoneri, M.; Ram, R.J. Raman spectroscopy of complex defined media: Biopharmaceutical applications. J. Raman Spectrosc. 2015, 46, 545–550. [Google Scholar] [CrossRef]
- Barrett, T.W. Laser Raman spectra of mono-, oligo- and polysaccharides in solution. Spectrochim. Acta Part A Mol. Spectrosc. 1981, 37, 233–239. [Google Scholar] [CrossRef]
- De Gelder, J.; De Gussem, K.; Vandenabeele, P.; Moens, L. Reference database of Raman spectra of biological molecules. J. Raman Spectrosc. 2007, 38, 1133–1147. [Google Scholar] [CrossRef]
- Kačuráková, M.; Mathlouthi, M. FTIR and laser-Raman spectra of oligosaccharides in water: Characterization of the glycosidic bond. Carbohydr. Res. 1996, 284, 145–157. [Google Scholar] [CrossRef]
- Rhiel, M.H.; Amrhein, M.I.; Marison, I.W.; Stockar, U. The Influence of Correlated Calibration Samples on the Prediction Performance of Multivariate Models Based on Mid-Infrared Spectra of Animal Cell Cultures. Anal. Chem. 2002, 74, 5227–5236. [Google Scholar] [CrossRef]
- Cassanas, G.; Morssli, M.; Fabregue, E.; Bardet, L. Vibrational spectra of lactic acid and lactates. J. Raman Spectrosc. 1991, 22, 409–413. [Google Scholar] [CrossRef]
- Pecul, M.; Rizzo, A.; Leszczynski, J. Vibrational Raman and Raman Optical Activity Spectra of d-Lactic Acid, d-Lactate, and d-Glyceraldehyde: Ab Initio Calculations. J. Phys. Chem. A 2002, 106, 11008–11016. [Google Scholar] [CrossRef]
- Kengne-Momo, R.P.; Daniel, P.; Lagarde, F.; Jeyachandran, Y.L.; Pilard, J.F.; Durand-Thouand, M.J.; Thouand, G. Protein interactions investigated by the Raman spectroscopy for biosensor applications. Int. J. Spectrosc. 2012, 2012, 462901. [Google Scholar] [CrossRef]
- Painter, P.C.; Koenig, J.L. Raman spectroscopic study of the structure of antibodies. Biopolymers 1975, 14, 457–468. [Google Scholar] [CrossRef]
- Carey, P.R. CHAPTER 4—Protein Conformation from Raman and Resonance Raman Spectra. In Biochemical Applications of Raman and Resonance Raman Spectroscopes; Carey, P.R., Ed.; Academic Press: Cambridge, MA, USA, 1982; pp. 71–98. [Google Scholar]









| Off-Line Variable | Latent Variable Selection | Calibration (RMSE) | Validation (RMSEP) | Calibration (R2) | Validation (R2) |
|---|---|---|---|---|---|
| Glucose (g L−1) | 7 | 0.19 | 0.38 | 0.97 | 0.88 |
| VCD (1 × 106 cells mL−1) | 7 | 1.78 | 3.49 | 0.99 | 0.96 |
| Lactate (g L−1) | 7 | 0.59 | 1.16 | 0.96 | 0.94 |
| Antibody (g L−1) | 7 | 0.17 | 0.09 | 0.99 | 0.94 |
| Statistical Measure of Fit | Calibration (T1) | Validation (P1) | Validation (P2) |
|---|---|---|---|
| Product concentration (mg mL−1) | |||
| R2 | 0.99 | 0.99 | 0.98 |
| RMSE/RMSEP | 0.16 | 1.09 | 3.62 |
| Monomer concentration (%) | |||
| R2 | 0.98 | 0.86 | 0.34 |
| RMSE/RMSEP | 1.09 | 4.27 | 13.68 |
| Variable | PLS Regression Peak (β) Wavenumber [cm−1] [DSP Model] | Reference Wavenumber of Analyte [cm−1] | Assignment | Reference | |||
|---|---|---|---|---|---|---|---|
| Protein **** (Fc-fusion *****) | start | max | end | ||||
| 757 | 758 | Trp | [72] | ||||
| 759 | [73] | ||||||
| 1003 | 1002 | Phe | [74] | ||||
| 1004 | [72] | ||||||
| 1005 | [73] | ||||||
| 1029 | - | - | - | ||||
| 1208 | 1210 | Tyr; Phe | |||||
| 1236 | 1239 | Amide III | [74] | ||||
| 1243 | [72] | ||||||
| 1337 | 1340 | Amide III | [73] | ||||
| 1447 | 1450 | CH2 def | [73] | ||||
| 1451 | [72] | ||||||
| 1553 | 1552 | Trp | [72] | ||||
| 1554 | [73] | ||||||
| 1673 | 1673 | Amide I | [74] | ||||
| Protein aggregate | 831 | 863 | 901 | 830; 850 | Tyr | [45] | |
| 878 | Tyr | [73] | |||||
| 1437 | 1448 | 1446 | - | Tyr | [45] | ||
| Tyr | [73] | ||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goldrick, S.; Umprecht, A.; Tang, A.; Zakrzewski, R.; Cheeks, M.; Turner, R.; Charles, A.; Les, K.; Hulley, M.; Spencer, C.; et al. High-Throughput Raman Spectroscopy Combined with Innovate Data Analysis Workflow to Enhance Biopharmaceutical Process Development. Processes 2020, 8, 1179. https://doi.org/10.3390/pr8091179
Goldrick S, Umprecht A, Tang A, Zakrzewski R, Cheeks M, Turner R, Charles A, Les K, Hulley M, Spencer C, et al. High-Throughput Raman Spectroscopy Combined with Innovate Data Analysis Workflow to Enhance Biopharmaceutical Process Development. Processes. 2020; 8(9):1179. https://doi.org/10.3390/pr8091179
Chicago/Turabian StyleGoldrick, Stephen, Alexandra Umprecht, Alison Tang, Roman Zakrzewski, Matthew Cheeks, Richard Turner, Aled Charles, Karolina Les, Martyn Hulley, Chris Spencer, and et al. 2020. "High-Throughput Raman Spectroscopy Combined with Innovate Data Analysis Workflow to Enhance Biopharmaceutical Process Development" Processes 8, no. 9: 1179. https://doi.org/10.3390/pr8091179
APA StyleGoldrick, S., Umprecht, A., Tang, A., Zakrzewski, R., Cheeks, M., Turner, R., Charles, A., Les, K., Hulley, M., Spencer, C., & Farid, S. S. (2020). High-Throughput Raman Spectroscopy Combined with Innovate Data Analysis Workflow to Enhance Biopharmaceutical Process Development. Processes, 8(9), 1179. https://doi.org/10.3390/pr8091179