Functionalized N-Pyridinylmethyl Engrafted Bisarylmethylidenepyridinones as Anticancer Agents

 , and
, and
Abstract
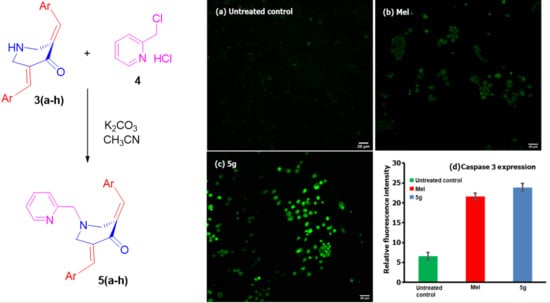
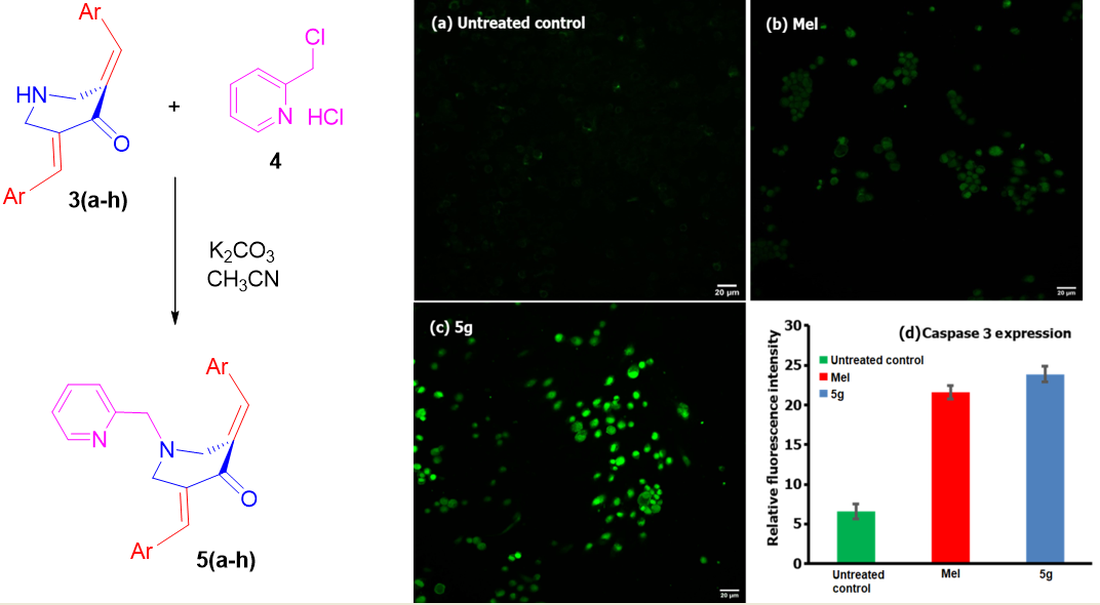
1. Introduction
2. Materials and Methods
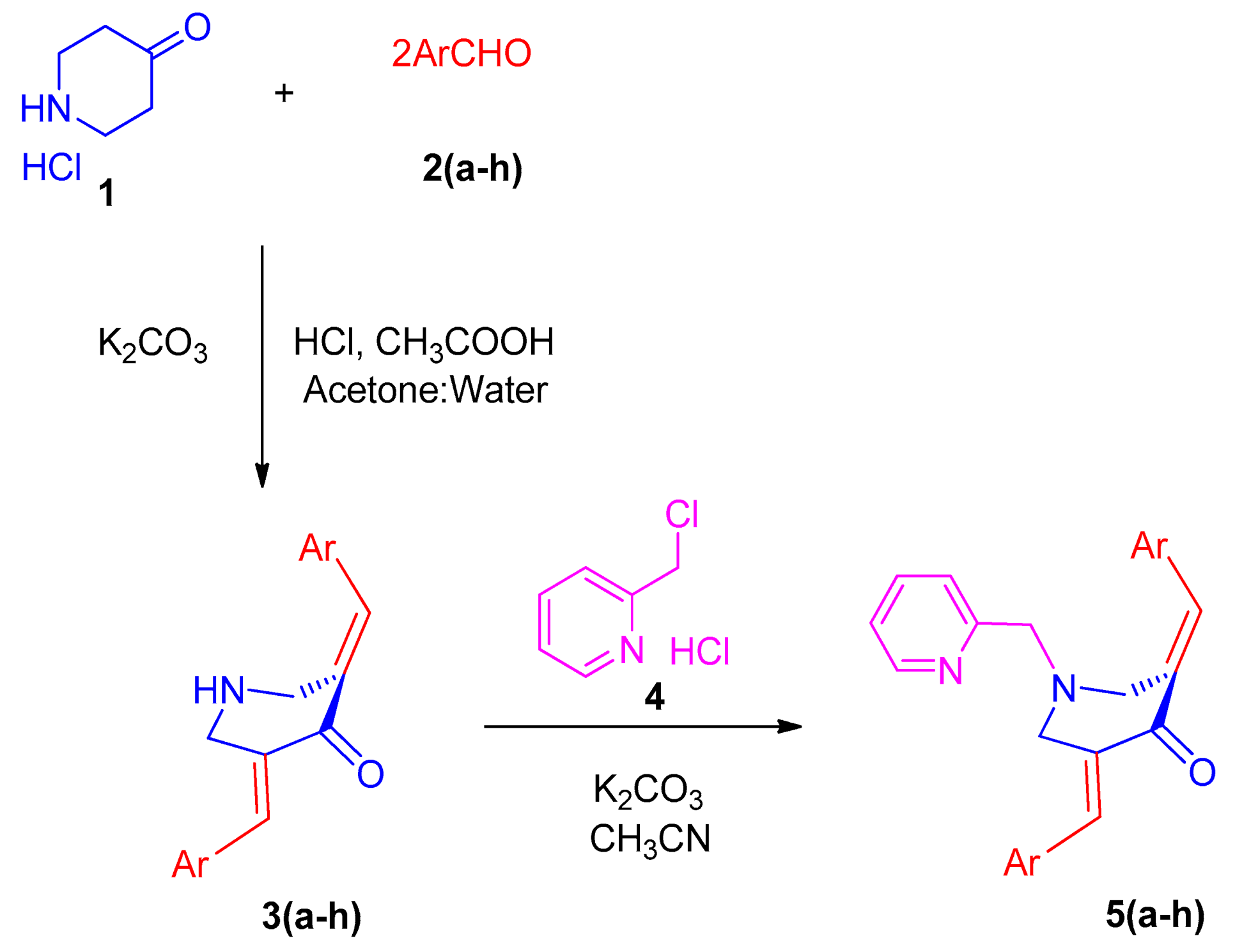
2.1. Chemistry
2.2. General Procedure for the Synthesis of N-Pyridinylmethyl-Bisarylmethylidenepyridinones 5(a–h)
2.2.1. N-1-(2-Pyridinylmethyl)-3,5-bis[(E)-Phenylmethylidene]Tetrahydro-4(1H)-Pyridinone (5a)
2.2.2. N-1-(2-Pyridinylmethyl)-3,5-bis[(E)-(2-Methylphenyl)Methylidene]Tetrahydro-4(1H)-Pyridinone (5b)
2.2.3. N-1-(2-Pyridinylmethyl)-3,5-bis[(E)-(2-Methoxylphenyl)Methylidene]Tetrahydro-4(1H)-Pyridinone (5c)
2.2.4. N-1-(2-Pyridinylmethyl)-3,5-bis[(E)-(3-Nitrophenyl)Methylidene]Tetrahydro-4(1H)-Pyridinone (5d)
2.2.5. N-1-(2-Pyridinylmethyl)-3,5-bis[(E)-(4-Methylphenyl)Methylidene]Tetrahydro-4(1H)-Pyridinone (5e)
2.2.6. N-1-(2-Pyridinylmethyl)-3,5-Bis[(E)-(4-Chlorophenyl)Methylidene]Tetrahydro-4(1H)-Pyridinone (5f)
2.2.7. N-1-(2-Pyridinylmethyl)-3,5-Bis[(E)-(4-Fluorophenyl)Methylidene]Tetrahydro-4(1H)-Pyridinone (5g)
2.2.8. N-1-(2-Pyridinylmethyl)-3,5-Bis[(E)-(2,4-Dichlorophenyl)Methylidene]Tetrahydro-4(1H)-Pyridinone (5h)
2.3. Biology
2.3.1. Cell Culture and Maintenance
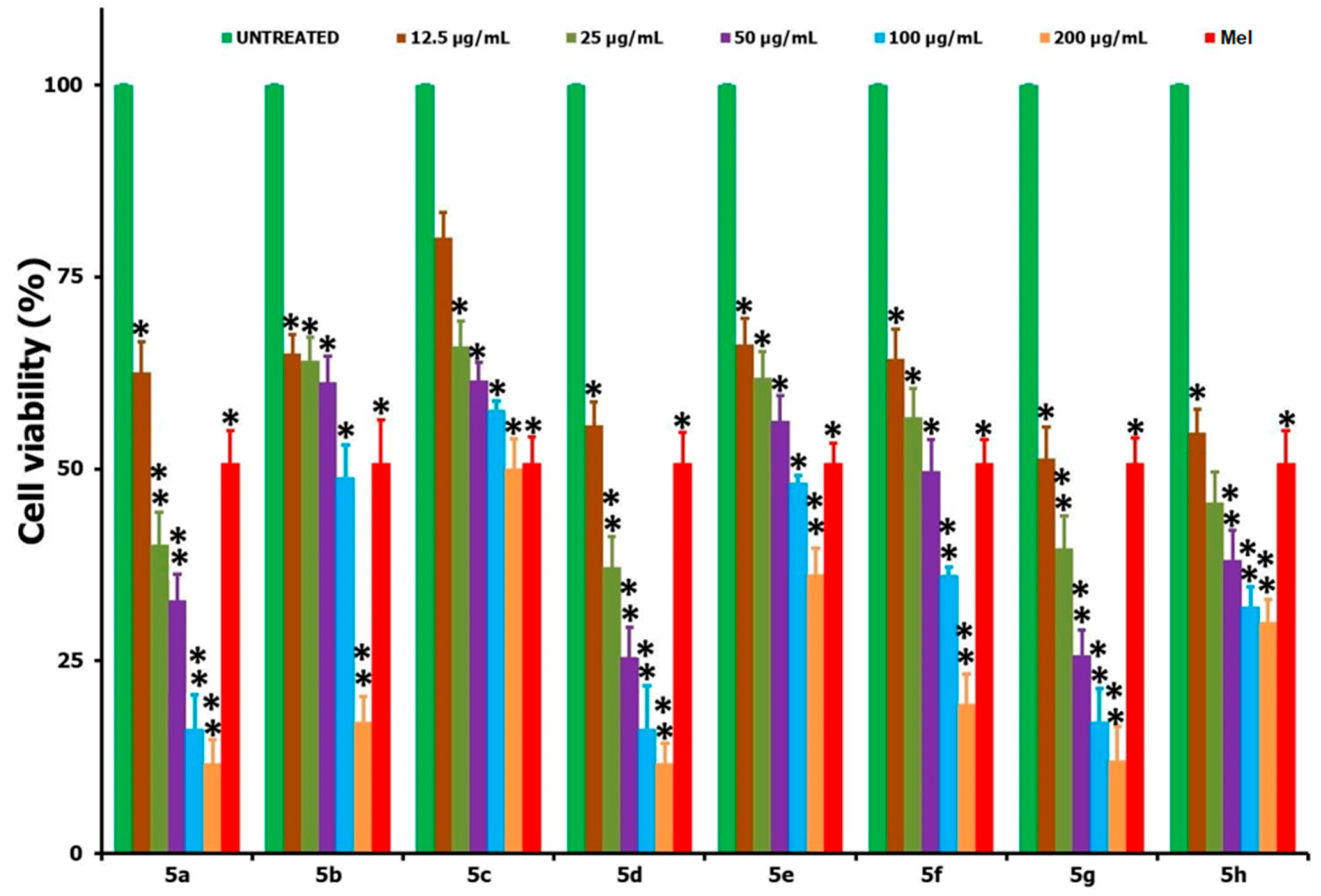
2.3.2. MTT Cytotoxicity Assay
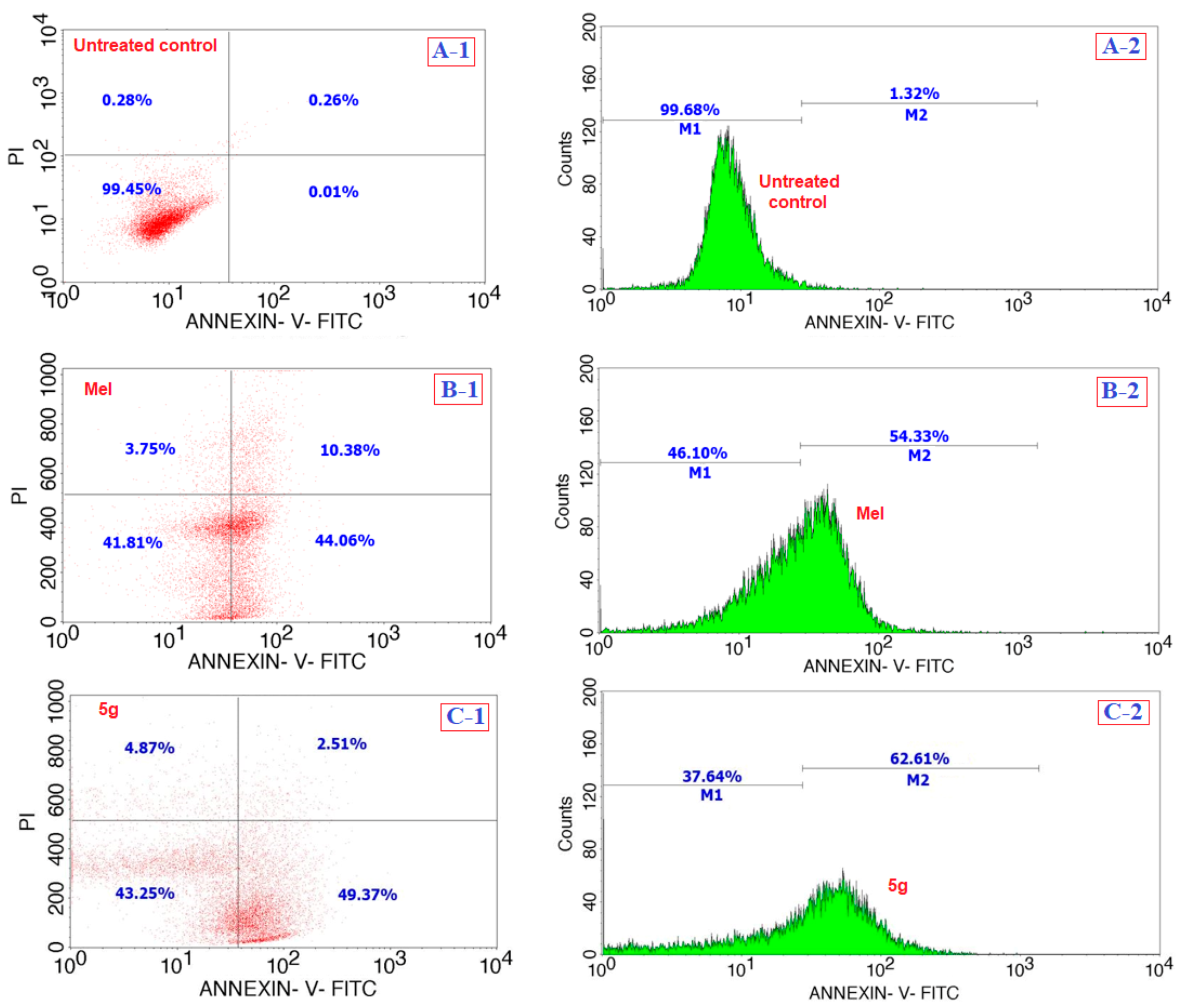
2.3.3. Apoptosis Assay
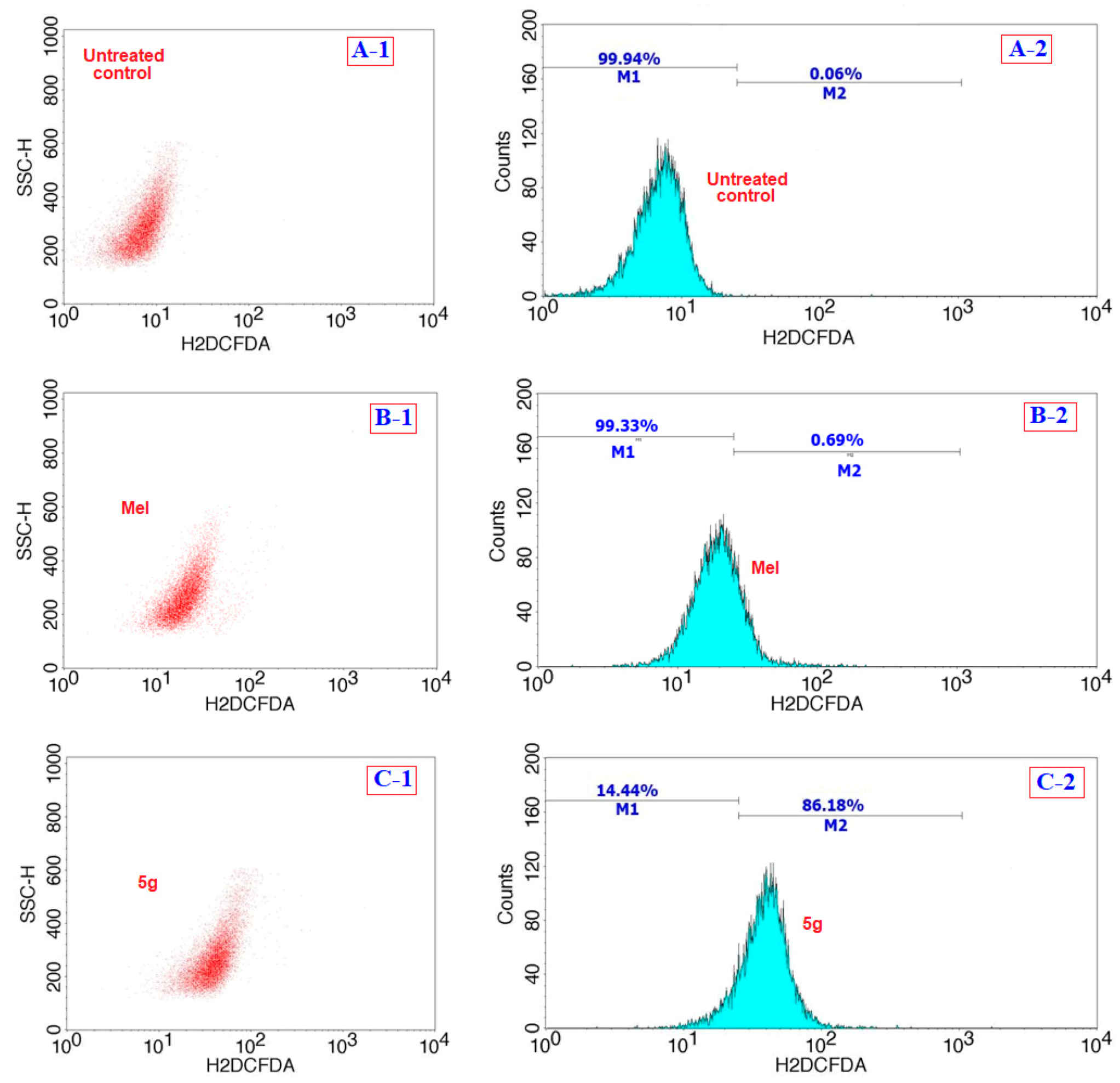
2.3.4. Oxidative Stress Assay
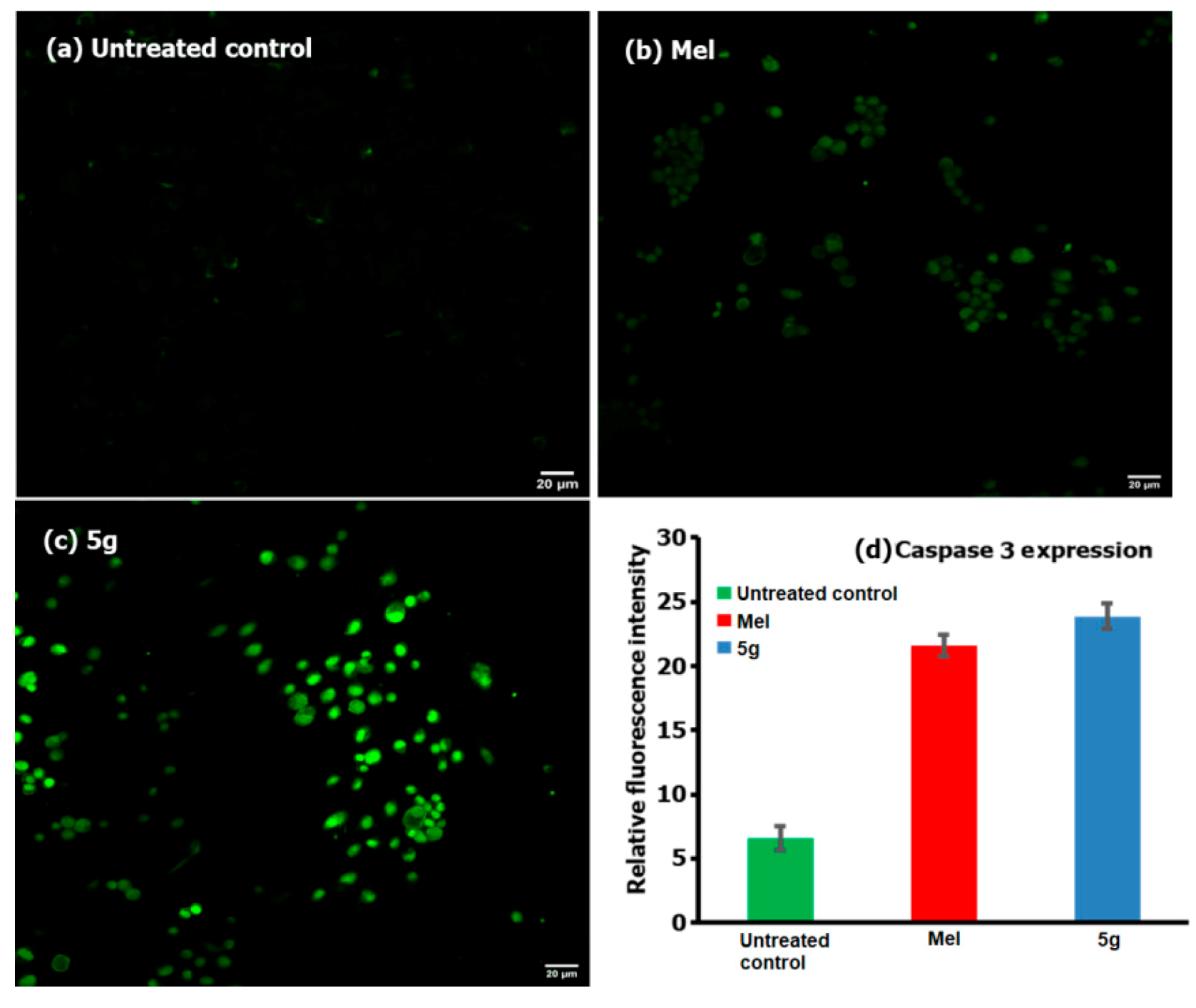
2.3.5. Caspase-3 Expression Assay
2.3.6. Statistical Analysis
3. Results and Discussion
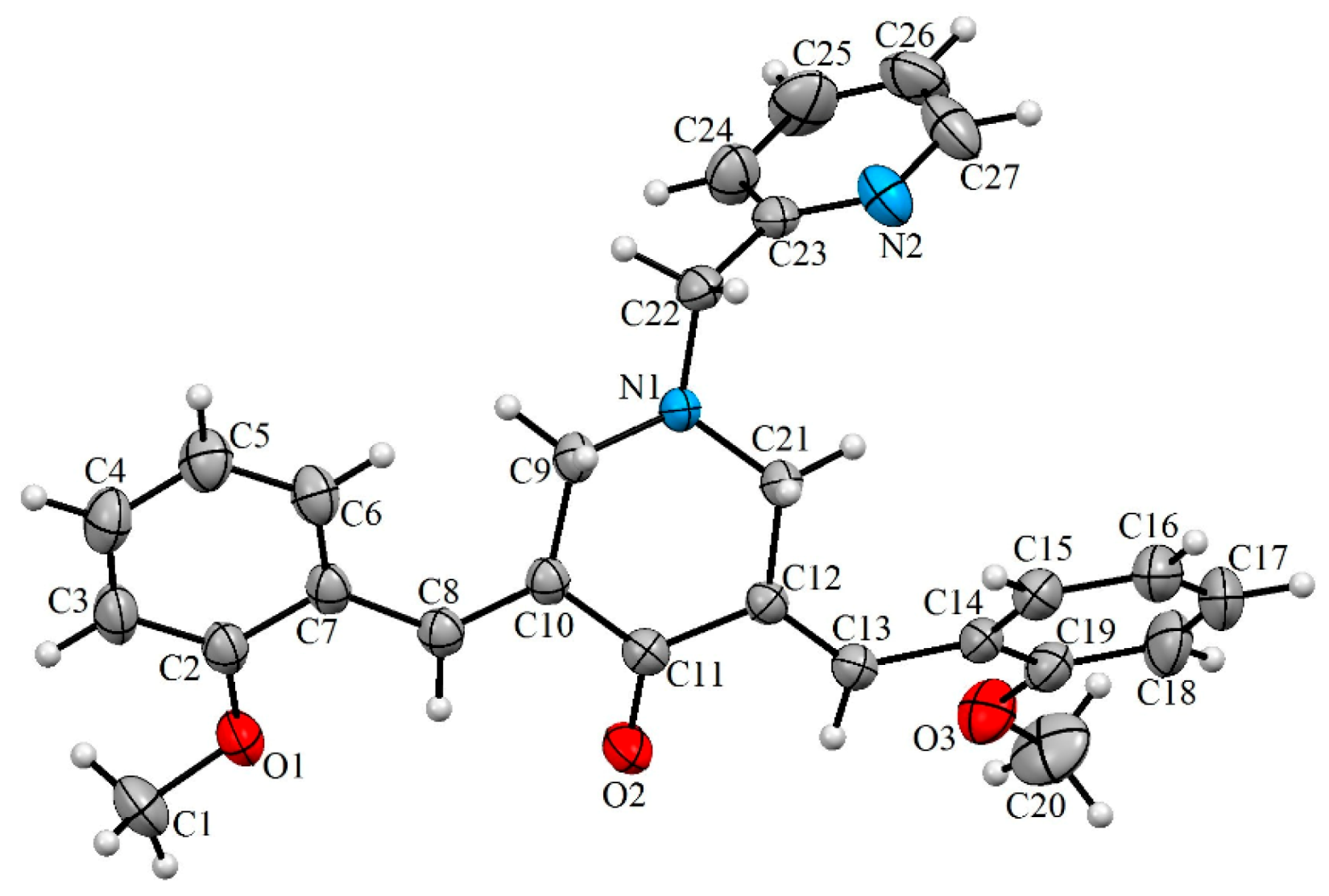
3.1. Chemistry
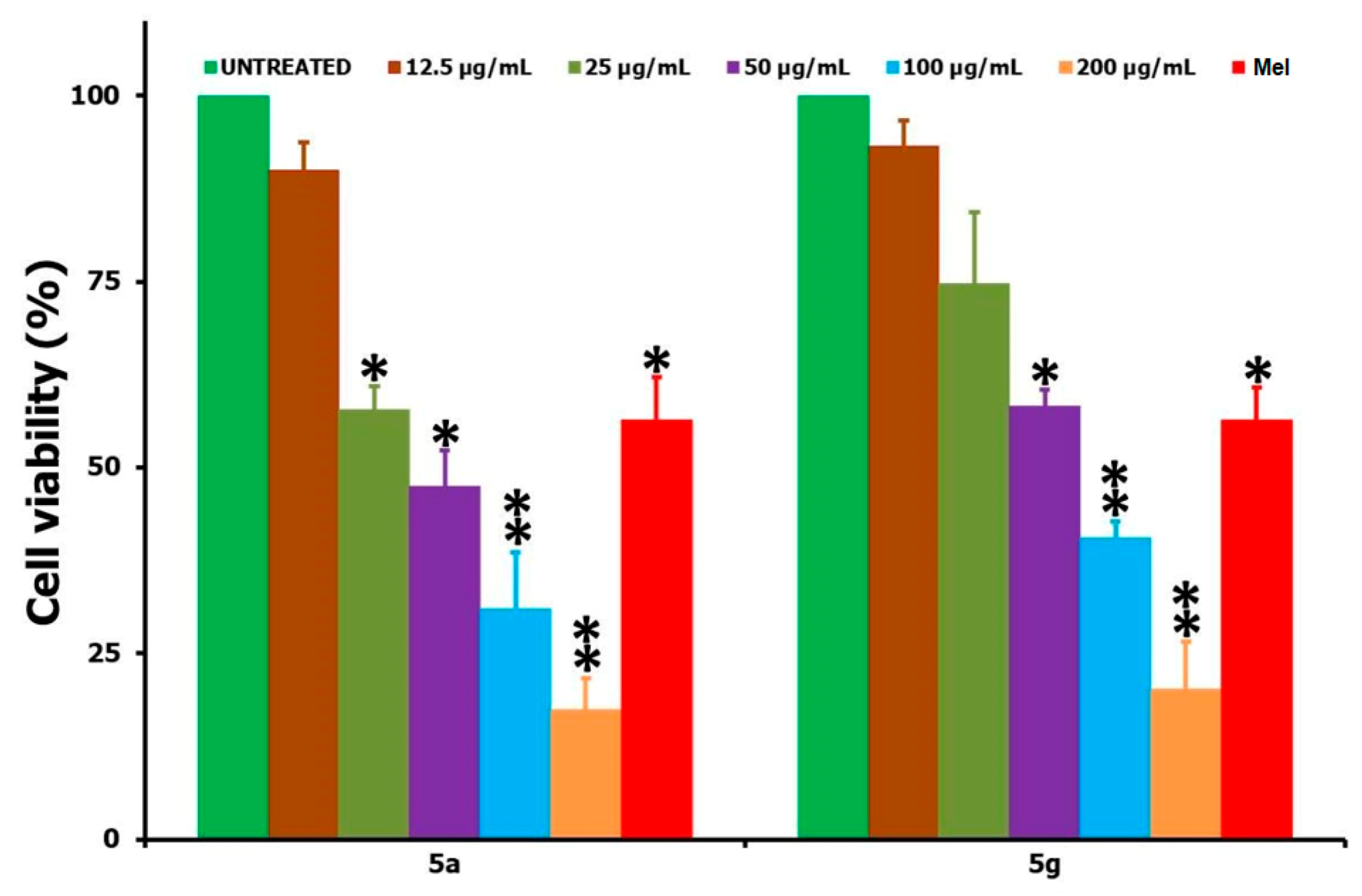
3.2. Biology
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Harris, C.C. Chemical and physical carcinogenesis: Advances and perspectives for the 1990s. Cancer Res. 1991, 51, 5023–5044. [Google Scholar]
- Wong, C.F.; Guminski, A.; Saunders, N.A.; Burgess, A.J. Exploiting novel cell cycle targets in the development of anticancer agents. Curr. Cancer Drug Targets 2005, 5, 85–102. [Google Scholar] [CrossRef] [PubMed]
- Lunardi, F.; Guzela, M.; Rodrigues, A.T.; Corrêa, R.; Eger-Mangrich, I.; Steindel, M.; Grisard, E.C.; Assreuy, J.; Calixto, J.B.; Santos, A.R. Trypanocidal and leishmanicidal properties of substitution-containing chalcones. Antimicrob. Agents Chemother. 2003, 47, 1449–1451. [Google Scholar] [CrossRef] [PubMed]
- Chimenti, F.; Fioravanti, R.; Bolasco, A.; Chimenti, P.; Secci, D.; Rossi, F.; Yanez, M.; Orallo, F.; Ortuso, F.; Alcaro, S. Chalcones: A Valid Scaffold for Monoamine Oxidases Inhibitors. J. Med. Chem. 2009, 52, 2818–2824. [Google Scholar] [CrossRef] [PubMed]
- Zarghi, A.; Zebardast, T.; Hakimion, F.; Shirazi, F.H.; Rao, P.N.; Knaus, E.E. Synthesis and biological evaluation of 1,3-diphenylprop-2-en-1-ones possessing a methanesulfonamido or an azido pharmacophore as cyclooxygenase-1/-2 inhibitors. Bioorg. Med. Chem. 2006, 14, 7044–7050. [Google Scholar] [CrossRef]
- Bag, S.; Ramar, S.; Degani, M.S. Synthesis and biological evaluation of α, β-unsaturated ketone as potential antifungal agents. Med. Chem. Res. 2009, 18, 309–316. [Google Scholar] [CrossRef]
- Das, U.; Pati, H.N.; Sakagami, H.; Hashimoto, K.; Kawase, M.; Balzarini, J.; De Clercq, E.; Dimmock, J.R. 3,5-Bis(benzylidene)-1-[3-(2-hydroxyethylthio)propanoyl]piperidin-4-ones: A novel cluster of potent tumor-selective cytotoxins. J. Med. Chem. 2011, 54, 3445–3449. [Google Scholar] [CrossRef][Green Version]
- Dimmock, J.R.; Kumar, P.; Chen, M.; Quail, J.W.; Yang, J.; Allen, T.M.; Kao, G.Y. Synthesis and cytotoxic evaluation of mesna adducts of some 1-aryl-4,4-dimethyl-5-(1-piperidino)-1-penten-3-one hydrochlorides. Pharmazie 1995, 50, 449–453. [Google Scholar] [CrossRef]
- Okey, A.B.; Harper, P.A. Chemical carcinogenesis. In Principles of Medical Pharmacology, 7th ed.; Galant, H., Grant, D.M., Mitchell, J., Eds.; Elsevier: Toronto, ON, Canada, 2007; p. 902. [Google Scholar]
- Cheng, C.C. Progress in Medicinal Chemistry, Volume 25; Ellis, G.P., Wesl, G.B., Eds.; Elsevier Science Publishers: Amsterdam, The Netherlands, 1988; pp. 35–83. [Google Scholar]
- Dimmock, J.R.; Erciyas, E.; Sidhu, K.K.; Luo, X.; Mezey, P.G.; Allen, T.M.; Murray, L. Charge densities of atoms of conjugated styryl ketones having activity against L1210 leukemia cells. Drug Des Deliv. 1990, 7, 45–49. [Google Scholar] [PubMed]
- Dimmock, J.R.; Wong, M.C.L. Bioactivities and potential uses in drug design of acyclic alpha, beta-unsaturated ketones. Can. J. Pharm. Sci. 1976, 11, 35–53. [Google Scholar]
- Dimmock, J.R.; Smith, L.M. Syntheses and evaluation of ketals, hemithioketals, and dithioketals of conjugated styryl ketones principally for antineoplastic activity. J. Pharm. Sci. 1980, 69, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Dimmock, J.R.; Nyathi, C.B.; Smith, P.J. Syntheses and bioactivities of 1-(hydroxyphenyl)-1-nonen-3-ones and related ethers and esters. J. Pharm. Sci. 1979, 68, 1216–1221. [Google Scholar] [CrossRef] [PubMed]
- Dimmock, J.R.; Kirkpatrick, D.L.; Webb, N.G.; Cross, B.M. Synthesis and evaluation of some alkoxy-, chloro-, and acyloxy-conjugated styryl ketones against P-388 lymphocytic leukemia and an examination of the metabolism and toxicological effects of 1-(m-ethoxymethyloxyphenyl)-1-nonen-3-one in rats. J. Pharm. Sci. 1982, 71, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Dimmock, J.R.; Arora, V.K.; Wonko, S.L.; Hamon, N.W.; Quail, J.W.; Jia, Z.; Warrington, R.C.; Fang, W.D.; Lee, J.S. 3,5-Bis-benzylidene-4-piperidones and related compounds with high activity towards P388 leukemia cells. Drug Des. Deliv. 1990, 6, 183–194. [Google Scholar]
- Dimmock, J.R.; Padmanilayam, M.P.; Puthucode, R.N.; Nazarali, A.J.; Motaganahalli, N.L.; Zello, G.A.; Quail, J.W.; Oloo, E.O.; Kraatz, H.B.; Prisciak, J.S.; et al. A conformational and structure-activity relationship study of cytotoxic 3,5-bis(arylidene)-4-piperidones and related N-acryloyl analogues. J. Med. Chem. 2001, 44, 586–593. [Google Scholar] [CrossRef]
- Pati, H.N.; Das, U.; Quail, J.W.; Kawase, M.; Sakagami, H.; Dimmock, J.R. Cytotoxic 3,5-bis(benzylidene)piperidin-4-ones and N-acyl analogs displaying selective toxicity for malignant cells. Eur. J. Med. Chem. 2008, 43, 1–7. [Google Scholar] [CrossRef]
- Dimmock, J.R.; Kandepu, N.M.; Nazarali, A.J.; Motaganahalli, N.L.; Kowalchuk, T.P.; Pugazhenthi, U.; Prisciak, J.S.; Quail, J.W.; Allen, T.M.; LeClerc, R.; et al. Sequential cytotoxicity: A theory evaluated using novel 2-[4-(3-aryl-2-propenoyloxy)phenylmethylene]cyclohexanones and related compounds. J. Med. Chem. 2000, 43, 3933–3940. [Google Scholar] [CrossRef]
- Tsutsui, K.; Komuro, C.; Ono, K.; Nishidai, T.; Shibamoto, Y.; Takahashi, M.; Abe, M. Chemosensitization by buthionine sulfoximine in vivo. Int. J. Radiat. Oncol. Biol. Phys. 1986, 12, 1183–1186. [Google Scholar] [CrossRef]
- Mitchell, J.B.; Russo, A. The role of glutathione in radiation and drug induced cytotoxicity. Br. J. Cancer Suppl. 1987, 8, 96–104. [Google Scholar]
- Brandes, L.J.; Queen, G.M.; LaBella, F.S. N,N-diethyl-2-[4-(phenylmethyl)phenoxy] ethanamine (DPPE) a chemopotentiating and cytoprotective agent in clinical trials: Interaction with histamine at cytochrome P450 3A4 and other isozymes that metabolize antineoplastic drugs. Cancer Chemother. Pharmacol. 2000, 45, 298–304. [Google Scholar] [CrossRef]
- Griffin, R.J.; Arris, C.E.; Bleasdale, C.; Boyle, F.T.; Calvert, A.H.; Curtin, N.J.; Dalby, C.; Kanugula, S.; Lembicz, N.K.; Newell, D.R.; et al. Resistance-modifying agents. 8. Inhibition of O(6)-alkylguanine-DNA alkyltransferase by O(6)-alkenyl-, O(6)-cycloalkenyl-, and O(6)-(2-oxoalkyl)guanines and potentiation of temozolomide cytotoxicity in vitro by O(6)-(1-cyclopentenylmethyl)guanine. J. Med. Chem. 2000, 43, 4071–4083. [Google Scholar] [CrossRef] [PubMed]
- Vahrmeijer, A.L.; van Dierendonck, J.H.; Schutrups, J.; van de Velde, C.J.; Mulder, G.J. Potentiation of the cytostatic effect of melphalan on colorectal cancer hepatic metastases by infusion of buthionine sulfoximine (BSO) in the rat: Enhanced tumor glutathione depletion by infusion of BSO in the hepatic artery. Cancer Chemother. Pharmacol. 1999, 44, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Al-thamili, D.M.; Almansour, A.I.; Arumugam, N.; Kansız, S.; Dege, N.; Soliman, S.M.; Azam, M.; Kumar, R.S. Highly functionalized N-1-(2-pyridinylmethyl)-3,5-bis[(E)-arylmethylidene]tetrahydro-4(1H)-pyridinones: Synthesis, characterization, crystal structure and DFT studies. J. Mol. Struct. 2020, 1222, 128940. [Google Scholar] [CrossRef]
- Mishra, T.; Arya, R.K.; Meena, S.; Joshi, P.; Pal, M.; Meena, B.; Upreti, D.K.; Rana, T.S.; Datta, D. Isolation, characterization and anticancer potential of cytotoxic triterpenes from Betula utilis bark. PLoS ONE 2016, 11, e0159430. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Compound | IC50 Values (µg/mL, 24 h) | |
|---|---|---|---|
| HepG2 Cells | L929 Cells | ||
| 1 |  | 13.5 ± 4.2 | 47.96 ± 3.2 |
| 2 |  | 82.33 ± 3.1 | NA |
| 3 |  | 180.87 ± 3.4 | NA |
| 4 |  | 126.05 ± 4.1 | NA |
| 5 |  | 102.15 ± 3.2 | NA |
| 6 |  | 56.72 ± 2.9 | NA |
| 7 |  | 12.06 ± 2.5 | 67.25 ± 4.3 |
| 8 |  | 14.76 ± 3.1 | NA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
M. Al-thamili, D.; Almansour, A.I.; Arumugam, N.; Mohammad, F.; Kumar, R.S. Functionalized N-Pyridinylmethyl Engrafted Bisarylmethylidenepyridinones as Anticancer Agents. Processes 2020, 8, 1154. https://doi.org/10.3390/pr8091154
M. Al-thamili D, Almansour AI, Arumugam N, Mohammad F, Kumar RS. Functionalized N-Pyridinylmethyl Engrafted Bisarylmethylidenepyridinones as Anticancer Agents. Processes. 2020; 8(9):1154. https://doi.org/10.3390/pr8091154
Chicago/Turabian StyleM. Al-thamili, Dhaifallah, Abdulrahman I. Almansour, Natarajan Arumugam, Faruq Mohammad, and Raju Suresh Kumar. 2020. "Functionalized N-Pyridinylmethyl Engrafted Bisarylmethylidenepyridinones as Anticancer Agents" Processes 8, no. 9: 1154. https://doi.org/10.3390/pr8091154
APA StyleM. Al-thamili, D., Almansour, A. I., Arumugam, N., Mohammad, F., & Kumar, R. S. (2020). Functionalized N-Pyridinylmethyl Engrafted Bisarylmethylidenepyridinones as Anticancer Agents. Processes, 8(9), 1154. https://doi.org/10.3390/pr8091154