Neural Differentiation Dynamics Controlled by Multiple Feedback Loops in a Comprehensive Molecular Interaction Network




Abstract
1. Introduction
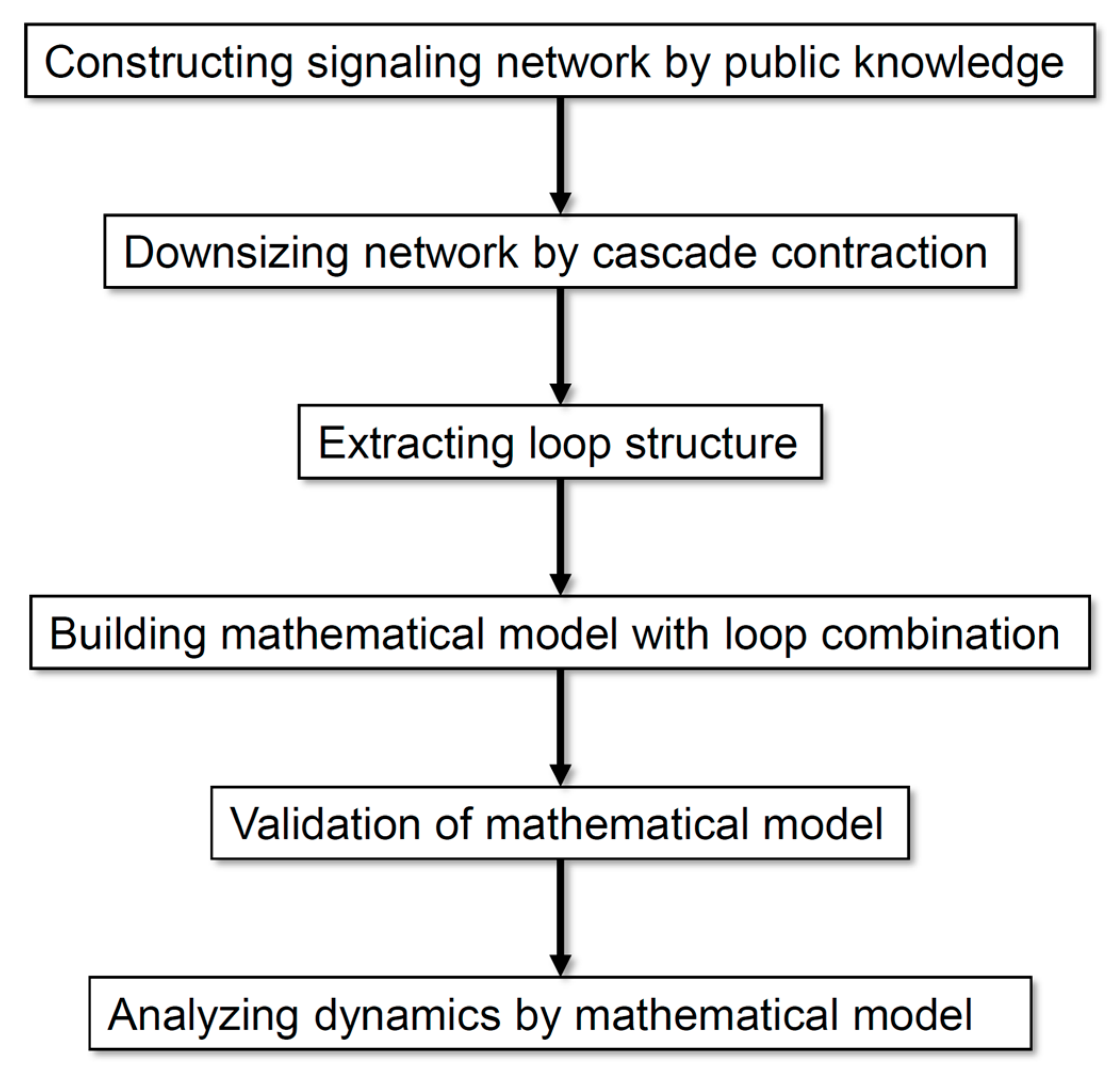
2. Materials and Methods
2.1. Construction of a Neuronal Differentiation Network
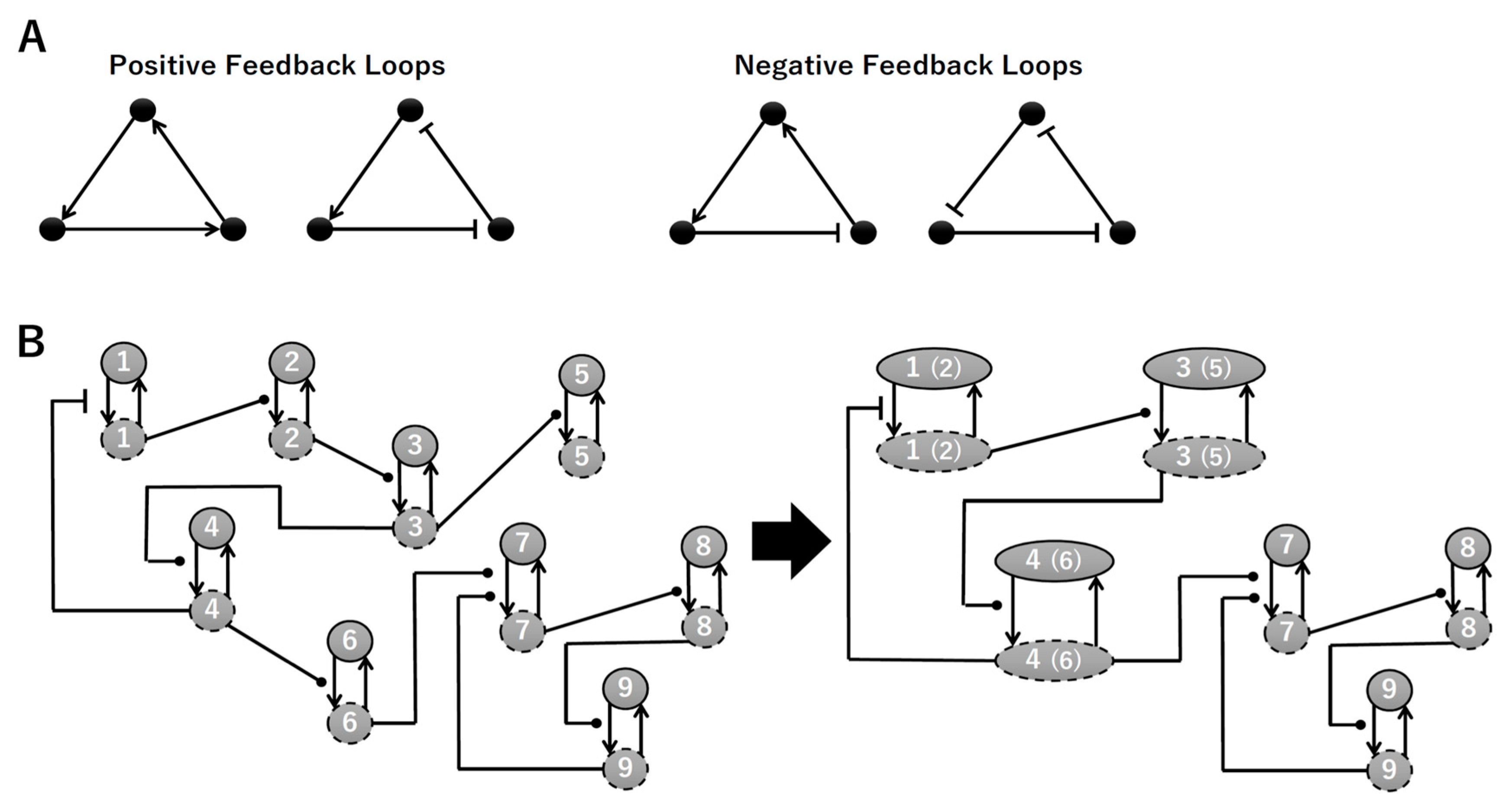
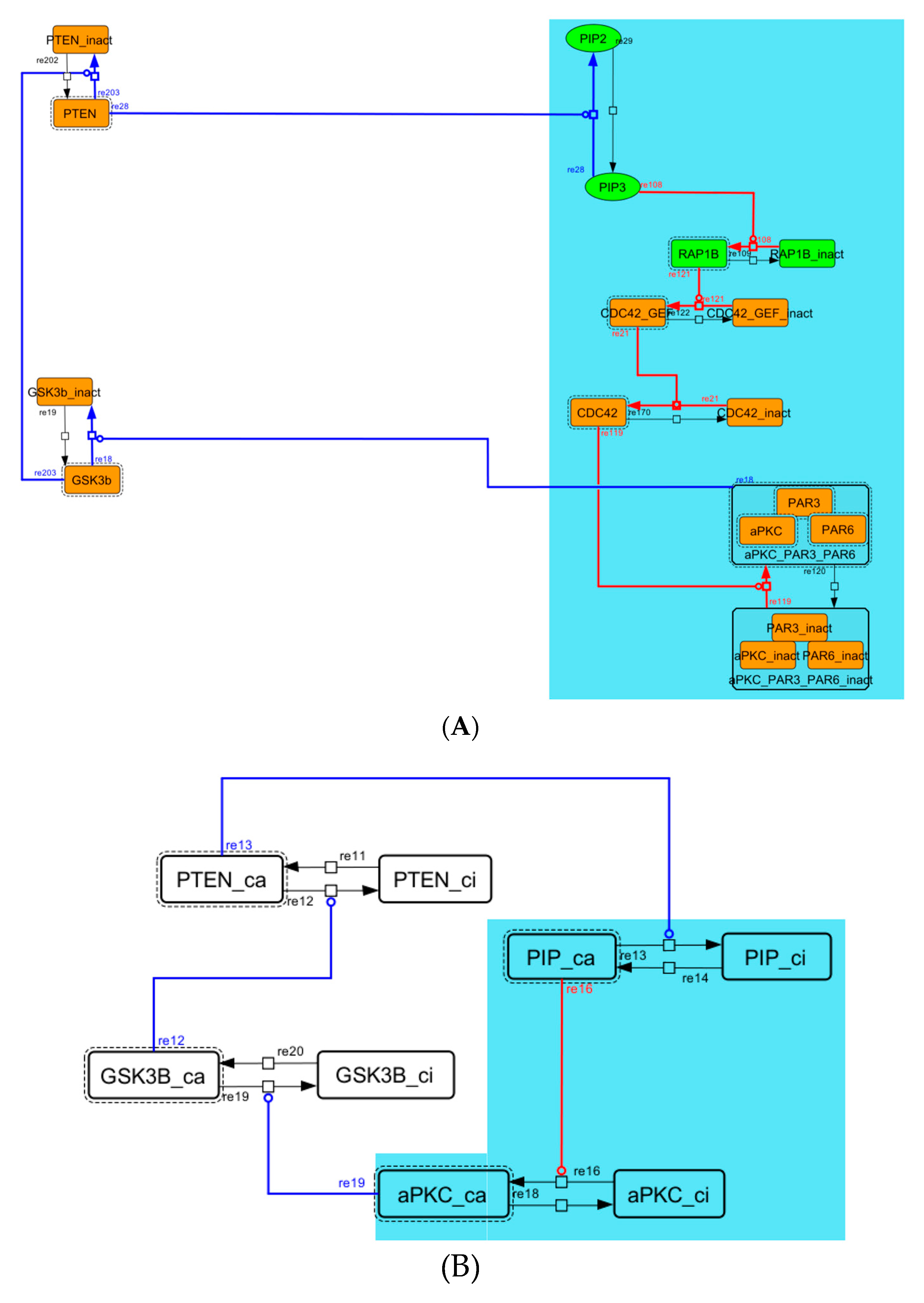
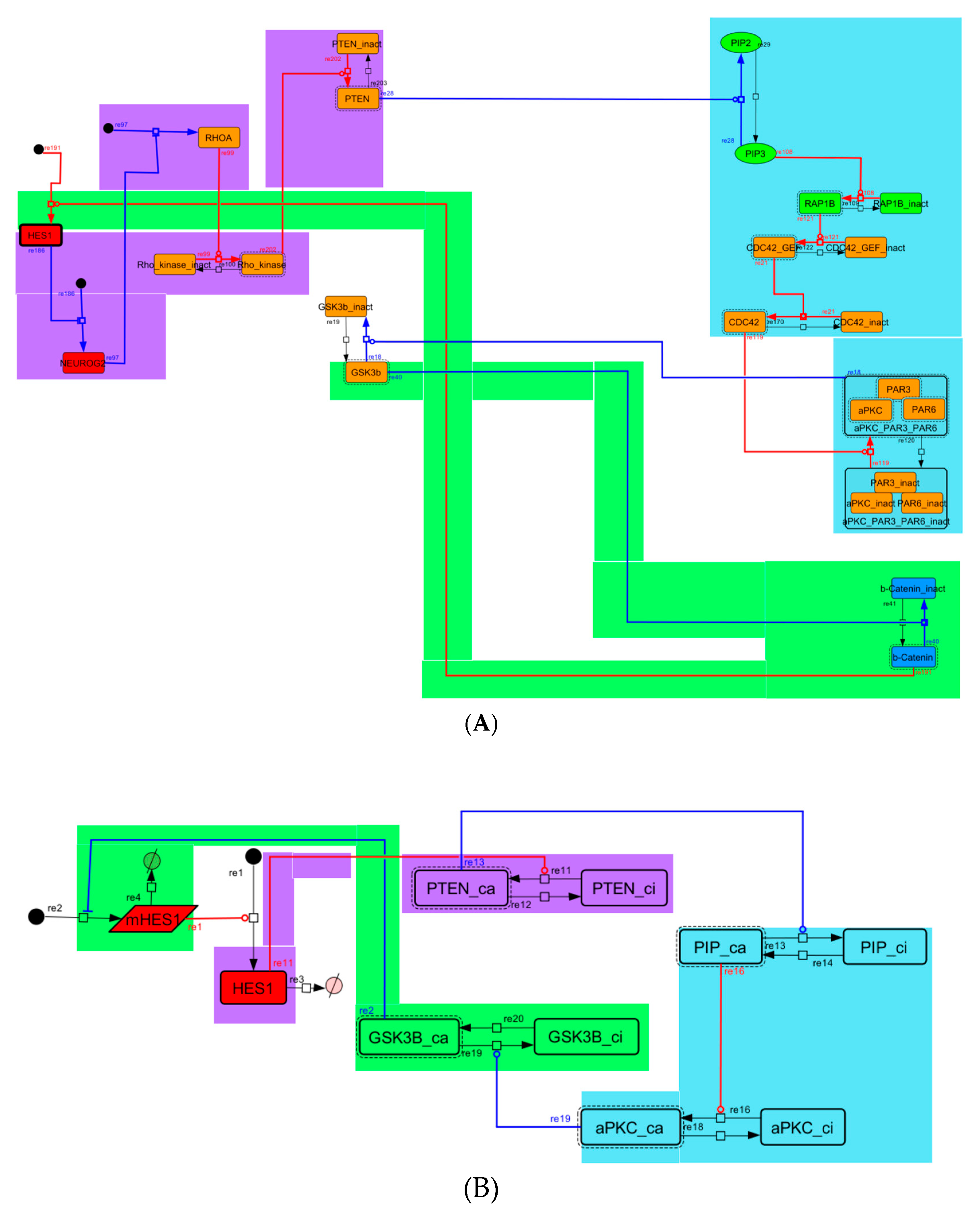
2.2. Contraction of the Network
2.3. Mathematical Model Construction
2.4. Simulation and Analysis
3. Results
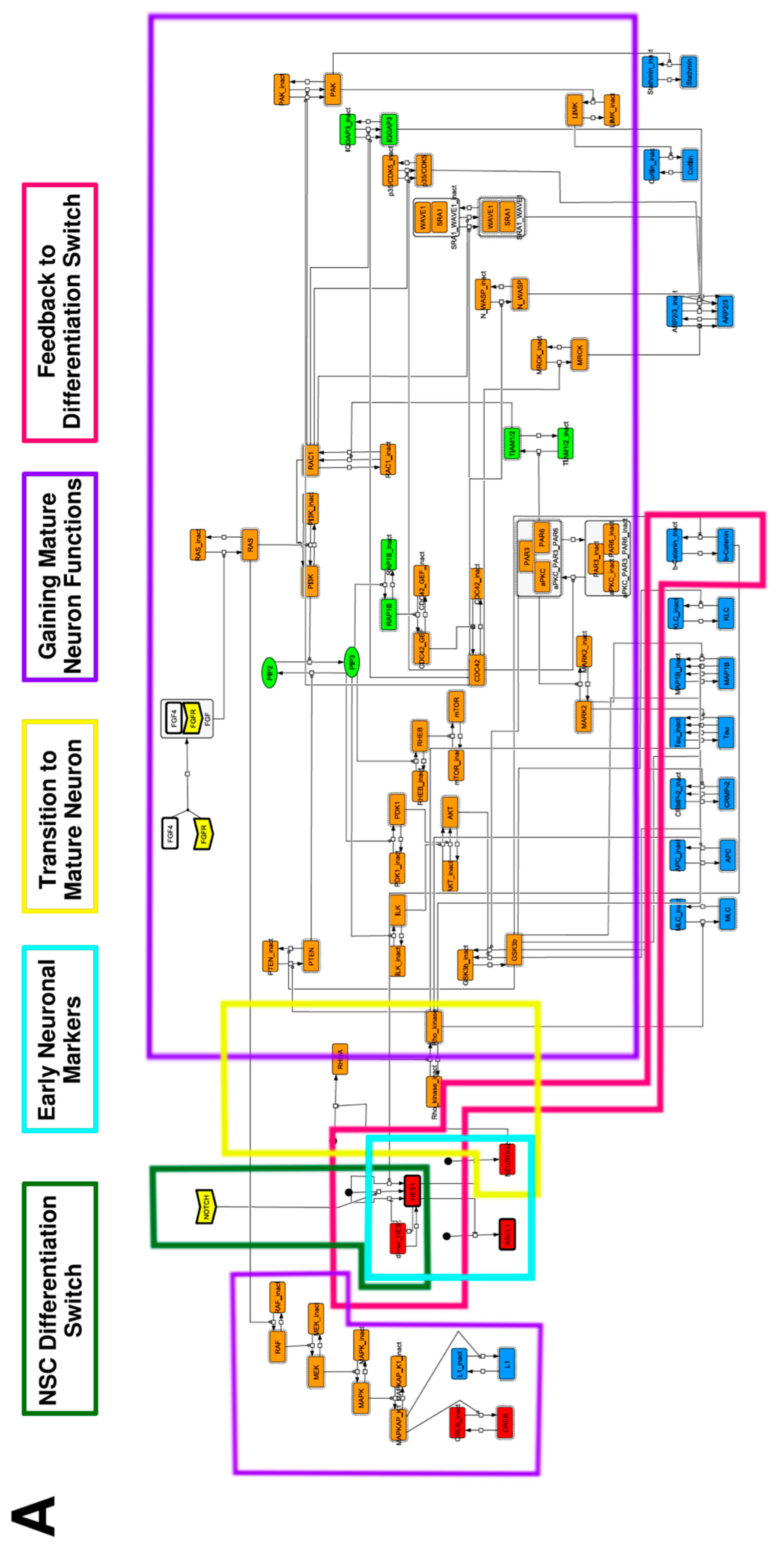
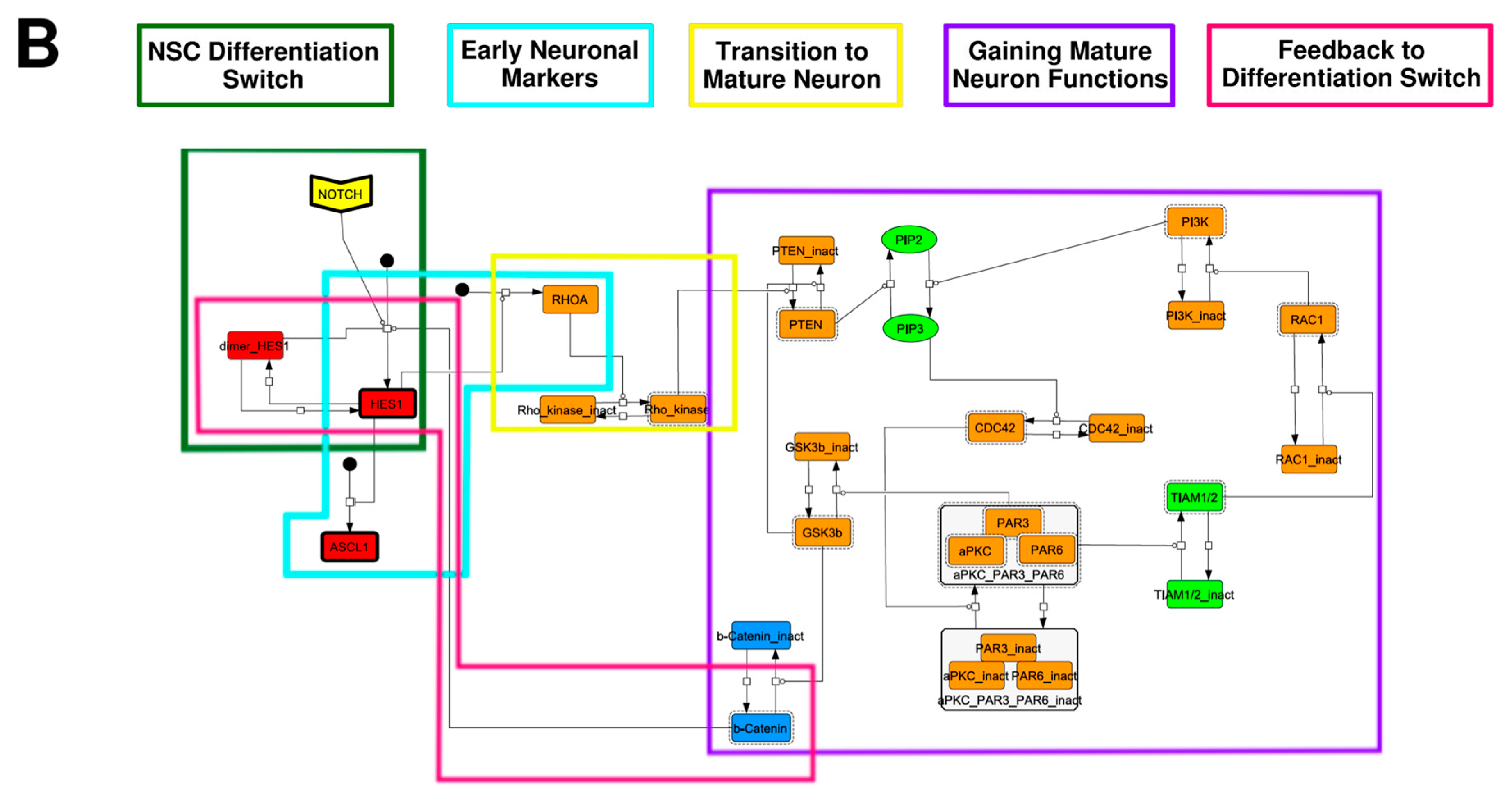
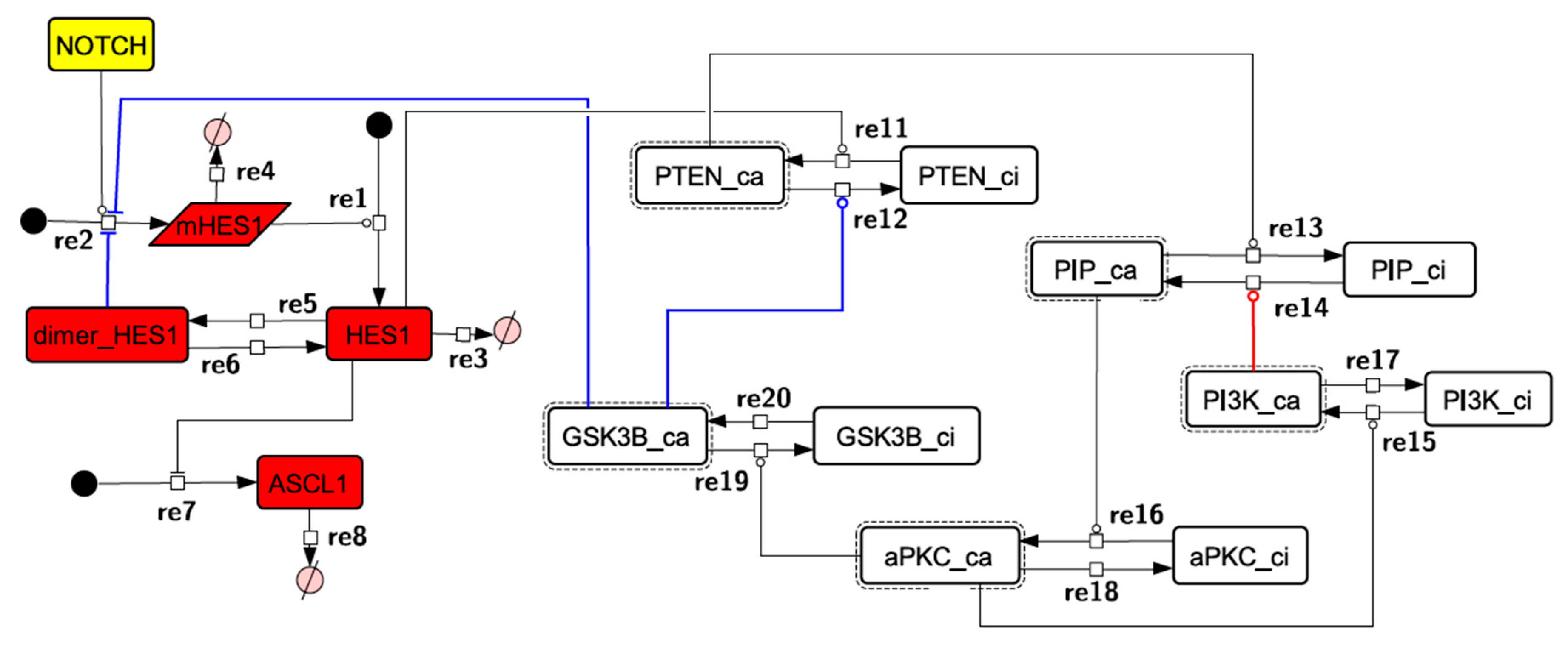
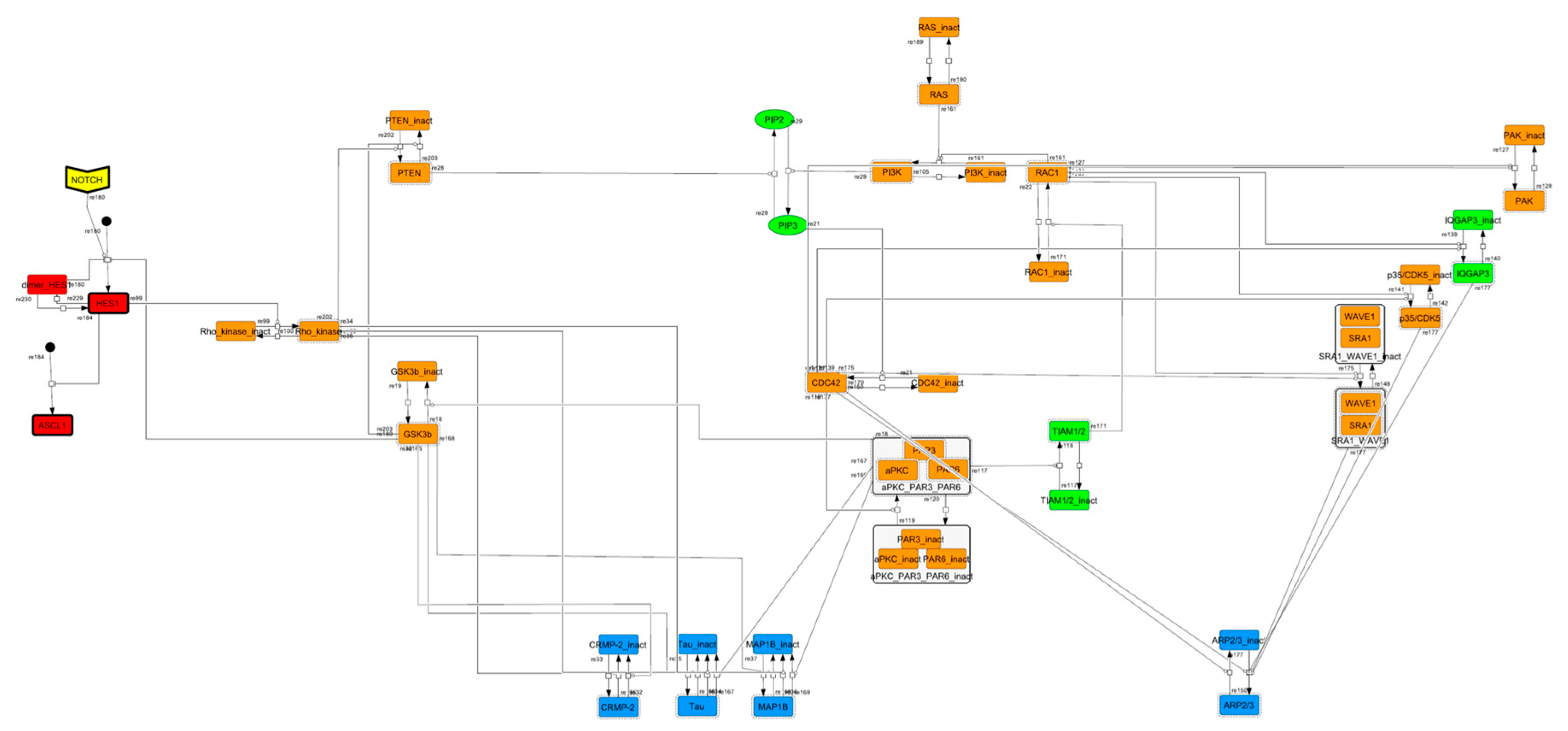
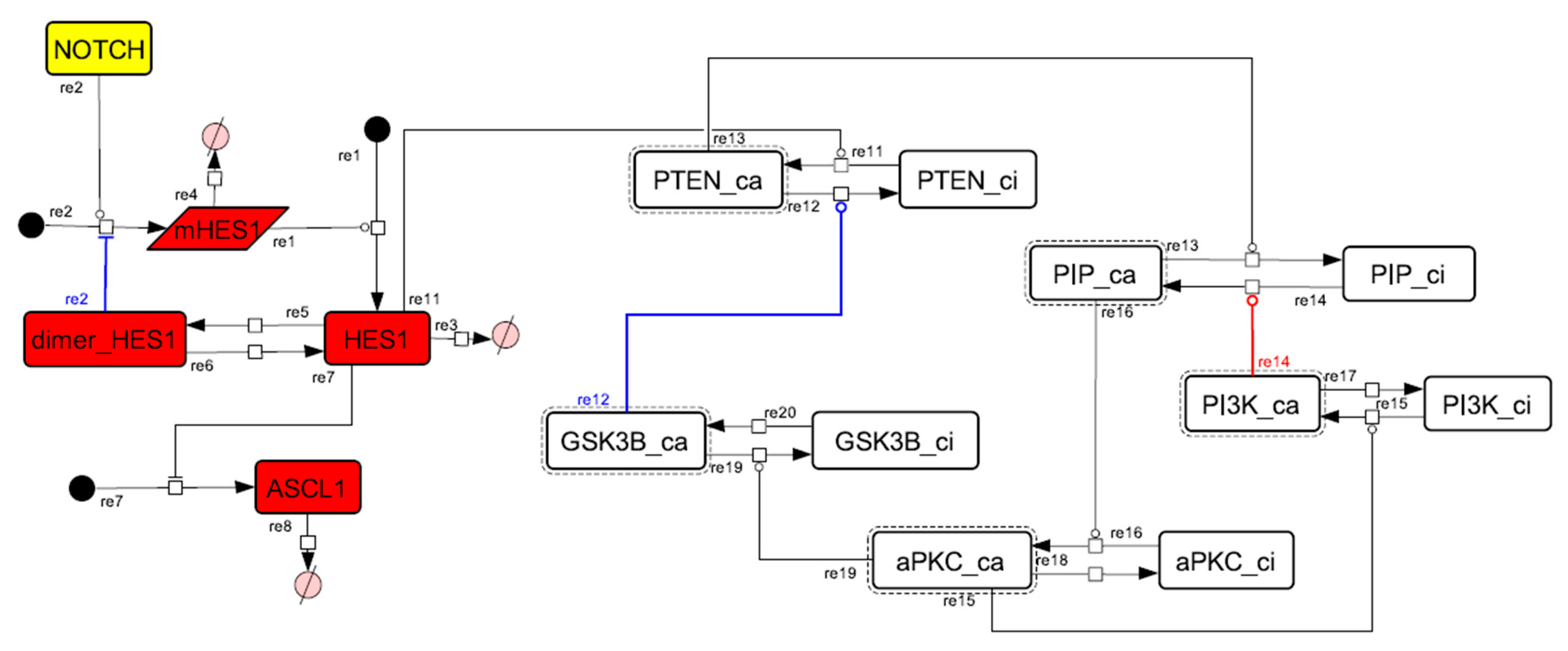
3.1. Signaling Network of Neuronal Differentiation
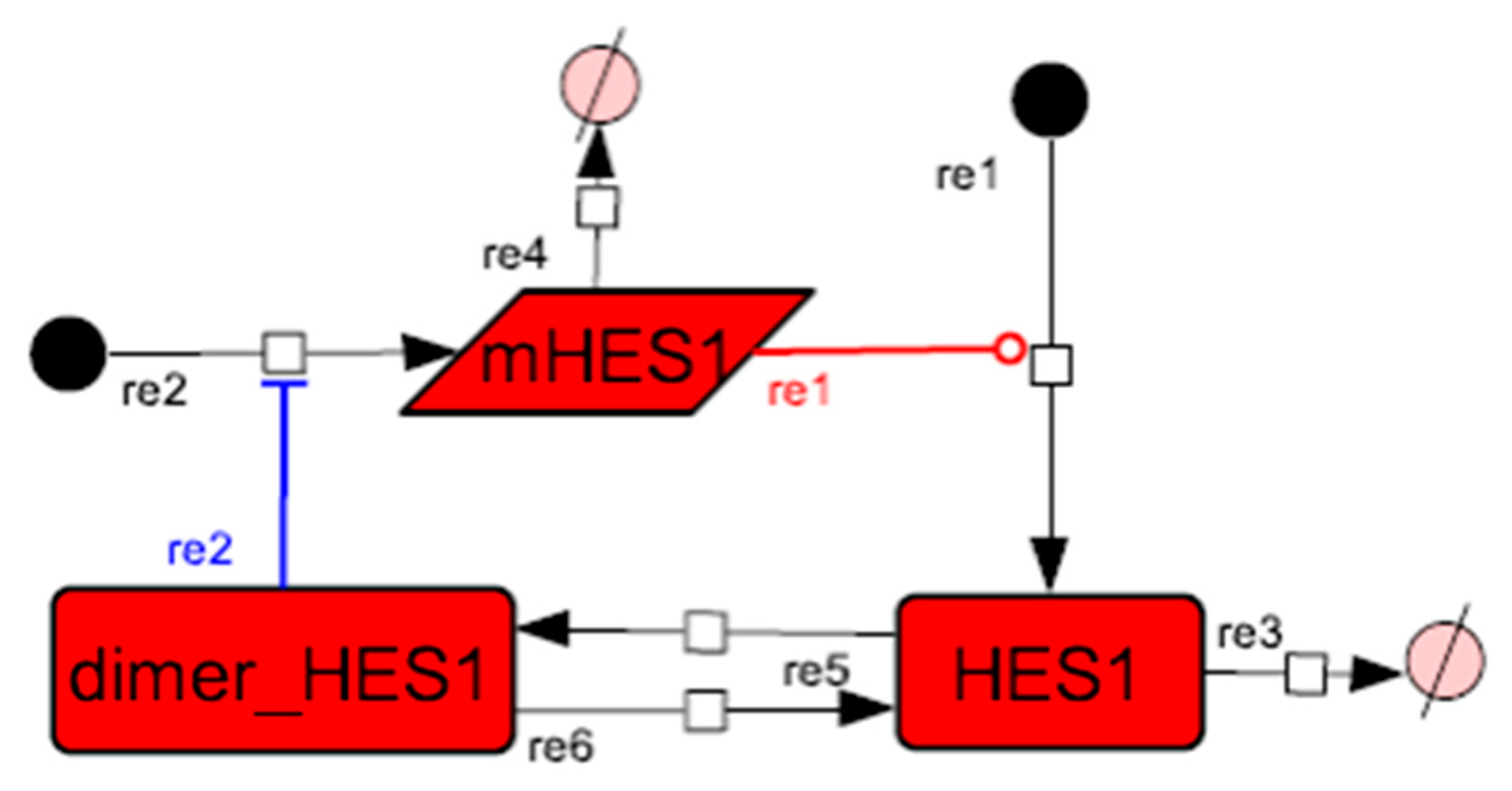
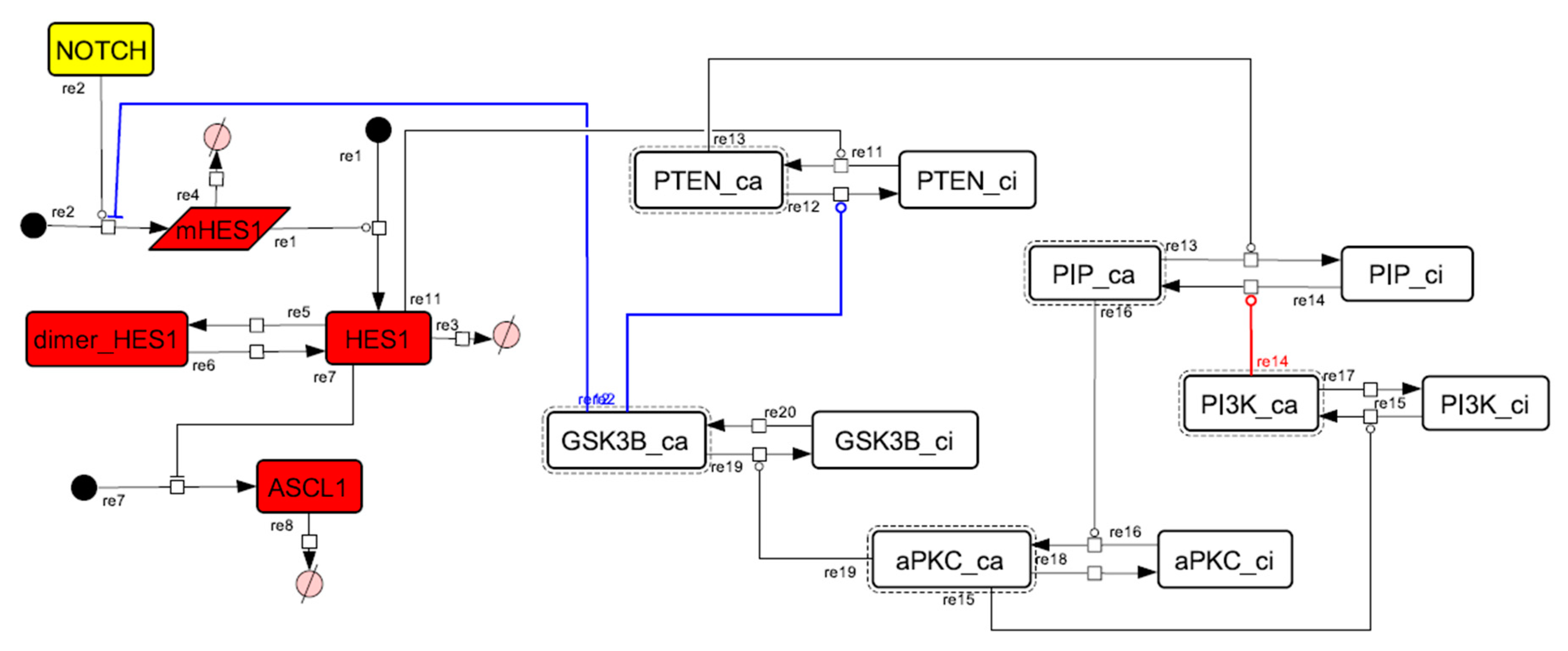
3.2. Mathematical Model of the Core Network
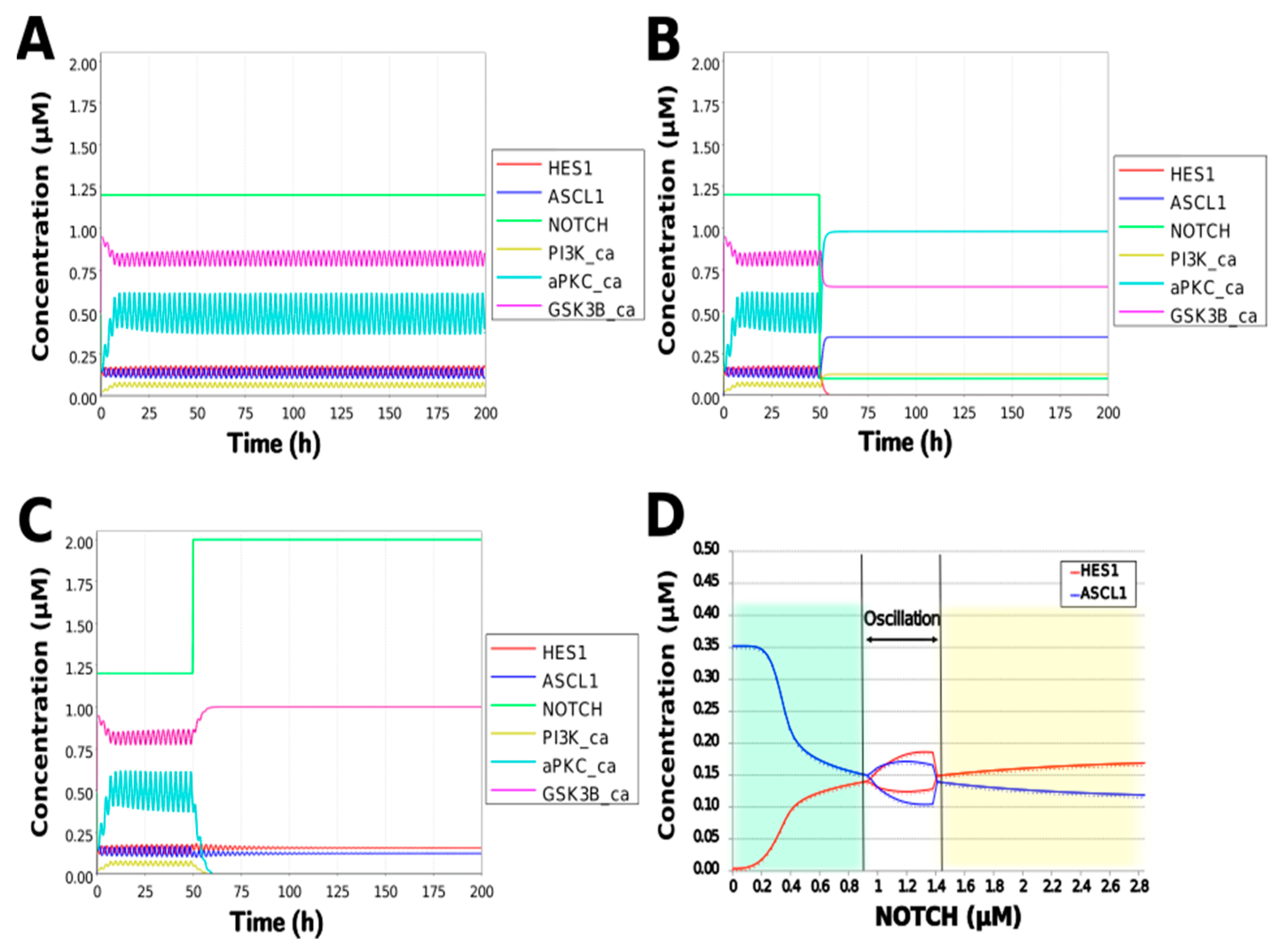
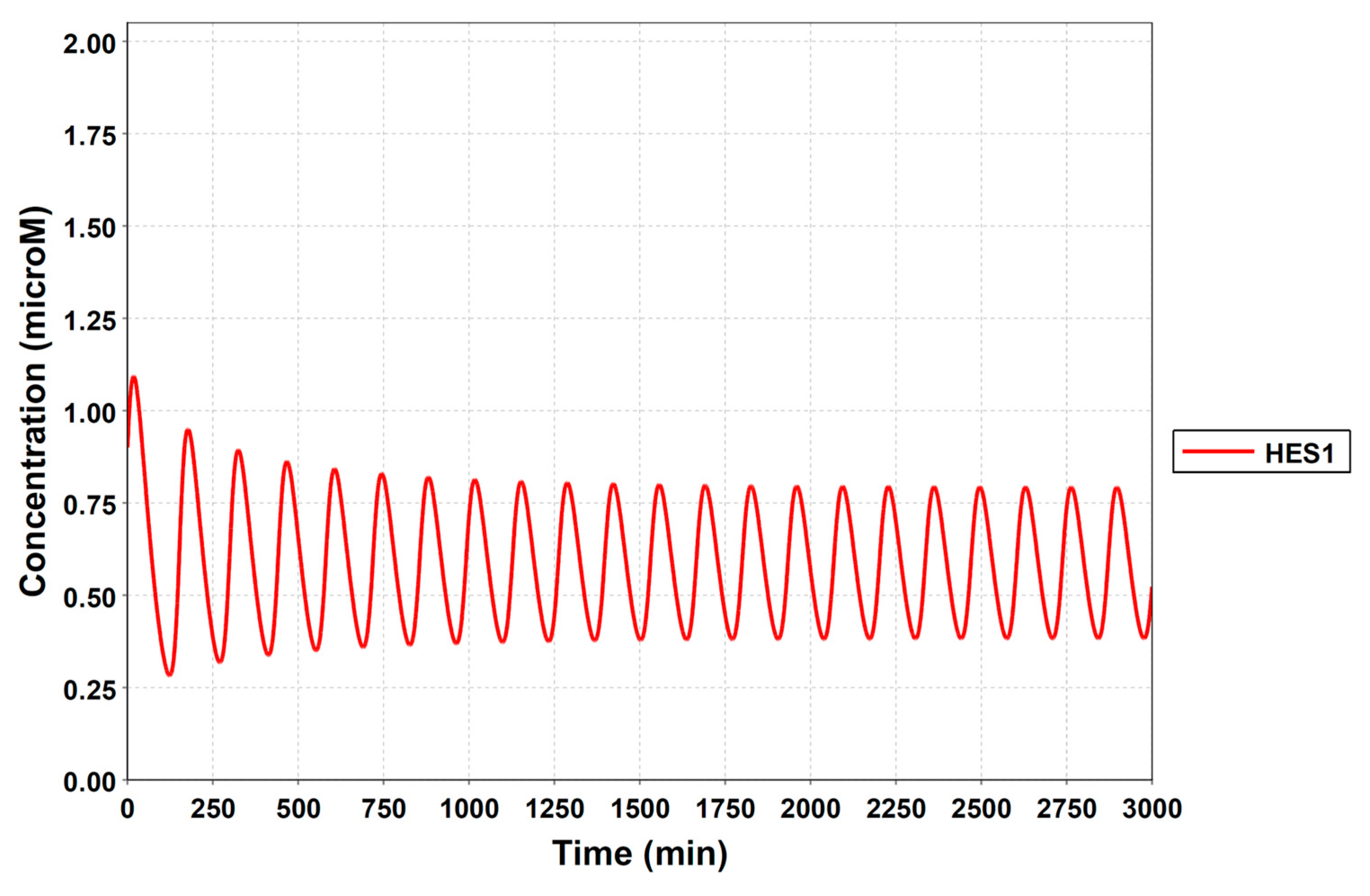
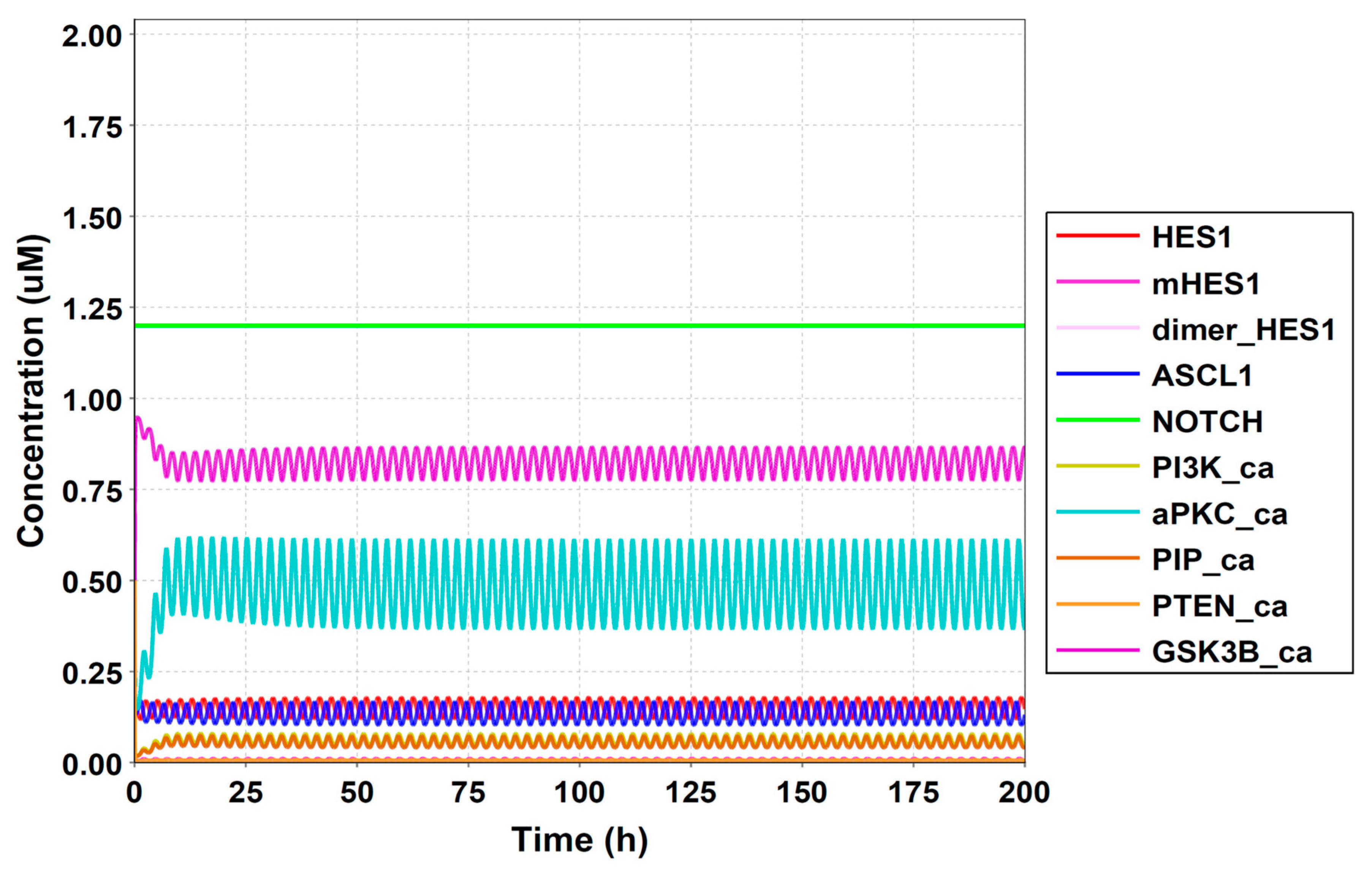
3.3. Simulation of the Oscillatory Dynamics
3.4. Model Validation
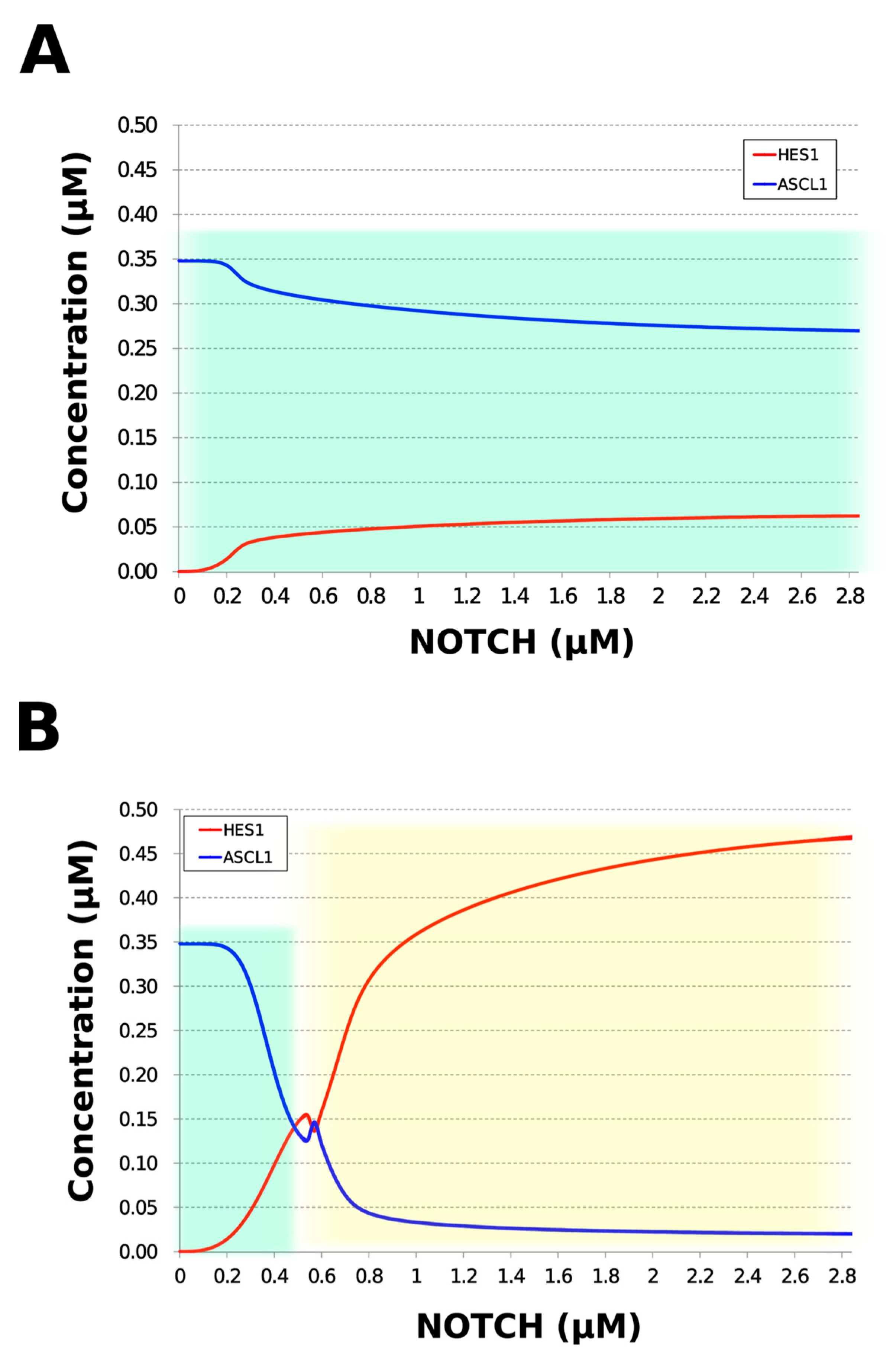
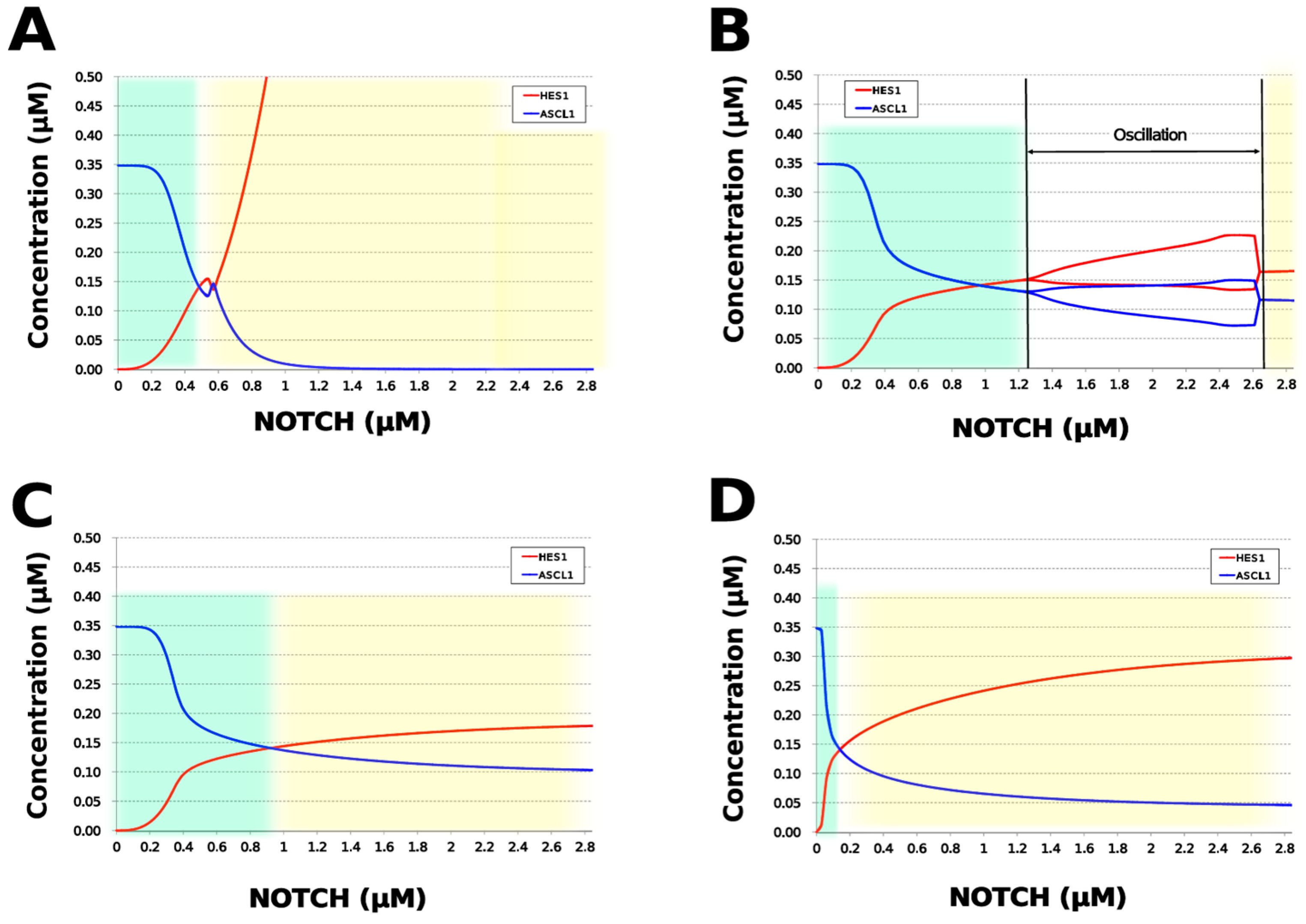
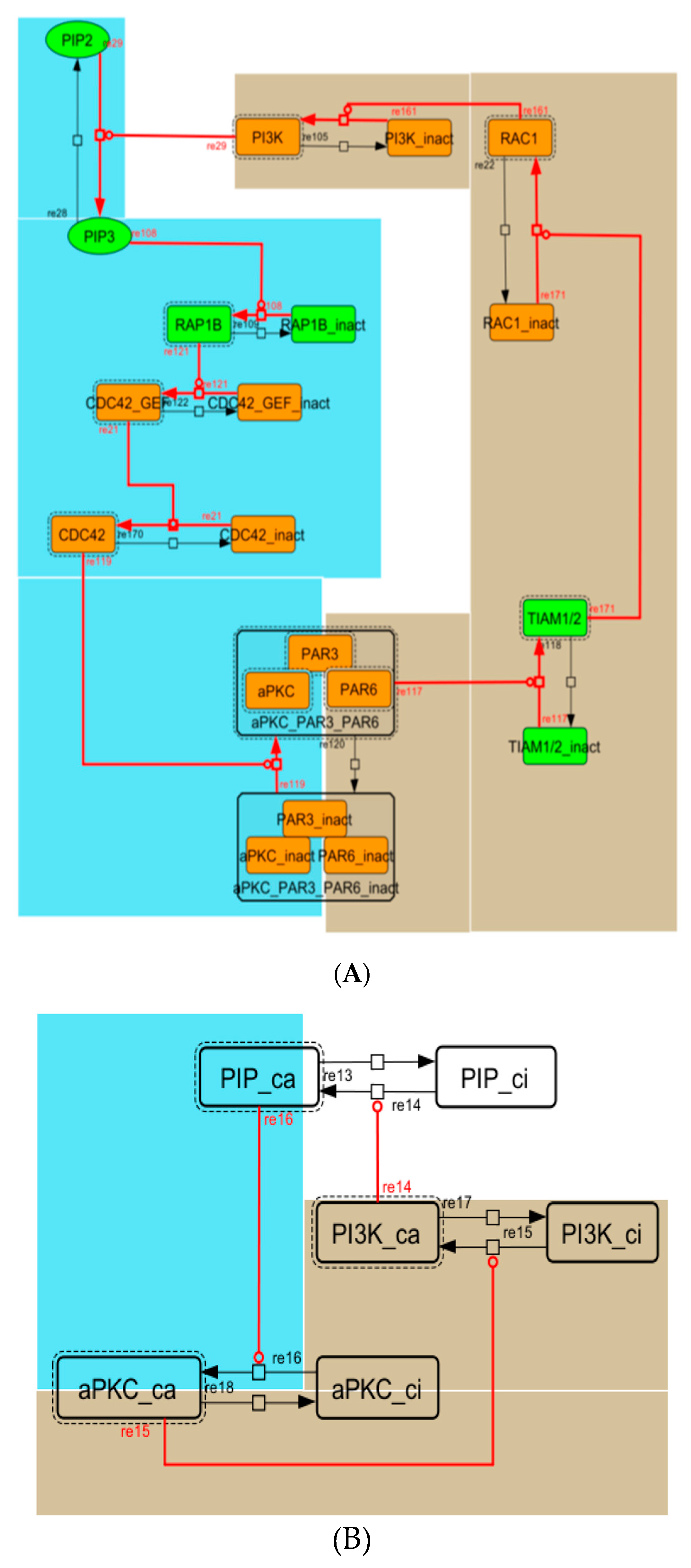
3.5. Model Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. Supporting Figures and Tables












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Node Name | Reason | Integrated To (Identifier in the Toy Model) |
|---|---|---|
| NEUROG2 | Cascade (whole signaling network) | Rho_kinase |
| MLC | Cascade (whole signaling network) | Rho_kinase |
| RhoA | Cascade (whole signaling network) | Rho_kinase |
| RAP1B | Cascade (whole signaling network) | PIP3 |
| CDC42_GEF | Cascade (whole signaling network) | PIP3 |
| Cofilin | Cascade (whole signaling network) | PAK |
| LIMK | Cascade (whole signaling network) | PAK |
| Stathmin | Cascade (whole signaling network) | PAK |
| N_WASP | Cascade (whole signaling network) | CDC42 |
| MRCK | Cascade (whole signaling network) | CDC42 |
| KLC | Cascade (whole signaling network) | GSK3B |
| APC | Cascade (whole signaling network) | GSK3B |
| b-catenin | Cascade (whole signaling network) | GSK3B |
| mTOR | Cascade (whole signaling network) | PIP3 |
| RHEB | Cascade (whole signaling network) | PIP3 |
| PDK1 | Cascade (whole signaling network) | PIP3 |
| ILK | Cascade (whole signaling network) | PIP3 |
| AKT | Cascade (whole signaling network) | PIP3 |
| L1 | Cascade (whole signaling network) | RAS |
| CREB | Cascade (whole signaling network) | RAS |
| MAPKAP_K1 | Cascade (whole signaling network) | RAS |
| MAPK | Cascade (whole signaling network) | RAS |
| MEK | Cascade (whole signaling network) | RAS |
| RAF | Cascade (whole signaling network) | RAS |
| MARK2 | Cascade (whole signaling network) | aPKC_PAR3_PAR6 |
| Arp2/3 | Feedback loop extraction | - |
| IQGAP3 | Feedback loop extraction | - |
| PAK | Feedback loop extraction | - |
| p35/CDK5 | Feedback loop extraction | - |
| SRA1_WAVE1 | Feedback loop extraction | - |
| MAP1B | Feedback loop extraction | - |
| Tau | Feedback loop extraction | - |
| CRMP-2 | Feedback loop extraction | - |
| RAS | Parameterization | - |
| Rho_kinase | Cascade (core network) | PTEN_ca (s22) |
| TIAM1/2 | Cascade (core network) | PI3K_ca (s20) |
| RAC1 | Cascade (core network) | PI3K_ca (s20) |
| PIP3 | Cascade (core network) | PIP_ca (s16) |
| GSK3B | Cascade (core network) | GSK3B_ca (s24) |
| PTEN | Cascade (core network) | PTEN_ca (s22) |
| aPKC_PAR3_PAR6 | Cascade (core network) | aPKC_ca (s18) |
| Equation No. | Differential equations |
|---|---|
| 1 | |
| 2 | |
| 3 |
References
- Karr, J.R.; Sanghvi, J.C.; Macklin, D.N.; Gutschow, M.V.; Jacobs, J.M.; Bolival, B., Jr.; Assad-Garcia, N.; Glass, J.I.; Covert, M.W. A whole-cell computational model predicts phenotype from genotype. Cell 2012, 150, 389–401. [Google Scholar] [CrossRef]
- Vignes, M.; Vandel, J.; Allouche, D.; Ramadan-Alban, N.; Cierco-Ayrolles, C.; Schiex, T.; Mangin, B.; de Givry, S. Gene regulatory network reconstruction using Bayesian networks, the Dantzig Selector, the Lasso and their meta-analysis. PLoS ONE 2011, 6, e29165. [Google Scholar] [CrossRef]
- Chai, L.E.; Loh, S.K.; Low, S.T.; Mohamad, M.S.; Deris, S.; Zakaria, Z. A review on the computational approaches for gene regulatory network construction. Comput. Biol. Med. 2014, 48, 55–65. [Google Scholar] [CrossRef]
- Park, Y.; Kellis, M. Deep learning for regulatory genomics. Nat. Biotechnol. 2015, 33, 825–826. [Google Scholar] [CrossRef]
- Karr, J.R.; Williams, A.H.; Zucker, J.D.; Raue, A.; Steiert, B.; Timmer, J.; Kreutz, C.; Wilkinson, S.; Allgood, B.A.; Bot, B.M.; et al. Summary of the DREAM8 Parameter Estimation Challenge: Toward Parameter Identification for Whole-Cell Models. PLoS Comput. Biol. 2015, 11, e1004096. [Google Scholar] [CrossRef]
- Itzkovitz, S.; Levitt, R.; Kashtan, N.; Milo, R.; Itzkovitz, M.; Alon, U. Coarse-graining and self-dissimilarity of complex networks. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2005, 71 Pt 2, 016127. [Google Scholar] [CrossRef]
- Kim, D.H.; Noh, J.D.; Jeong, H. Scale-free trees: The skeletons of complex networks. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2004, 70 Pt 2, 046126. [Google Scholar] [CrossRef]
- Kim, J.R.; Kim, J.; Kwon, Y.K.; Lee, H.Y.; Heslop-Harrison, P.; Cho, K.H. Reduction of complex signaling networks to a representative kernel. Sci. Signal. 2011, 4, ra35. [Google Scholar] [CrossRef]
- Milo, R.; Shen-Orr, S.; Itzkovitz, S.; Kashtan, N.; Chklovskii, D.; Alon, U. Network motifs: Simple building blocks of complex networks. Science 2002, 298, 824–827. [Google Scholar] [CrossRef]
- Riccione, K.A.; Smith, R.P.; Lee, A.J.; You, L. A synthetic biology approach to understanding cellular information processing. ACS Synth. Biol. 2012, 1, 389–402. [Google Scholar] [CrossRef][Green Version]
- Louvi, A.; Artavanis-Tsakonas, S. Notch signalling in vertebrate neural development. Nat. Rev. Neurosci. 2006, 7, 93–102. [Google Scholar] [CrossRef]
- Monk, N.A. Oscillatory expression of Hes1, p53, and NF-kappaB driven by transcriptional time delays. Curr. Biol. 2003, 13, 1409–1413. [Google Scholar] [CrossRef]
- Zeiser, S.; Müller, J.; Liebscher, V. Modeling the Hes1 oscillator. J. Comput. Biol. 2007, 14, 984–1000. [Google Scholar] [CrossRef]
- Imayoshi, I.; Isomura, A.; Harima, Y.; Kawaguchi, K.; Kori, H.; Miyachi, H.; Fujiwara, T.; Ishidate, F.; Kageyama, R. Oscillatory control of factors determining multipotency and fate in mouse neural progenitors. Science 2013, 342, 1203–1208. [Google Scholar] [CrossRef]
- Bai, G.; Sheng, N.; Xie, Z.; Bian, W.; Yokota, Y.; Benezra, R.; Kageyama, R.; Guillemot, F.; Jing, N. Id sustains Hes1 expression to inhibit precocious neurogenesis by releasing negative autoregulation of Hes1. Dev. Cell 2007, 13, 283–297. [Google Scholar] [CrossRef]
- Kageyama, R.; Ohtsuka, T.; Hatakeyama, J.; Ohsawa, R. Roles of bHLH genes in neural stem cell differentiation. Exp. Cell Res. 2005, 306, 343–348. [Google Scholar] [CrossRef]
- Kageyama, R.; Ohtsuka, T.; Kobayashi, T. Roles of Hes genes in neural development. Dev. Growth Differ. 2008, 50 (Suppl. S1), S97–S103. [Google Scholar] [CrossRef]
- Seki, T.; Sawamoto, K.; Parent, J.M.; Alvarez-Buylla, A. (Eds.) Neurogenesis in the Adult Brain; Springer: Tokyo, Japan, 2011; Volume 1. [Google Scholar]
- Roybon, L.; Mastracci, T.L.; Ribeiro, D.; Sussel, L.; Brundin, P.; Li, J.Y. GABAergic differentiation induced by Mash1 is compromised by the bHLH proteins Neurogenin2, NeuroD1, and NeuroD2. Cereb. Cortex 2010, 20, 1234–1244. [Google Scholar] [CrossRef]
- Bhat, K.M.; Maddodi, N.; Shashikant, C.; Setaluri, V. Transcriptional regulation of human MAP2 gene in melanoma: Role of neuronal bHLH factors and Notch1 signaling. Nucleic Acids Res. 2006, 34, 3819–3832. [Google Scholar] [CrossRef]
- Arimura, N.; Kaibuchi, K. Neuronal polarity: From extracellular signals to intracellular mechanisms. Nat. Rev. Neurosci. 2007, 8, 194–205. [Google Scholar] [CrossRef]
- Hand, R.; Bortone, D.; Mattar, P.; Nguyen, L.; Heng, J.I.; Guerrier, S.; Boutt, E.; Peters, E.; Barnes, A.P.; Parras, C.; et al. Phosphorylation of Neurogenin2 specifies the migration properties and the dendritic morphology of pyramidal neurons in the neocortex. Neuron 2005, 48, 45–62. [Google Scholar] [CrossRef]
- Shimizu, T.; Kagawa, T.; Inoue, T.; Nonaka, A.; Takada, S.; Aburatani, H.; Taga, T. Stabilized beta-catenin functions through TCF/LEF proteins and the Notch/RBP-Jkappa complex to promote proliferation and suppress differentiation of neural precursor cells. Mol. Cell Biol. 2008, 28, 7427–7441. [Google Scholar] [CrossRef]
- Schaefer, C.F.; Anthony, K.; Krupa, S.; Buchoff, J.; Day, M.; Hannay, T.; Buetow, K.H. PID: The Pathway Interaction Database. Nucleic Acids Res. 2009, 37, D674–D679. [Google Scholar] [CrossRef]
- Kelder, T.; van Iersel, M.P.; Hanspers, K.; Kutmon, M.; Conklin, B.R.; Evelo, C.T.; Pico, A.R. WikiPathways: Building research communities on biological pathways. Nucleic Acids Res. 2012, 40, D1301–D1307. [Google Scholar] [CrossRef]
- Funahashi, A.; Matsuoka, Y.; Jouraku, A.; Morohashi, M.; Kikuchi, N.; Kitano, H. CellDesigner 3.5: A Versatile Modeling Tool for Biochemical Networks. Proc. IEEE 2008, 96, 1254–1265. [Google Scholar] [CrossRef]
- Kitano, H.; Funahashi, A.; Matsuoka, Y.; Oda, K. Using process diagrams for the graphical representation of biological networks. Nat. Biotechnol. 2005, 23, 961–966. [Google Scholar] [CrossRef]
- Dräger, A.; Hassis, N.; Supper, J.; Schröder, A.; Zell, A. SBMLsqueezer: A CellDesigner plug-in to generate kinetic rate equations for biochemical networks. BMC Syst. Biol. 2008, 2, 39. [Google Scholar] [CrossRef]
- Milo, R.; Jorgensen, P.; Moran, U.; Weber, G.; Springer, M. BioNumbers--the database of key numbers in molecular and cell biology. Nucleic Acids Res. 2010, 38, D750–D753. [Google Scholar] [CrossRef]
- Bar-Even, A.; Noor, E.; Savir, Y.; Liebermeister, W.; Davidi, D.; Tawfik, D.S.; Milo, R. The moderately efficient enzyme: Evolutionary and physicochemical trends shaping enzyme parameters. Biochemistry 2011, 50, 4402–4410. [Google Scholar] [CrossRef]
- Legewie, S.; Herzel, H.; Westerhoff, H.V.; Blüthgen, N. Recurrent design patterns in the feedback regulation of the mammalian signalling network. Mol. Syst. Biol. 2008, 4, 190. [Google Scholar] [CrossRef]
- Hoops, S.; Sahle, S.; Gauges, R.; Lee, C.; Pahle, J.; Simus, N.; Singhal, M.; Xu, L.; Mendes, P.; Kummer, U. COPASI-A COmplex PAthway SImulator. Bioinformatics 2006, 22, 3067–3074. [Google Scholar] [CrossRef]
- Machné, R.; Finney, A.; Müller, S.; Lu, J.; Widder, S.; Flamm, C. The SBML ODE Solver Library: A native API for symbolic and fast numerical analysis of reaction networks. Bioinformatics 2006, 22, 1406–1407. [Google Scholar] [CrossRef]
- Petzold, L. Automatic selection of methods for solving stiff and nonstiff systems of ordinary differential equations. SIAM J. Sci. Stat. Comput. 1983, 4, 136–148. [Google Scholar] [CrossRef]
- Eberhardt, M.; Lai, X.; Tomar, N.; Gupta, S.; Schmeck, B.; Steinkasserer, A.; Schuler, G.; Vera, J. Third-kind encounters in biomedicine: Immunology meets mathematics and informatics to become quantitative and predictive. Methods Mol. Biol. 2016, 1386, 135–179. [Google Scholar]
- Trinh, H.C.; Le, D.H.; Kwon, Y.K. PANET: A GPU-based tool for fast parallel analysis of robustness dynamics and feed-forward/feedback loop structures in large-scale biological networks. PLoS ONE 2014, 9, e103010. [Google Scholar] [CrossRef]
- Patra, S.; Mohapatra, A. Application of dynamic expansion tree for finding large network motifs in biological networks. PeerJ. 2019, 7, e6917. [Google Scholar] [CrossRef]
- Hirata, H.; Yoshiura, S.; Ohtsuka, T.; Bessho, Y.; Harada, T.; Yoshikawa, K.; Kageyama, R. Oscillatory expression of the bHLH factor Hes1 regulated by a negative feedback loop. Science 2002, 298, 840–843. [Google Scholar] [CrossRef]
- Kageyama, R.; Ohtsuka, T.; Kobayashi, T. The Hes gene family: Repressors and oscillators that orchestrate embryogenesis. Development 2007, 134, 1243–1251. [Google Scholar] [CrossRef]
- Foltz, D.R.; Santiago, M.C.; Berechid, B.E.; Nye, J.S. Glycogen synthase kinase-3beta modulates notch signaling and stability. Curr. Biol. 2002, 12, 1006–1011. [Google Scholar] [CrossRef]
- Guha, S.; Cullen, J.P.; Morrow, D.; Colombo, A.; Lally, C.; Walls, D.; Redmond, E.M.; Cahill, P.A. Glycogen synthase kinase 3 beta positively regulates Notch signaling in vascular smooth muscle cells: Role in cell proliferation and survival. Basic Res. Cardiol. 2011, 106, 773–785. [Google Scholar] [CrossRef]
- Jin, Y.H.; Kim, H.; Oh, M.; Ki, H.; Kim, K. Regulation of Notch1/NICD and Hes1 expressions by GSK-3alpha/beta. Mol. Cells 2009, 27, 15–19. [Google Scholar] [CrossRef]
- Kim, W.Y.; Wang, X.; Wu, Y.; Doble, B.W.; Patel, S.; Woodgett, J.R.; Snider, W.D. GSK-3 is a master regulator of neural progenitor homeostasis. Nat. Neurosci. 2009, 12, 1390–1397. [Google Scholar] [CrossRef]
- Ye, F.; Chen, Y.; Hoang, T.; Montgomery, R.L.; Zhao, X.H.; Bu, H.; Hu, T.; Taketo, M.M; van Es, J.H.; Clevers, H.; et al. HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the beta-catenin-TCF interaction. Nat. Neurosci. 2009, 12, 829–838. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, Z.; Shu, H.; Liu, S.; Song, Y.; Qiu, K.; Yang, H. The modulatory effects of bHLH transcription factors with the Wnt/beta-catenin pathway on differentiation of neural progenitor cells derived from neonatal mouse anterior subventricular zone. Brain Res. 2010, 1315, 1–10. [Google Scholar] [CrossRef]
- Peignon, G.; Durand, A.; Cacheux, W.; Ayrault, O.; Terris, B.; Laurent-Puig, P.; Shroyer, N.F.; Van Seuningen, I.; Honjo, T.; Perret, C.; et al. Complex interplay between b-catenin signalling and Notch effectors in intestinal tumorigenesis. Gut 2011, 60, 166–176. [Google Scholar] [CrossRef]
- Liu, Z.H.; Dai, X.M.; Du, B. Hes1: A key role in stemness, metastasis and multidrug resistance. Cancer Biol. Ther. 2015, 16, 353–359. [Google Scholar] [CrossRef]
- Chasman, D.; Fotuhi Siahpirani, A.; Roy, S. Network-based approaches for analysis of complex biological systems. Curr. Opin. Biotechnol. 2016, 39, 157–166. [Google Scholar] [CrossRef]
- McLeay, R.C.; Bailey, T.L. Motif Enrichment Analysis: A unified framework and an evaluation on ChIP data. BMC Bioinform. 2010, 11, 165. [Google Scholar] [CrossRef]
- Morimoto, M.; Nishinakamura, R.; Saga, Y.; Kopan, R. Different assemblies of Notch receptors coordinate the distribution of the major bronchial Clara, ciliated and neuroendocrine cells. Development 2012, 139, 4365–4373. [Google Scholar] [CrossRef]
- Ramos, C.; Rocha, S.; Gaspar, C.; Henrique, D. Two Notch ligands, Dll1 and Jag1, are differently restricted in their range of action to control neurogenesis in the mammalian spinal cord. PLoS ONE 2010, 5, e15515. [Google Scholar] [CrossRef]
- Cantone, M.; Küspert, M.; Reiprich, S.; Lai, X.; Eberhardt, M.; Göttle, P.; Beyer, F.; Azim, K.; Küry, P.; Wegner, M.; et al. A gene regulatory architecture that controls region-independent dynamics of oligodendrocyte differentiation. Glia 2019, 67, 825–843. [Google Scholar] [CrossRef] [PubMed]
- Hikichi, T.; Matoba, R.; Ikeda, T.; Watanabe, A.; Yamamoto, T.; Yoshitake, S.; Tamura-Nakano, M.; Kimura, T.; Kamon, M.; Shimura, M.; et al. Transcription factors interfering with dedifferentiation induce cell type-specific transcriptional profiles. Proc. Natl. Acad. Sci. USA 2013, 110, 6412–6417. [Google Scholar] [CrossRef] [PubMed]
- Real, C.; Glavieux-Pardanaud, C.; Le Douarin, N.M.; Dupin, E. Clonally cultured differentiated pigment cells can dedifferentiate and generate multipotent progenitors with self-renewing potential. Dev. Biol. 2006, 300, 656–669. [Google Scholar] [CrossRef]
- Ionescu, C.; Lopes, A.; Copot, D.; Machado, J.A.T.; Bates, J.H.T. The role of fractional calculus in modeling biological phenomena: A review. Commun. Nonlinear Sci. Numer. Simul. 2017, 51, 141–159. [Google Scholar] [CrossRef]
- Kopelman, R. Fractal reaction kinetics. Science 1988, 241, 1620–1626. [Google Scholar] [CrossRef] [PubMed]
- Schnell, S.; Turner, T.E. Reaction kinetics in intracellular environments with macromolecular crowding: Simulations and rate laws. Prog. Biophys. Mol. Biol. 2004, 85, 235–260. [Google Scholar] [CrossRef]
- Hiroi, N.; Lu, J.; Iba, K.; Tabira, A.; Yamashita, S.; Okada, Y.; Flamm, C.; Oka, K.; Köhler, G.; Funahashi, A. Physiological environment induces quick response-slow exhaustion reactions. Front. Physiol. 2011, 2, 50. [Google Scholar] [CrossRef]
- Hiroi, N.; Klann, M.; Iba, K.; Heras Ciechomski, P.D.; Yamashita, S.; Tabira, A.; Okuhara, T.; Kubojima, T.; Okada, Y.; Oka, K.; et al. From microscopy data to in silico environments for in vivo-oriented simulations. EURASIP J. Bioinform. Syst. Biol. 2012, 2012, 7. [Google Scholar] [CrossRef]
- van Groningen, T.; Akogul, N.; Westerhout, E.M.; Chan, A.; Hasselt, N.E.; Zwijnenburg, D.A.; Broekmans, M.; Stroeken, P.; Haneveld, F.; Hooijer, G.K.J.; et al. A NOTCH feed-forward loop drives reprogramming from adrenergic to mesenchymal state in neuroblastoma. Nat. Commun. 2019, 10, 1530. [Google Scholar] [CrossRef]








| Equation No. | Equation |
|---|---|
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 |
| Parameter (unit) | 2.5 h period | Minimum | Maximum | Parameter Description |
|---|---|---|---|---|
| (h−1) | 0.99 | 0.69 | 2.45 | Degradation rate constant of reaction 3 for substrate s3 |
| (h−1) | 1.29 | 1.03 | 2.28 | Degradation rate constant of reaction 4 for substrate s2 |
| (µM·h−1) | 0.4074 | 0.37 | 0.65 | Dimerization rate constant of reaction 5 |
| (h−1) | 2.3 | 0.61 | 2.49 | Dissociation rate constant of reaction 6 |
| (h−1) | 31.2 | <0.001 | >100 | Degradation rate constant of reaction 8 for substrate s5 |
| (h−1) | 141.6 | 108 | 146 | Turnover number of reaction 11 |
| (h−1) | 132.6 | 130 | 174 | Turnover number of reaction 12 |
| (h−1) | 209.4 | 162 | 216 | Turnover number of reaction 13 |
| (h−1) | 132 | 128 | 173 | Turnover number of reaction 14 |
| (h−1) | 132 | 128 | 173 | Turnover number of reaction 15 |
| (h−1) | 174 | 171 | 238 | Turnover number of reaction 16 |
| (h−1) | 132 | 95 | 183 | Turnover number of reaction 19 |
| (µM·h−1) | 361.2 | 183 | 526 | Maximal transcription rate of reaction 2 |
| (µM·h−1) | 25.74 | 13.1 | 31.6 | Mass action constant of reaction 1 |
| (µM·h−1) | 10.86 | <0.001 | >100 | Maximal transcription rate of reaction 7 |
| (µM) | 50.0 | 48.9 | 65.5 | Michaelis–Menten constant of reaction 11 for substrate s15 |
| (µM) | 1.62 | 1.26 | 1.66 | Michaelis–Menten constant of reaction 12 for substrate s22 |
| (µM) | 0.21 | 0.21 | 0.29 | Michaelis–Menten constant of reaction 13 for substrate s16 |
| (µM) | 28.4 | 21.6 | 28.9 | Michaelis–Menten constant of reaction 14 for substrate s17 |
| (µM) | 12.7 | 9.4 | 13.1 | Michaelis–Menten constant of reaction 15 for substrate s21 |
| (µM) | 0.45 | 0.22 | 0.49 | Michaelis–Menten constant of reaction 16 for substrate s19 |
| (µM) | 1.2 | 1.18 | 1.61 | Michaelis–Menten constant of reaction 17 for substrate s20 |
| (µM) | 0.91 | 0.89 | 1.52 | Michaelis–Menten constant of reaction 18 for substrate s18 |
| (µM) | 9.0 | 6.2 | 12.8 | Michaelis–Menten constant of reaction 19 for substrate s24 |
| (µM) | 0.62 | 0.38 | 0.88 | Michaelis–Menten constant of reaction 20 for substrate s23 |
| (µM) | 0.04 | 0.029 | 0.048 | Half-maximal inhibitory concentration of substrate s24 in reaction 2 |
| (µM) | 0.0023 | <0.001 | 0.0025 | Half-maximal inhibitory concentration of substrate s9 in reaction 2 |
| (µM) | 0.116 | <0.001 | >100 | Half-maximal inhibitory concentration of substrate s3 in reaction 7 |
| (µM) | 2.5 | 2.18 | 3.17 | Half-maximal effective concentration of substrate s24 in reaction 2 |
| (µM·h−1) | 88.2 | 67.9 | 90.9 | Maximal reaction rate constant of reaction 17 |
| (µM·h−1) | 14.52 | 10.7 | 14.9 | Maximal reaction rate constant of reaction 18 |
| (µM·h−1) | 24.0 | 17.9 | 33.3 | Maximal reaction rate constant of reaction 20 |
| 2 | 2 | 2 | Inhibition coefficient of reaction 2 for substrate s24 | |
| 5 | 5 | >10 | Inhibition coefficient of reaction 2 for substrate s9 | |
| 2 | 1 | >10 | Inhibition coefficient of reaction 7 for substrate s3 | |
| 3 | 2 | 3 | Hill coefficient of reaction 2 for substrate s11 |
| Equation No. | Equation |
|---|---|
| 1’ | |
| 3’ |
| Equation No. | Equation |
|---|---|
| 2’ | |
| 2″ | |
| 5’ | |
| 6’ | |
| 7’ | |
| 8’ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iwasaki, T.; Takiguchi, R.; Hiraiwa, T.; Yamada, T.G.; Yamazaki, K.; Hiroi, N.F.; Funahashi, A. Neural Differentiation Dynamics Controlled by Multiple Feedback Loops in a Comprehensive Molecular Interaction Network. Processes 2020, 8, 166. https://doi.org/10.3390/pr8020166
Iwasaki T, Takiguchi R, Hiraiwa T, Yamada TG, Yamazaki K, Hiroi NF, Funahashi A. Neural Differentiation Dynamics Controlled by Multiple Feedback Loops in a Comprehensive Molecular Interaction Network. Processes. 2020; 8(2):166. https://doi.org/10.3390/pr8020166
Chicago/Turabian StyleIwasaki, Tsuyoshi, Ryo Takiguchi, Takumi Hiraiwa, Takahiro G. Yamada, Kazuto Yamazaki, Noriko F. Hiroi, and Akira Funahashi. 2020. "Neural Differentiation Dynamics Controlled by Multiple Feedback Loops in a Comprehensive Molecular Interaction Network" Processes 8, no. 2: 166. https://doi.org/10.3390/pr8020166
APA StyleIwasaki, T., Takiguchi, R., Hiraiwa, T., Yamada, T. G., Yamazaki, K., Hiroi, N. F., & Funahashi, A. (2020). Neural Differentiation Dynamics Controlled by Multiple Feedback Loops in a Comprehensive Molecular Interaction Network. Processes, 8(2), 166. https://doi.org/10.3390/pr8020166