1. Introduction
Fuel cells (FCs) have attracted much attention due to limited energy resources and environmental factors worldwide [
1]. Conversion of the chemical energy of the fuel into electrical energy occurs in one stage in FCs. The advantages of FCs are undeniable compared to traditional energy sources. These include zero emissions into the atmosphere, low working temperature, and high efficiency. It is customary to use hydrogen as fuel in a FC. Hydrogen has the largest energy reserve per unit weight. However, difficulties arise when storing hydrogen. The most effective form for transporting and storing this chemical element is a liquid state. However, gas can be converted to liquid form only at a temperature of −253 °C, which requires special containers, equipment, and considerable financial costs.
FC with direct oxidation of liquid fuel is an alternative to hydrogen FC. Liquid fuel (ethanol and methanol) have several advantages over hydrogen: ease of transportation, storage, use, and low cost [
2]. Ethanol is a renewable biofuel derived from biomass by fermentation [
3]. This is why in recent years, more and more attention has been paid to direct ethanol fuel cells (DEFCs). The performance of fuel cells using ethanol as fuel is not inferior to the performance of methanol fuel cells [
4].
The efficiency of DEFCs is determined by parameters such as the properties of the polymer electrolyte membrane, gas diffusion layer, structural decisions, the appropriate temperature, and balance in the system. However, the most important factor is the activity of the catalyst. Platinum on carbon support (Pt/C) is commonly used as the catalyst in FC. It is well known that the surface of the catalyst is a critical factor influencing its performance. Therefore, high surface area materials have been developed. Typically, the active phase is dispersed on a conductive support such as carbon. However, pure platinum is not the most efficient anodic catalyst for DEFCs [
5].
The ethanol oxidation reaction (EOR) at the platinum consists of parallel reactions. For successful ethanol electrooxidation, the catalyst must adsorb the ethanol molecule, contributing to the dissociation of the C–C and C–H bonds. This will lead to the formation of adsorbed intermediates [
6]. In addition, it must alleviate the reaction of the adsorbed intermediates with some oxygen containing particles to form carbon dioxide, formic, or acetic acids. Thus, a balance between the O-containing particles and adsorbed organic molecules should be maintained on the surface of the catalyst. This problem can be solved by increasing the electrolyte pH. This will lead to a change of EOR kinetics [
7]. However, the most effective is to use a two-component catalyst including the alloys of platinum with oxygen adsorbing metals (Ir [
8], Rh [
9]) or using metal oxides (NiO [
10], SnO
2 [
11], TiO
2 [
12]) as a catalyst support. In this case, the ethanol molecules adsorb on the platinum surface, while the oxygen adsorbing metal or metal oxides provide the presence of O-containing particles in the reaction zone.
In this study, we used the pulse alternating current (PAC) technique for the synthesis of Pt/C, PtMOn/C (M = Ni, Sn, Ti), and PtX/C (X = Rh, Ir) catalyst systems and compared their electrocatalytic properties in the ethanol oxidation reaction (EOR).
2. Materials and Methods
The synthesis of Pt, NiO, SnO2, TiO2, Rh, and PtIr nanoparticles occurred under the pulse alternating current (duty cycle 25%). The ratio of the current density between the anodic and cathodic pulses was 1:1. The PtMOn/C (M = Ni, Sn, Ti) and the PtRh/C catalysts were prepared in two stages. In the first stage, two metal (Ni, Sn, Ti, Rh) electrodes (thickness of 0.3 mm) were immersed in an electrolyzer with a Vulcan XC-72 carbon support suspended in 2 M NaOH (for Ti and Ni) or a 2 M NaCl (for Sn and Rh) aqueous solution. The suspension was permanently stirred and cooled to 45–50 °C. The metal electrodes were connected to an alternating current source. The formation of MOn particles and Rh nanoparticles occurred under the influence of PAC on the electrodes. The as-prepared MOn/C or Rh/C powders were collected by filtration and washed with distilled water. In the second stage, two Pt electrodes were immersed in the suspension of MOn/C or Rh/C support in a 2 M NaOH aqueous solution. PAC was applied to the electrodes again and platinum nanoparticles were dispersed into the electrolyte. The prepared PtMOn/C (M = Ni, Sn, Ti) and PtRh/C catalysts catalyst was rinsed with H2O to a neutral pH. Catalysts were dried at 80 °C until a constant weight. Pt/C and PtIr/C catalysts were obtained using Pt and PtIr (alloy) electrodes in a carbon (Vulcan XC-72) suspension in 2 M NaOH in one stage. The metal phase loading in the catalyst or metal oxide loading in the catalyst support was determined by the weight loss of the metal electrodes (Pt, Ni, Sn, Ti, Rh, PtIr) and the results of thermogravimetry. The results of thermogravimetry showed a correlation with the theoretical composition of the catalysts, calculated from the weight loss of the metal electrodes (Pt, Ni, Sn, Ti, Rh, and PtIr) during synthesis.
The x-ray powder diffraction (XRD) studies were performed with an ARL X’TRA powder diffractometer, Thermo Scientific, (Cu K
α target, λ = 1.5418 Å). XRD data were collected in the 2θ range of 20°–85° using step scan mode. The average grain size was determined using the Scherrer equation:
where
D is the volume averaged grain size;
λ is the x-ray wavelength; FWHM is the full width at half maximum of the diffraction peak;
K = 0.89 is the dimensionless particle shape coefficient (Scherrer constant); and
θ is a diffraction angle. The unit cell parameters (a) were determined with the UnitCell program using all peaks in the x-ray diffraction pattern.
Transmission electron microscopy (TEM) images were obtained with a JEM-2100 unit (200 kV). Suspension of a catalyst sample in hexane was prepared to study the obtained catalysts by TEM. The sample for analysis was prepared by depositing a droplet of the suspension onto an amorphous carbon-coated copper grid.
All electrochemical measurements were performed at room temperature using a standard three-electrode electrochemical cell and a P-45-X potentiostat (Elins, Russia). Electrochemical properties of the prepared catalyst systems were investigated by cyclic voltammetry (CV). A glassy carbon plate served as a working electrode, which was coated with “catalytic ink” prepared using 10 wt% Nafion dispersion, according to the technique [
13]. A Pt wire was an auxiliary electrode, and an Ag/AgCl electrode was a reference electrode. All potentials presented in this work are quoted versus a reversible hydrogen electrode (RHE). The electrochemically active surface area (ECSA) was evaluated via CO-stripping. The CO adsorption was performed in a pre-deaerated background electrolyte (0.5 M H
2SO
4) at E = 0.3 V vs. RHE. The ECSA was calculated from the charge value used for CO
ads oxidation, taking into account that the oxidation of a carbon monoxide monolayer on Pt needs 420 µC·cm
−2 [
14]. The electrocatalytic properties were investigated in the 0.5 M C
2H
5OH + 0.5 M H
2SO
4 solution over a potential range between 0.05 and 1.3 V (RHE).
The long-term cyclic stability of the synthesized Pt/C, PtMOn/C (M = Ni, Sn, Ti) and PtX/C (X = Rh, Ir) catalysts was investigated by cycling in 0.5 M H2SO4 over a potential range of 0.05–1.3 V vs. RHE.
3. Results and Discussion
The metal or metal oxide content in the catalyst system was controlled by the time of synthesis based on the preliminarily found dispersion rates for the Pt, Ni, Sn, Ti, Rh, and PtIr electrodes. The dispersion rates were determined experimentally and amounted to 21, 38, 45, 50, 12, and 19 mg·cm
−2·h
−1 for Pt, Ni, Sn, Ti, Rh, and PtIr, respectively. The compositions of the obtained catalysts are presented in
Table 1.
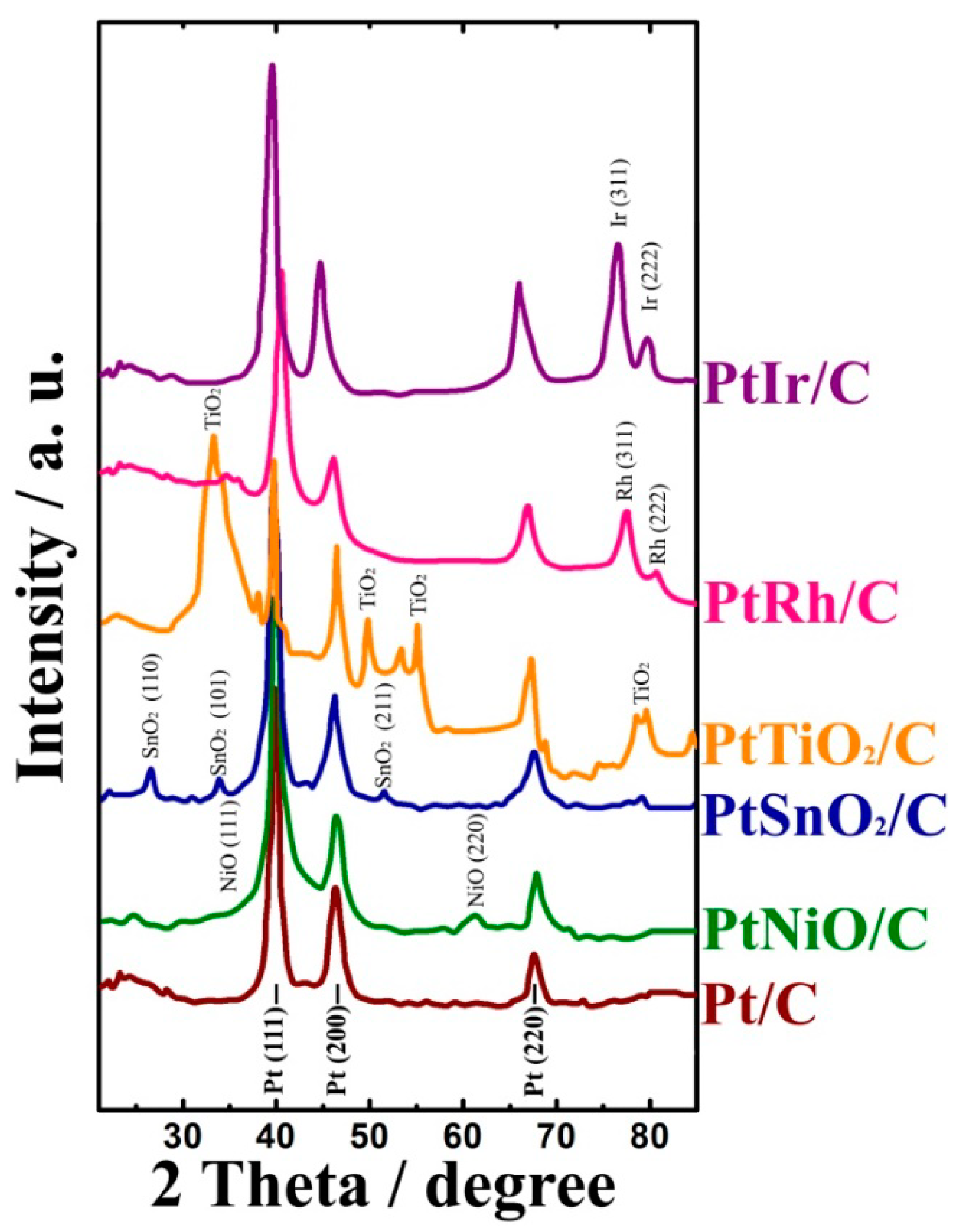
The prepared materials were investigated by XRD. The XRD patterns of the Pt/C, PtRh/C, PtIr/C, and Pt/MO
n/C catalysts synthesized via the PAC-technique are shown in
Figure 1.
XRD analysis showed the presence of a face-centered cubic structure Pt (Fm3m (No. 225)) in all of the catalyst samples. The analysis of the XRD patterns indicated the presence of titanium dioxide as anatase and rutile, tin dioxide with a tetragonal structure of SnO
2 (P42/mnm (No. 136)), and a nickel oxide cubic structure (Fm3m, JCPDS 75-0269) for the PtTiO
2/C, PtSnO
2/C, and PtNiO/C samples, respectively. Diffraction peaks characteristic for Rh and Ir were similarly observed in the PtRh/C and PtIr/C catalysts, respectively. The average size of crystallites in the synthesized catalysts was calculated from the data of the x-ray diffraction analysis (
Figure 1,
Table 1).
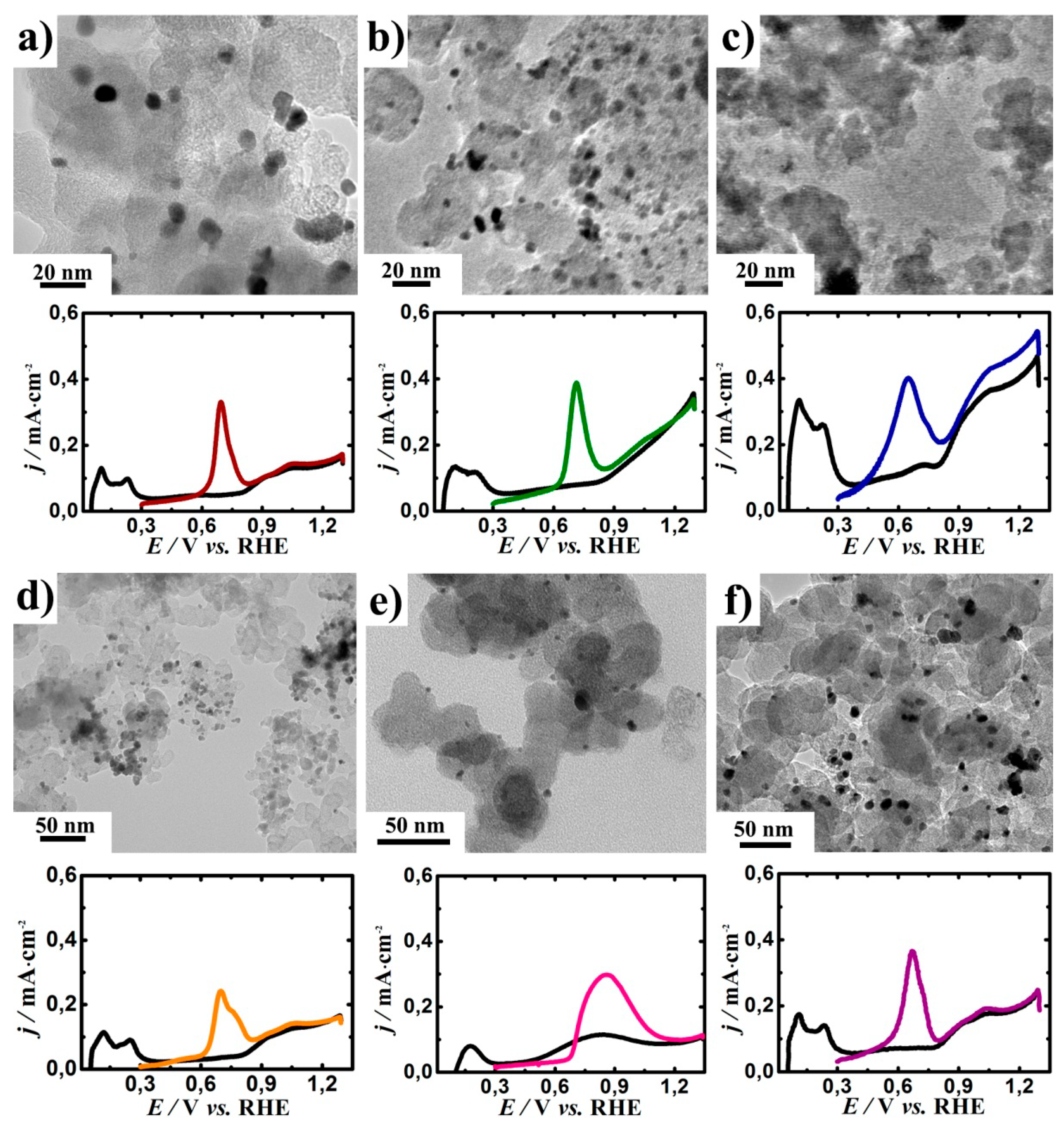
Figure 2 depicts the TEM image of the as-prepared catalysts. The surface average particle sizes presented in
Table 1 were obtained by measuring the sizes of 200 particles randomly selected from the TEM images. Transmission electron microscopy studies of the Pt/C, PtRh/C, PtIr/C, and PtMO
n/C catalysts revealed that the Pt, Rh, and PtIr (alloys) particles had a uniform distribution over the carbon surface. In addition, the platinum nanoparticles had a uniform distribution over the surface of the metal oxides. However, increased Pt agglomeration was seen from the TEM images of the PtSnO
2/C (
Figure 2c).
The ECSA is one of the essential characteristics of electrocatalysts. The surface area of porous materials is typically estimated by a method based on physical nitrogen adsorption [
15]. It is worth noting that the method is not suitable for the determination of the ECSA of platinum electrocatalysts, because in addition to the surface area of Pt nanoparticles undergoing the electrochemical process, it also considers the area of a carbon support that serves as an electron conductor in the catalyst. The ECSA of Pt-containing catalysts can usually be evaluated by means of electrochemical approaches [
16] that take into account the real conditions of electrochemical processes. CV was used to obtain the ECSA of the catalysts.
Figure 2 shows the anodic scan of the CV curve of the obtained catalysts in the 0.5 M H
2SO
4 electrolyte with a CO adsorbed ad-layer. The calculated values for the ECSA of the studied catalysts were about 14 ± 5 m
2/g
Pt (
Table 1).
The important parameters for the DEFC anode catalysts are the overpotential for CO
ad and ethanol oxidation. The Pt/C electrocatalyst showed the onset potential for the CO oxidation of 0.585 V. For all the other studied catalytic systems, except for PtRh/C and PtNiO/C, the presence of the second component resulted in reducing the overpotential for CO oxidation. For PtTiO
2/C and PtSnO
2/C, the CO oxidation peaks shifted by 0.110 V and 0.143 V to the cathode region, respectively (
Table 1,
Figure 2).
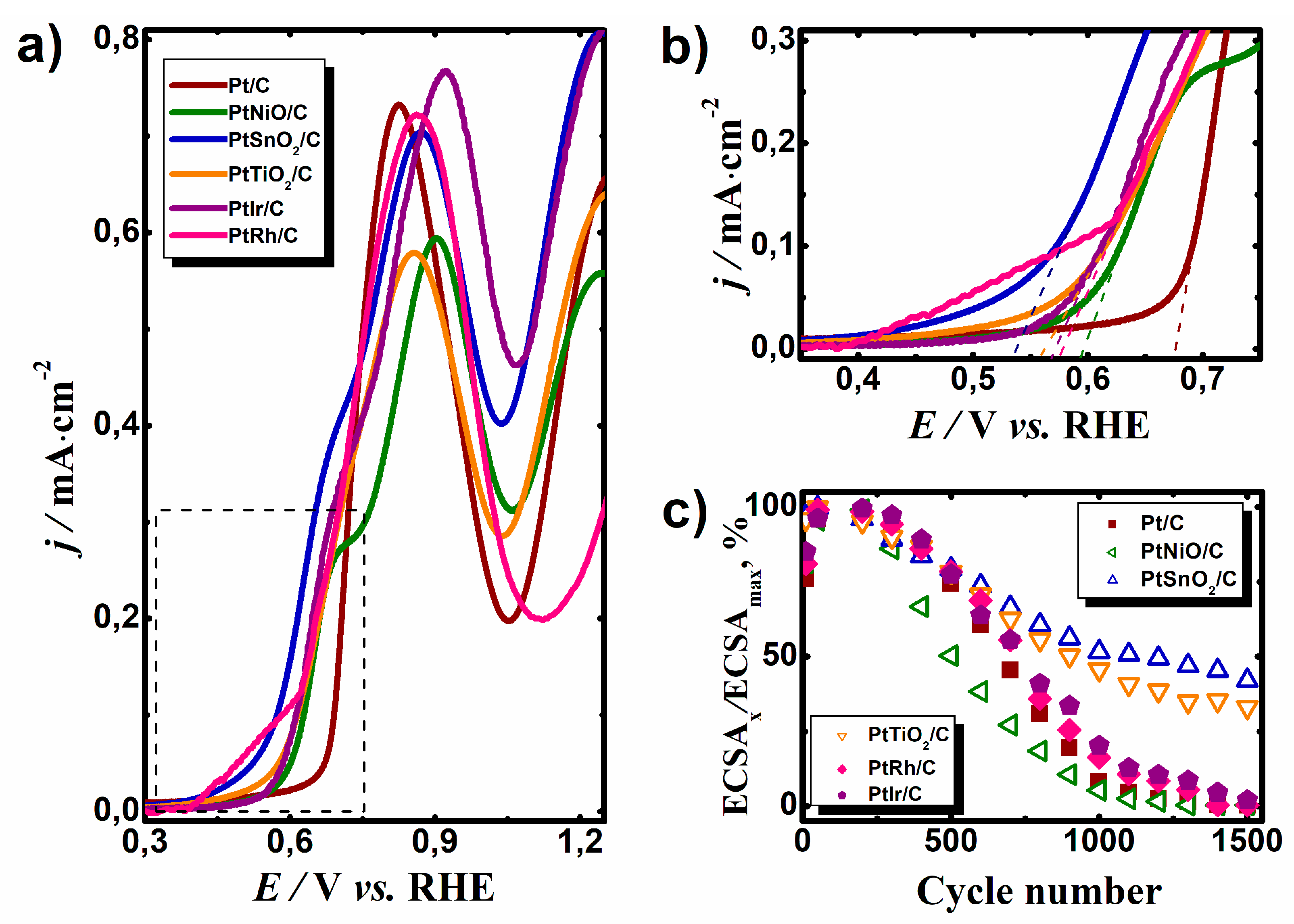
The as-prepared catalysts were also tested for the EOR (
Figure 3a,b). The CV curves of the platinum nanoparticles supported on carbon (Pt/C) and hybrid metal oxide–carbon (PtNiO/C, PtSnO
2/C, and PtTiO
2/C) as well as bimetallic catalysts (PtRh/C and PtIr/C) were recorded in 0.5 M C
2H
5OH + 0.5 M H
2SO
4 solution. The presence of the metal oxide in the support or second metal in the bimetallic catalytic system caused a cathodic shift of the onset potential of the EOR (
Table 1). The maximum effect was observed for the PtSnO
2/C system where the onset potential of the EOR shifted toward negative potential values by 0.158 V. The positive effect of metal oxide in support of the catalyst for CO
ad and ethanol oxidation reaction can be explained by the theory of bifunctional catalysis [
17]. In addition, electronic ligand effects are also possible [
18]. Numerous studies have shown that the introduction of Sn in the composition of the catalyst for the oxidation of organic compounds has a positive effect on the process. A positive effect was observed due to earlier and easier adsorption of OH
ads on the surface of SnO
2 [
19,
20]. Rhodium displayed a higher current in the potential region of 0.4–0.6 V compared to the other catalysts, which can be due to the adsorption of O-containing species on the Rh surface (
Figure 2f) and a little part is related to the oxidation of alcohol. The cathodic shift of the onset potential for EOR on the PtIr/C catalyst may be associated with the modification of the electronic structure of the PtIr alloy compared to Pt [
21]. As a result, the adsorption energy of O-containing particles required for the complete oxidation of ethanol chemisorption products decreased.
The stability of the synthesized catalysts was investigated by cycling in 0.5 M H
2SO
4 over a potential range of 0.05–1.3 V vs. RHE.
Figure 3c shows the dependence ECSA of the catalysts, which was normalized to the maximum ECSA of catalysts from the cycle numbers. For the first 100 cycles, the value of the ECSA increased due to the purification of the platinum surface. However, after the 300th cycle, a decrease in the ECSA of all samples was observed. After a long-term cyclic stability test, the largest decrease in ECSA was observed in the Pt/C, PtNiO/C, PtRh/C, and PtIr/C samples. The fact is that the stability of the electrocatalyst in the FC is largely dependent on the stability of the catalyst support. As electrocatalysts work under very hard conditions, in the presence of the active component of the catalyst, carbon support oxidation occurs, which causes a loss of active component contact with the carbon support and thus the active component is out of the reaction zone [
22]. It is assumed that the introduction of a metal oxide in the support will have a positive impact on the electrocatalyst’s durability. The catalyst based on the hybrid support containing SnO
2 or TiO
2 was significantly more stable. The value of the ECSA after 1500 cycles decreased to 58% and 40% for PtSnO
2/C and PtTiO
2/C, respectively. The behavior of the PtNiO/C electrocatalyst was the same as the Pt/C catalyst, but the rate of catalyst degradation was somewhat higher than that of Pt/C, which is probably due to the solubility of nickel oxide in the acidic medium.
4. Conclusions
In conclusion, the Pt/C, PtMOn/C (M = Ni, Sn, Ti) and PtX/C (X = Rh, Ir) catalyst systems were successfully prepared by using the pulse alternating current (PAC) technique. The XRD patterns of the samples showed the presence of diffraction peaks characteristic for Pt, NiO, SnO2, TiO2, Rh, and Ir. The TEM images indicate that the Pt, Rh, and PtIr (alloys) particles had a uniform distribution over the carbon surface in the Pt/C, PtRh/C, PtIr/C, and PtMOn/C (M = Ni, Sn, Ti) catalysts. When SnO2/C and TiO2/C were present in the supports of the catalyst, the onset potentials for carbon monoxide oxidation shifted in the cathodic direction by 0.143 V and 0.110 V, respectively, compared to the Pt/C catalyst.
All the obtained PtMOn/C and PtX/C catalyst systems revealed a better EOR performance compared to the conventional platinum catalyst. The addition of a second element to Pt or use of a hybrid support can evidently improve the EOR efficiency. A remarkable positive affecting shift of the onset potential for the EOR was observed as follows: PtSnO2/C > PtTiO2/C ≈ PtIr/C ≈ PtNiO/C > PtRh/C ≈ Pt/C. The addition of SnO2 to the Pt/C catalyst led to a decrease in the onset potential and significantly facilitated the EOR. In addition, the catalyst based on the hybrid support containing SnO2 was significantly more stable than the Pt/C catalyst.
Thereby, the PtSnO2/C catalyst prepared by the PAC technique could be considered as a promising anode catalyst for DEFCs. In this way, this study contributed to the development of fuel cell technology with the direct oxidation of liquid fuels.





{kind=link}
{kind=link}
{kind=link}