Mathematical Modeling of the Production of Elastomers by Emulsion Polymerization in Trains of Continuous Reactors

Abstract
1. Introduction
- -
- The effect of impurities in the process was included.
- -
- The PSD was modeled by using a population balance approach for the continuous distribution N(v,t)dv, which represents the number of particles having a volume between v and v + dv at time t, resulting in a partial differential equation (PDE). This equation was approximately solved by discretizing the v domain into small subdomains ∆v and converting the PDE into a set of ODEs.
- -
- -
- The possibility of considering side streams fed to reactors along the CSTR train was included.
2. Mathematical Model
2.1. Monomer Mass Balances
2.2. Rate of Polymerization
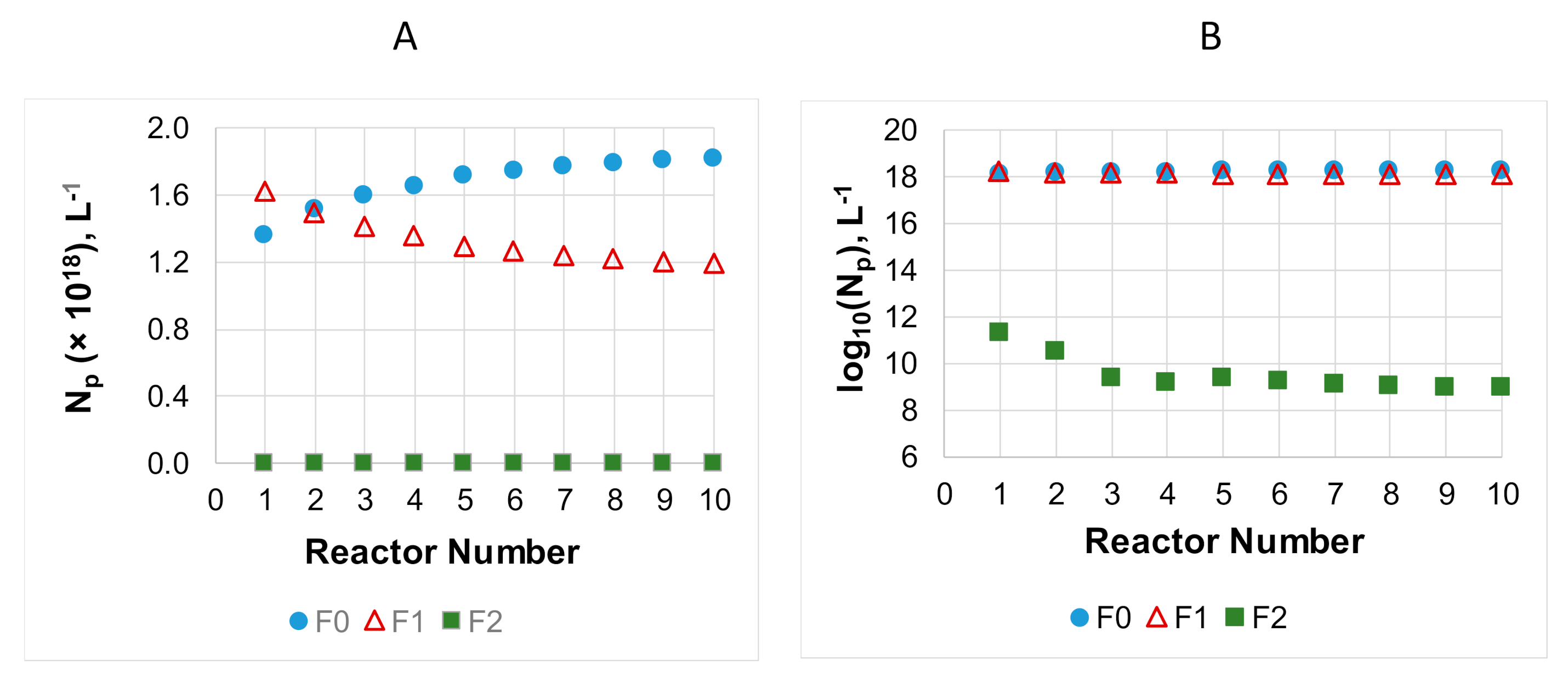
2.3. Population Balance Equations for Particles and Calculation of and
2.4. Radical Entry and Exit Coefficients
2.5. Mass Balances of Species
2.6. Monomer Partitioning
2.7. Total Mass Balance
2.8. Molecular Weight Distribution
2.9. Number of Branches
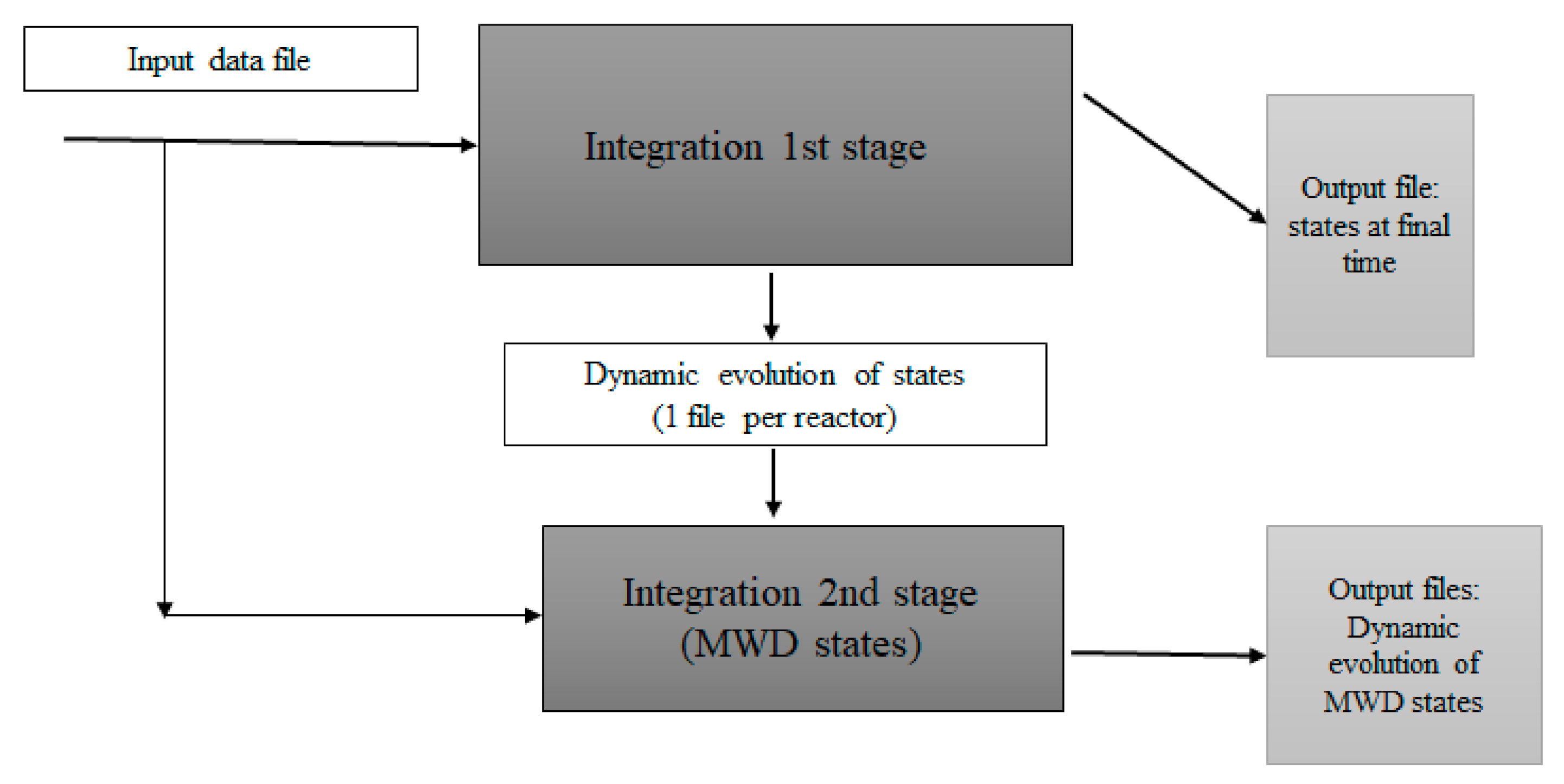
2.10. Strategy of Solution
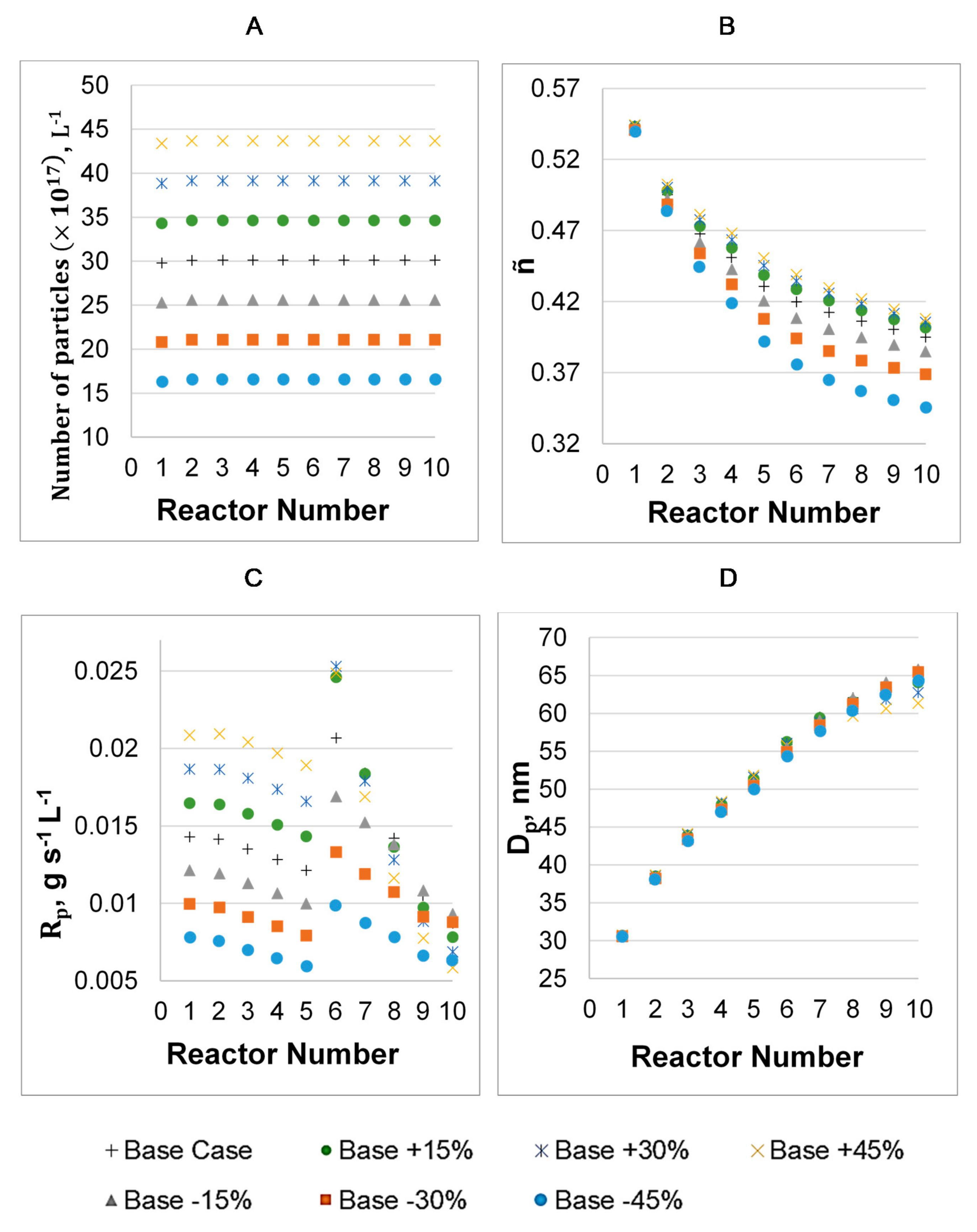
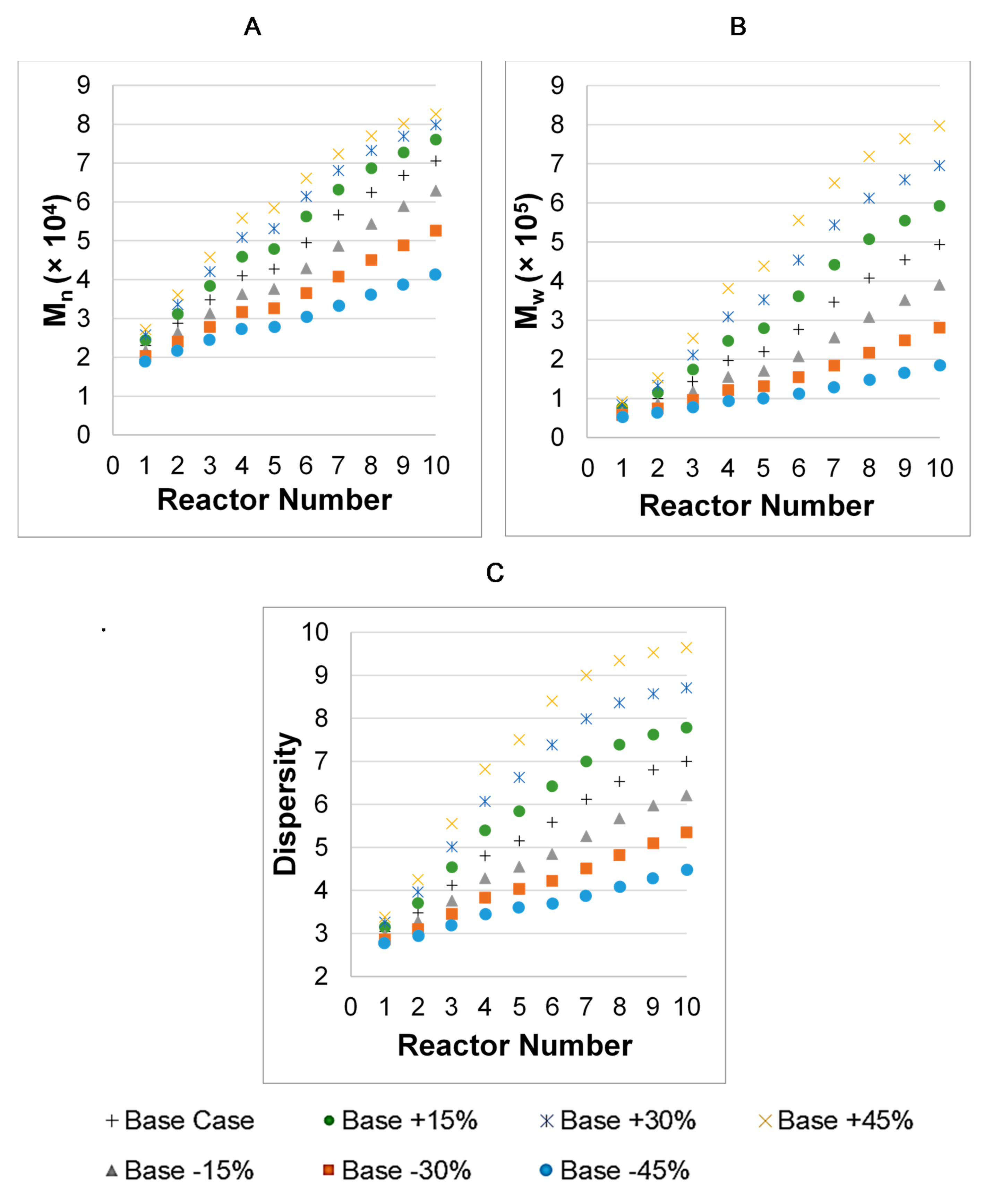
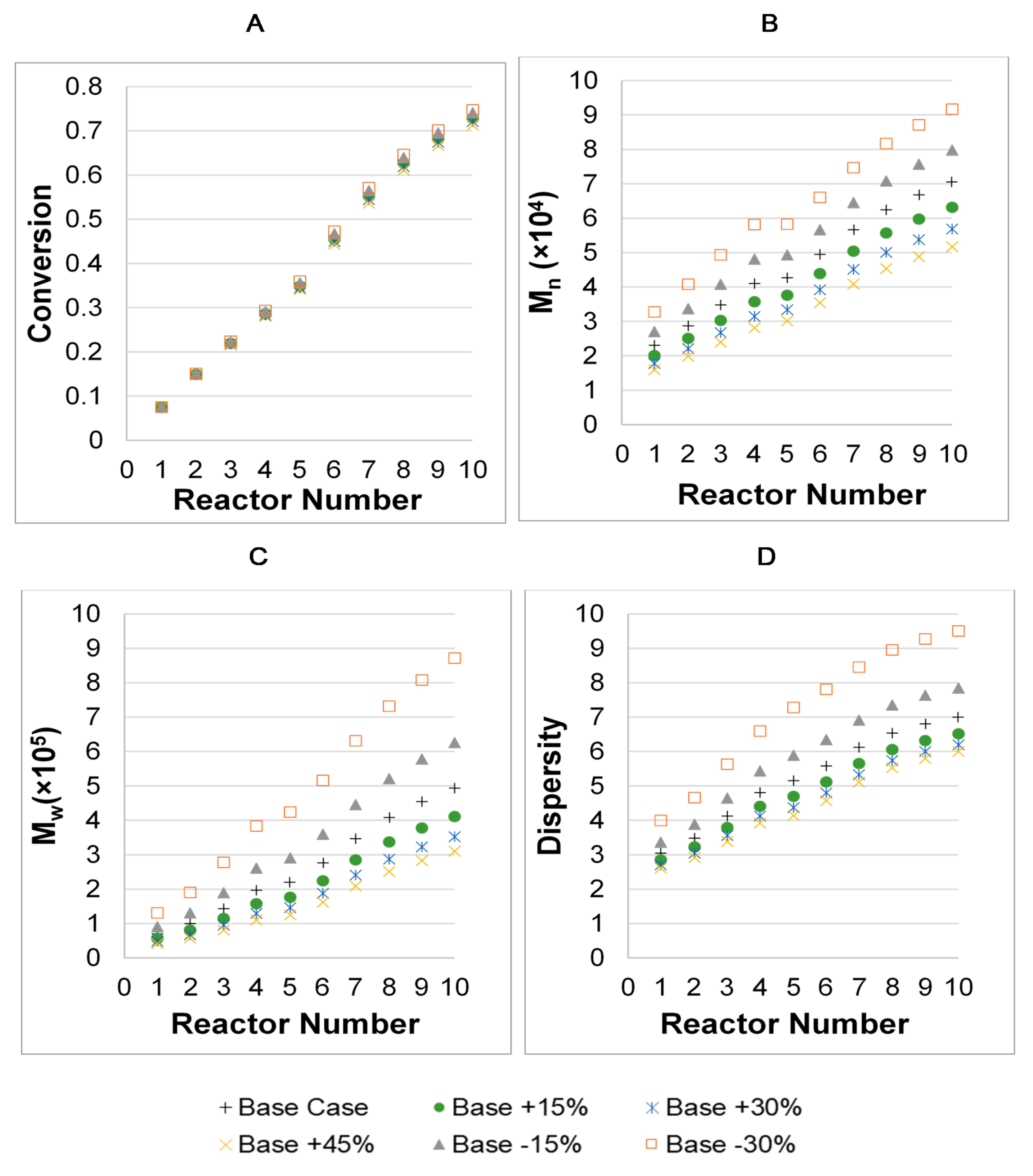
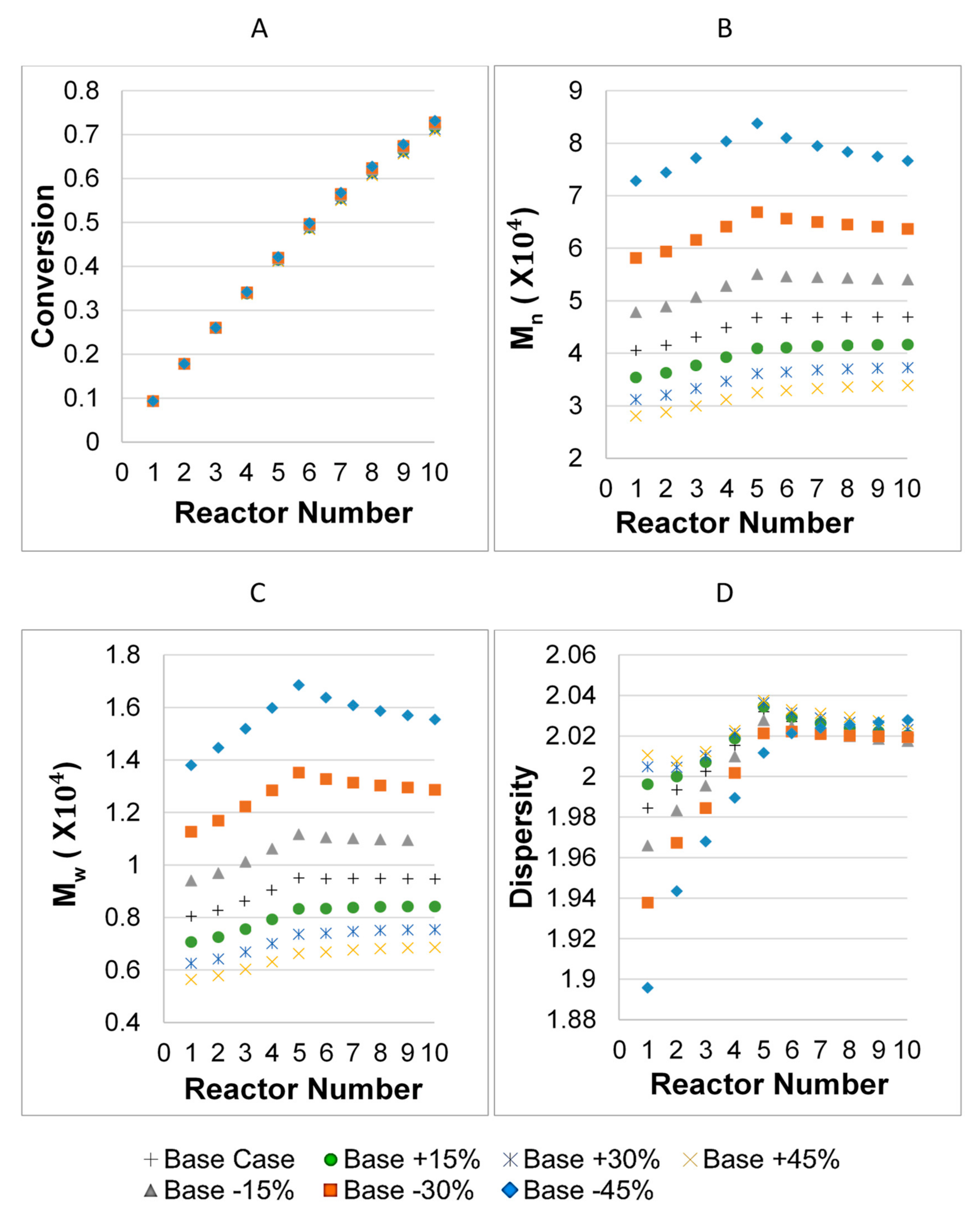
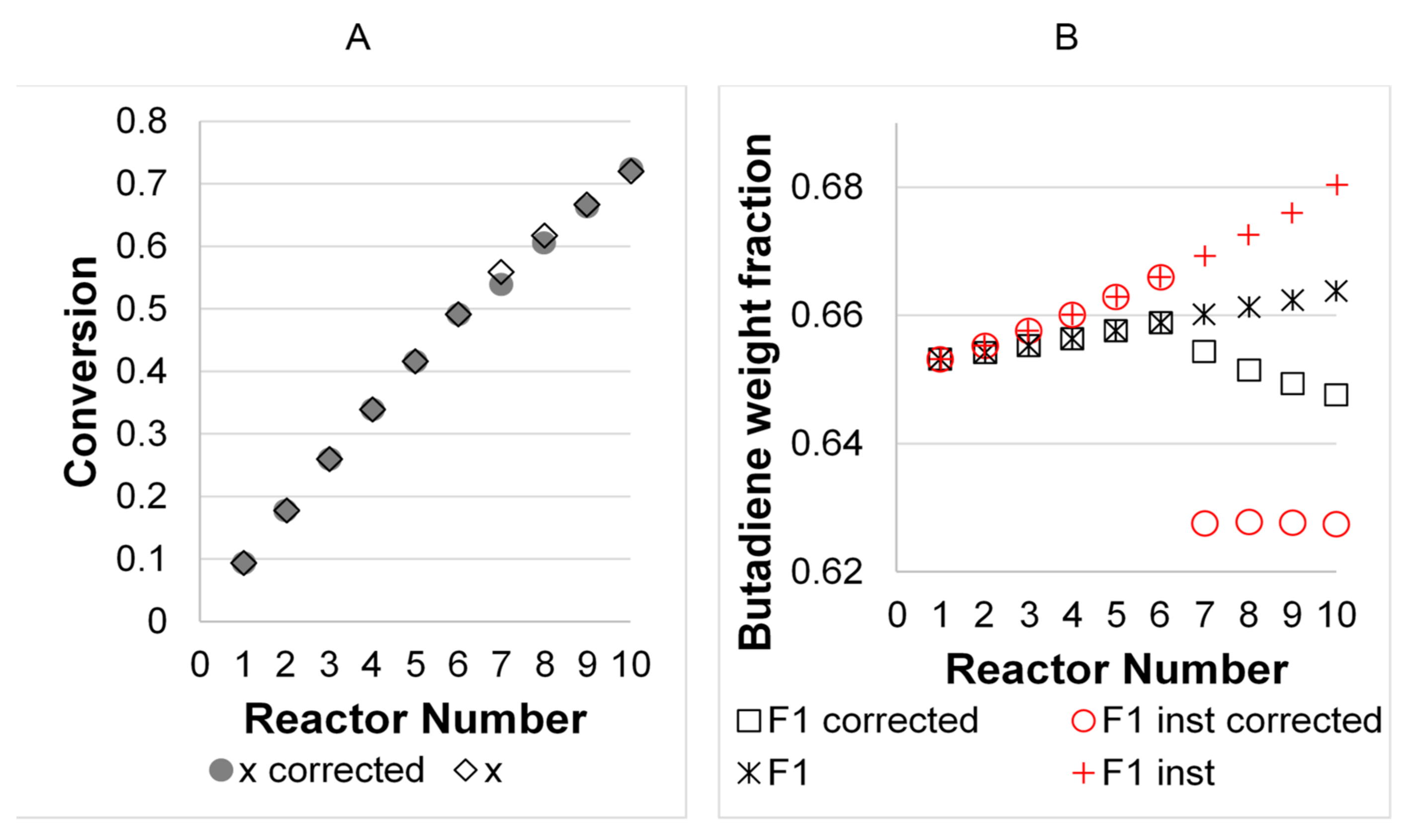
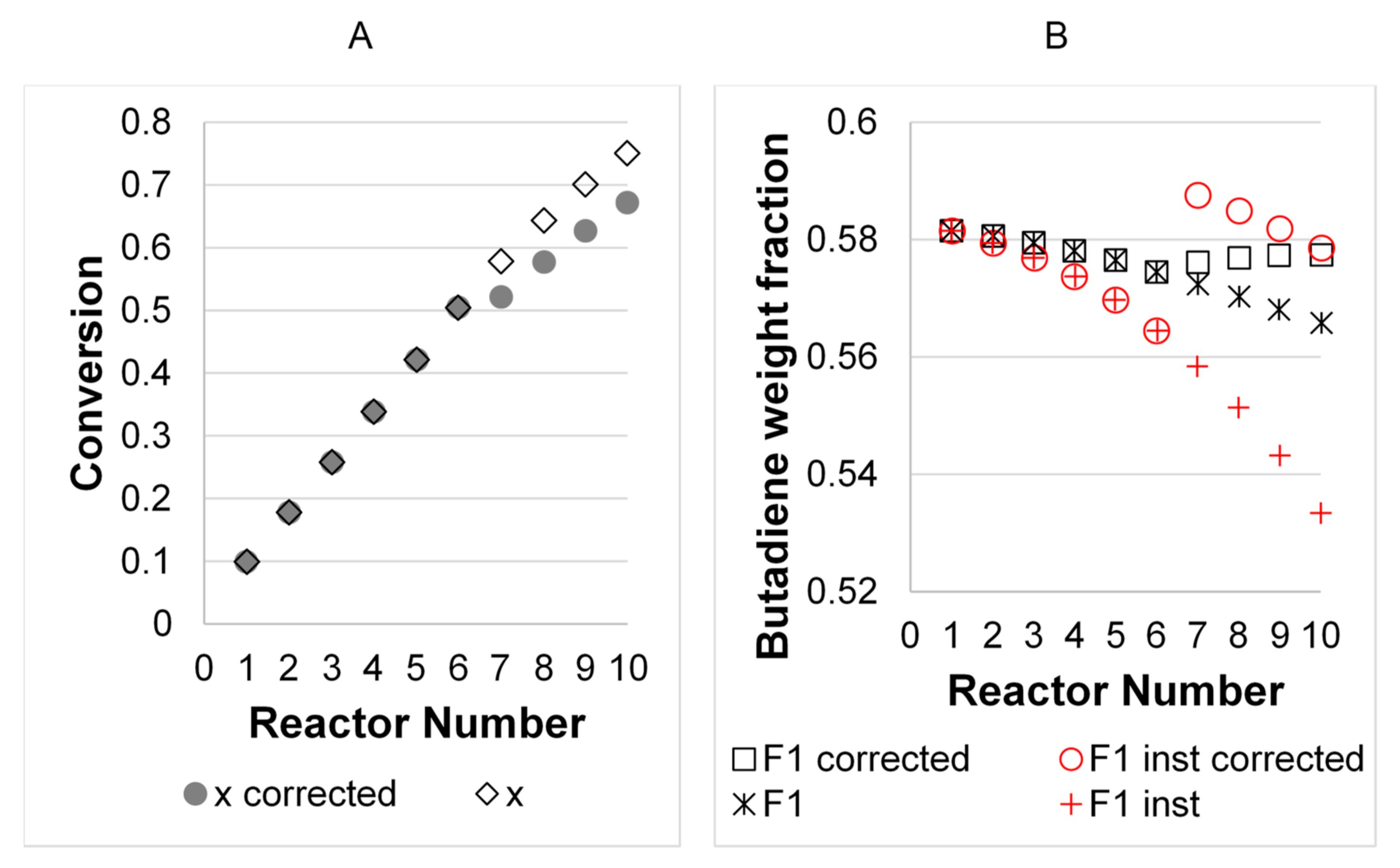
3. Results
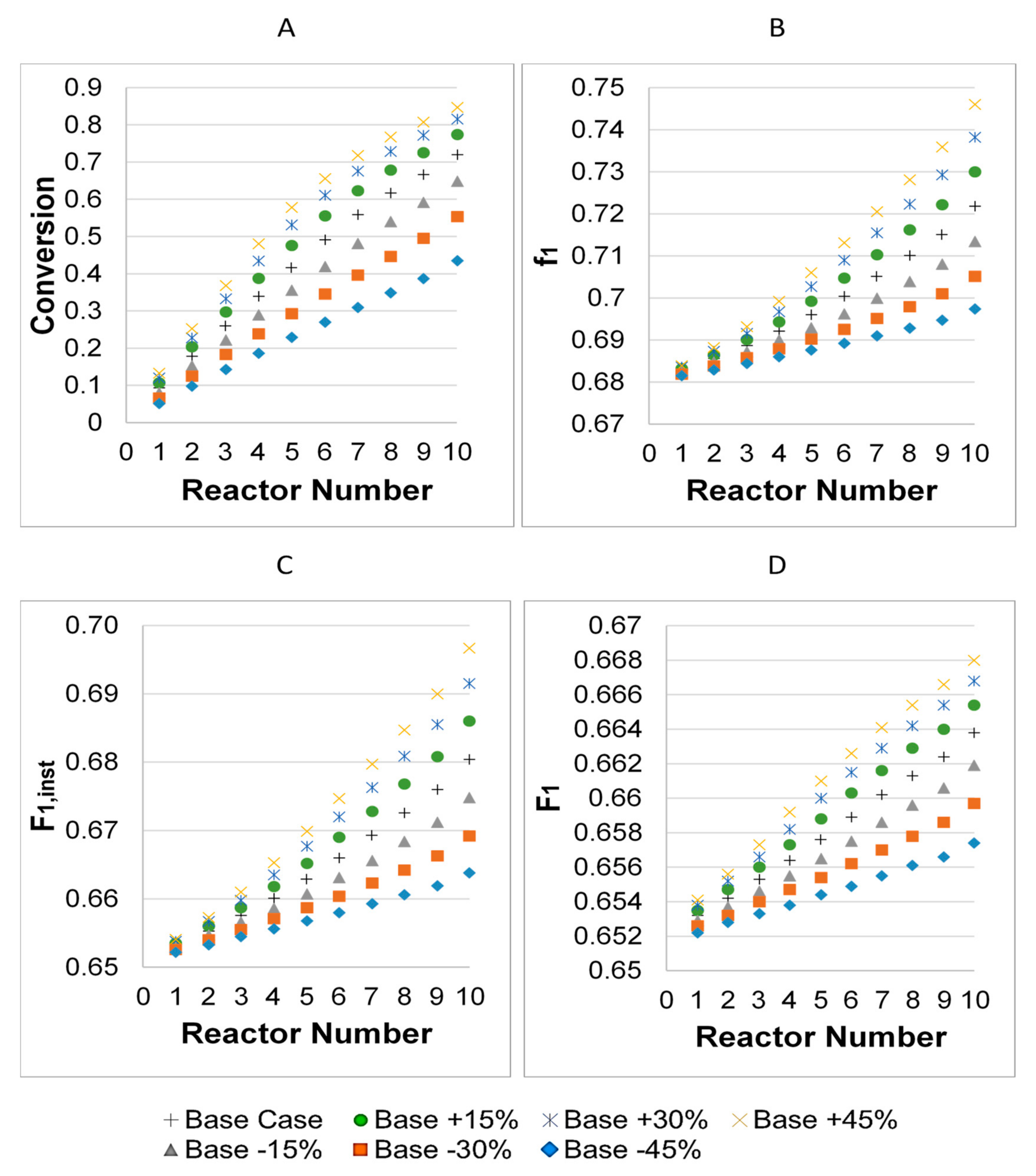
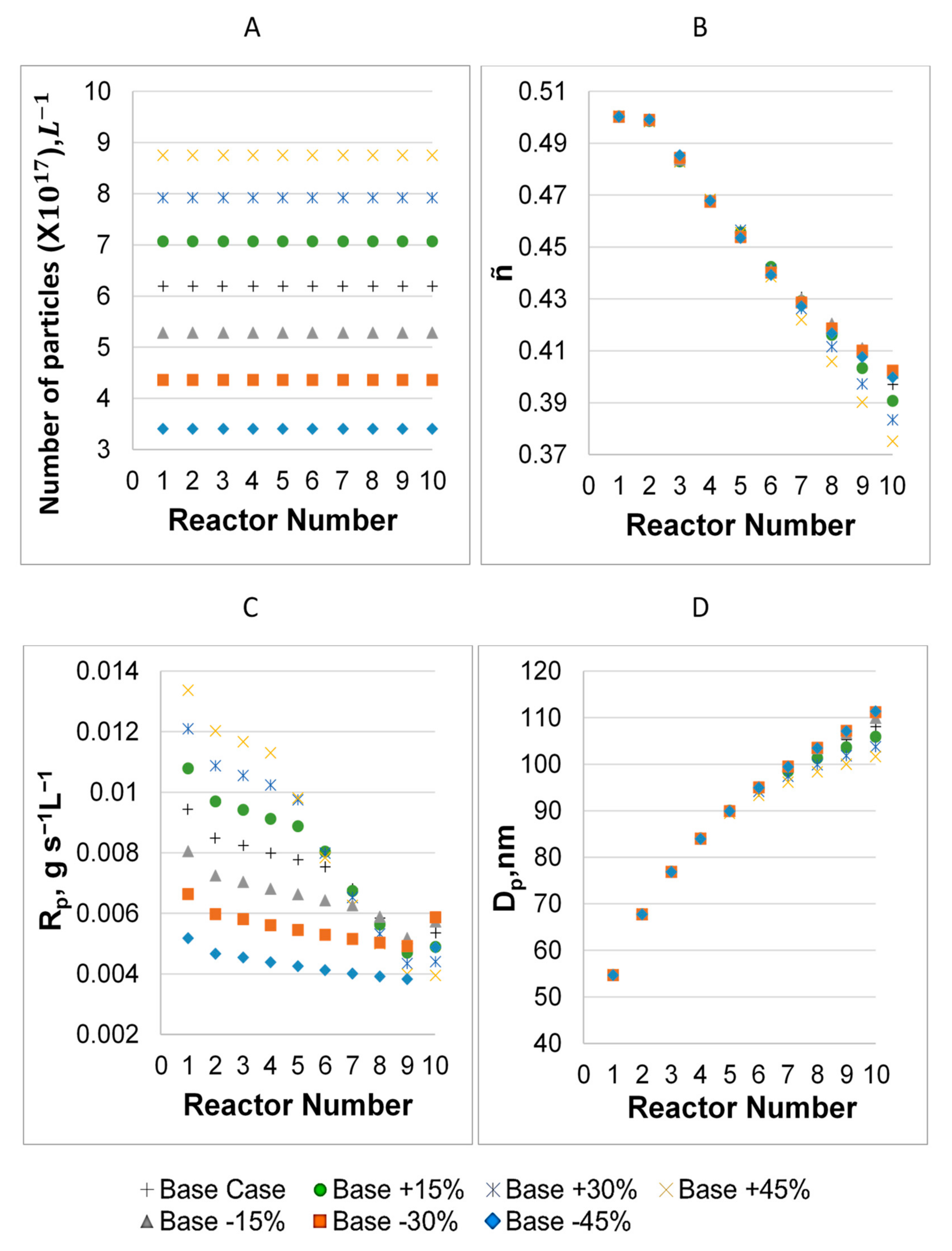
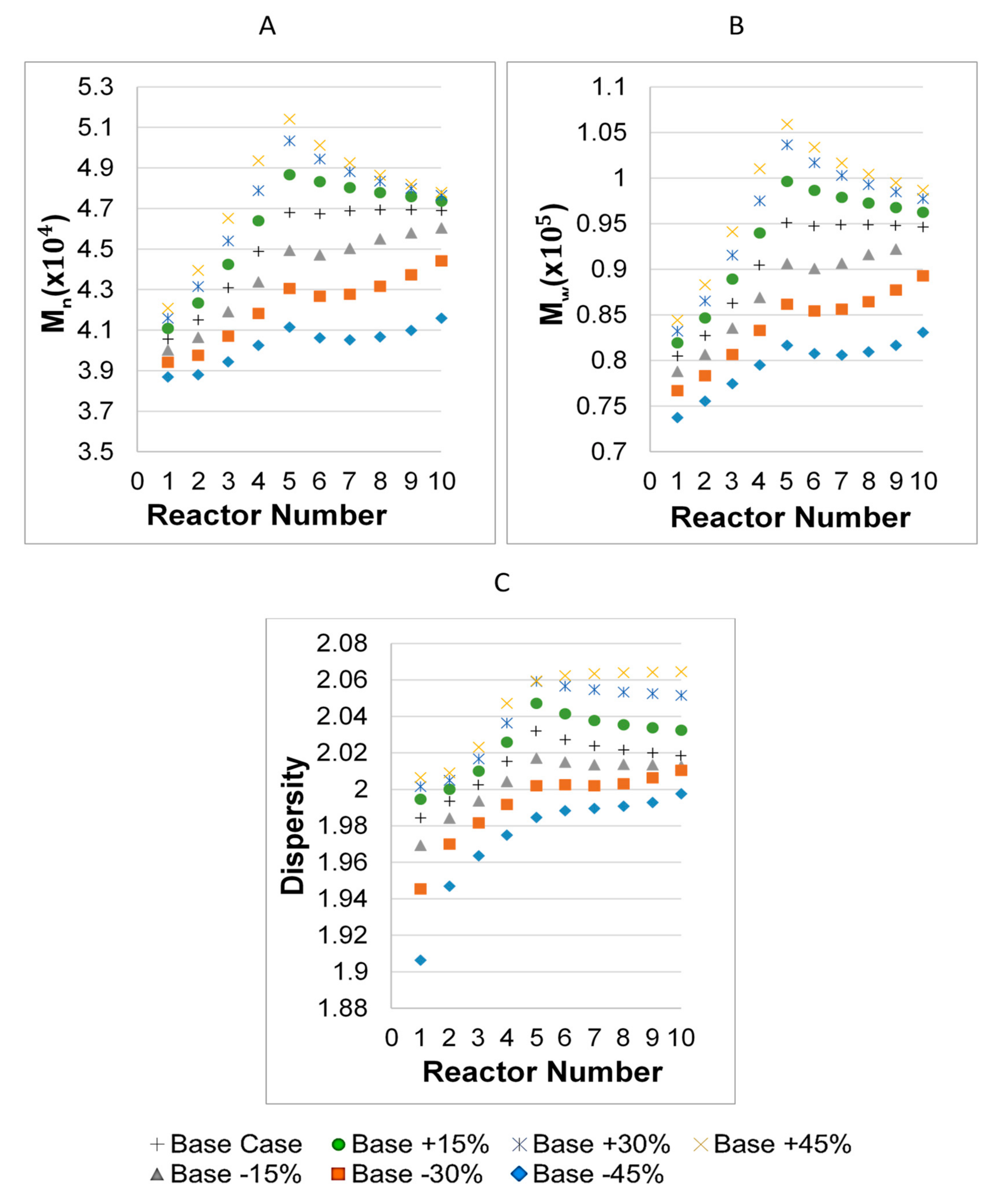
3.1. Parameter Values
- -
- Kinetic parameters (): These were based on independent kinetic data (e.g., the kinetic constants of the redox initiation system, as discussed above) or assumed similar to values published for chemically similar systems; therefore, it is expected that little error could be introduced through these parameters.
- -
- Surfactant-related parameters (: These were also estimated by assuming similar values to those of chemically similar systems, which vary within relatively narrow ranges, so no significant errors are expected associated with these estimations.
- -
- Desorption-related coefficients (: These parameters enter into the expressions of the desorption coefficient (see Supplementary Material Equations (S1h)–(S1i)) and do not have strong influence on the final responses, according to our experience.
- -
- Partition coefficients (: These parameters were inferred from known values of the solubilities of the respective components in water and organic media, so it is expected that the estimations used were not far from the real values. Indirect evidence of the adequacy of these estimations came from the behavior observed in the model for responses affected by these parameters, such as the copolymer composition (NBR case) and the molecular weights, which were close to experimental values.
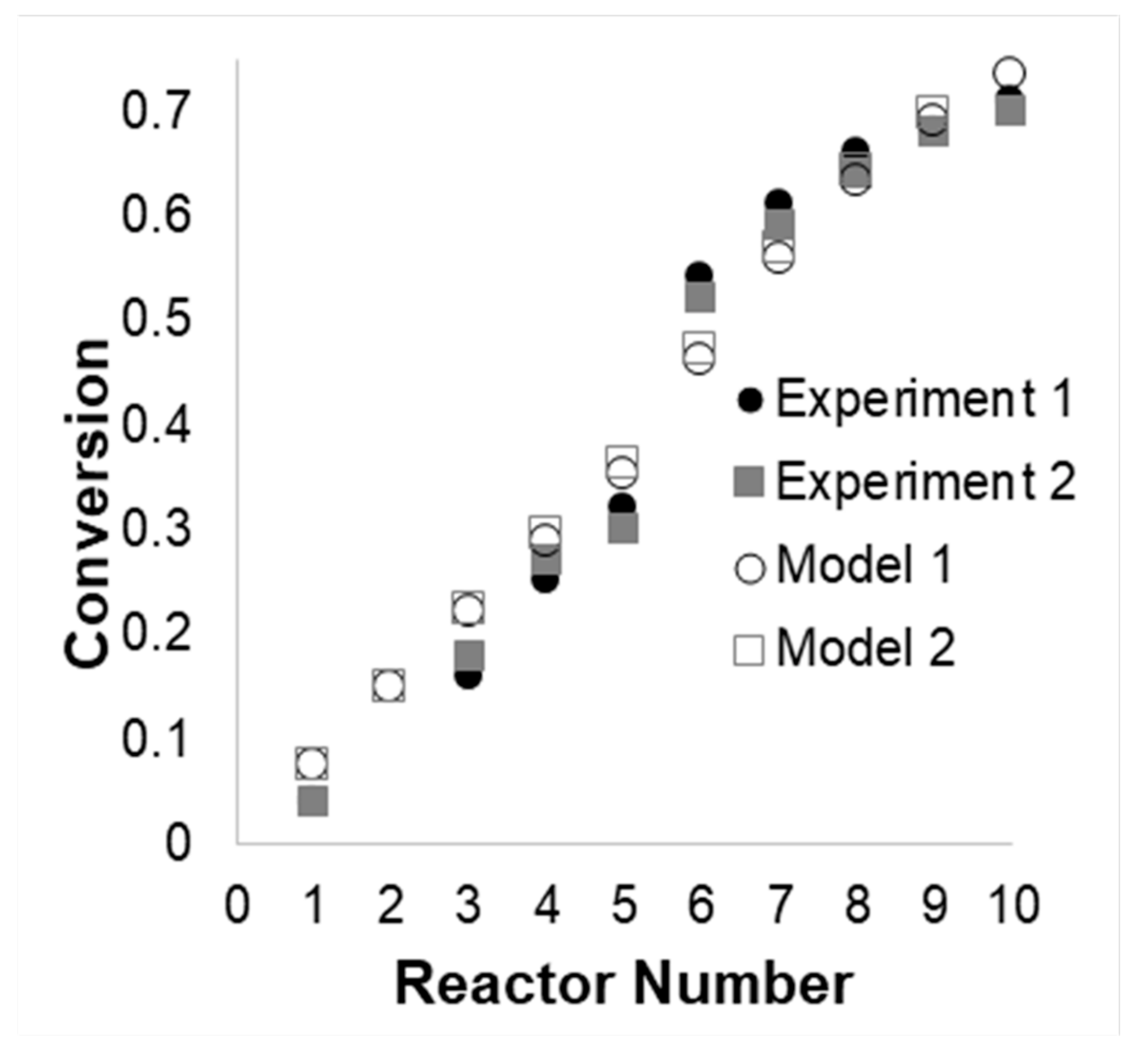
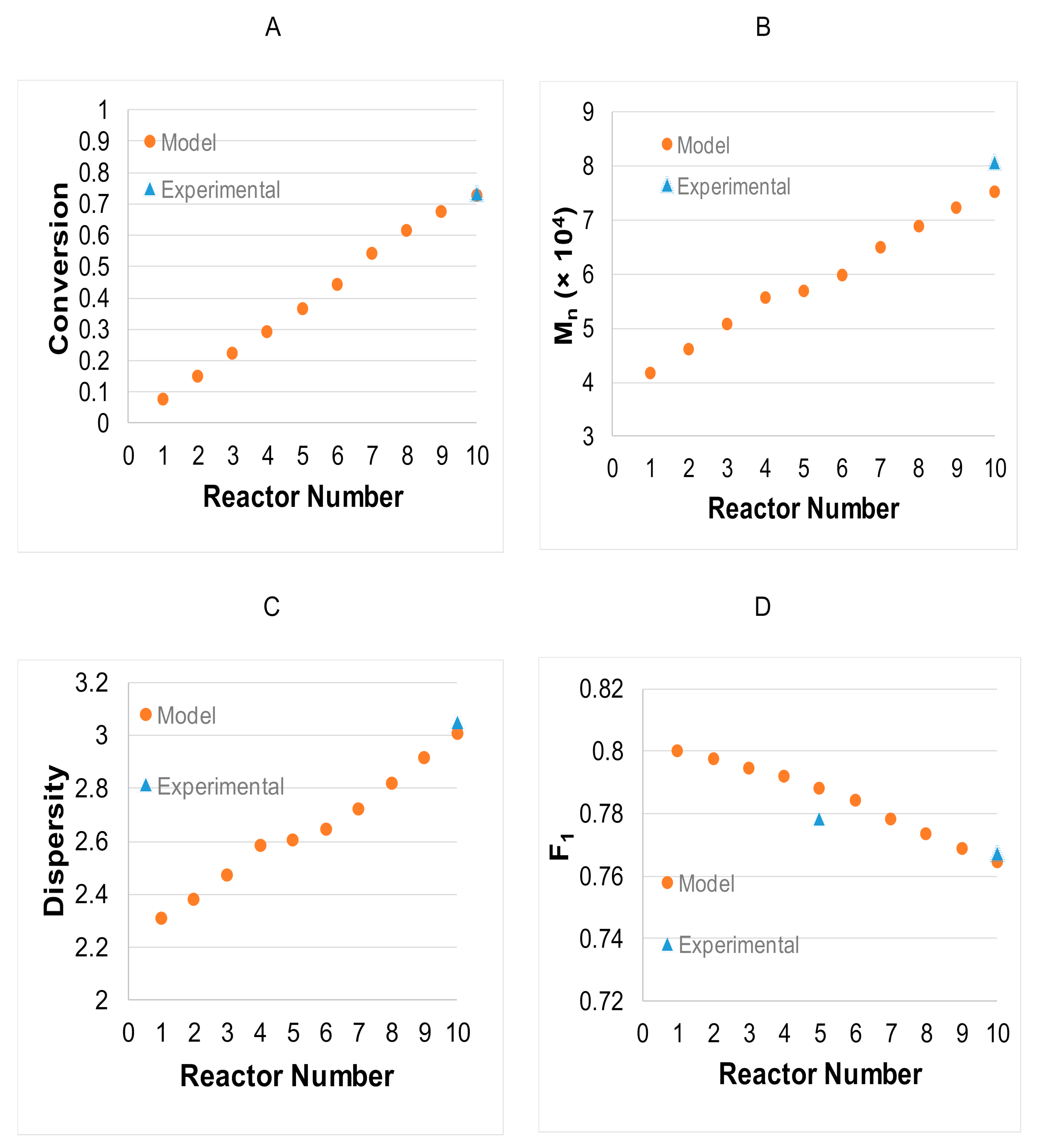
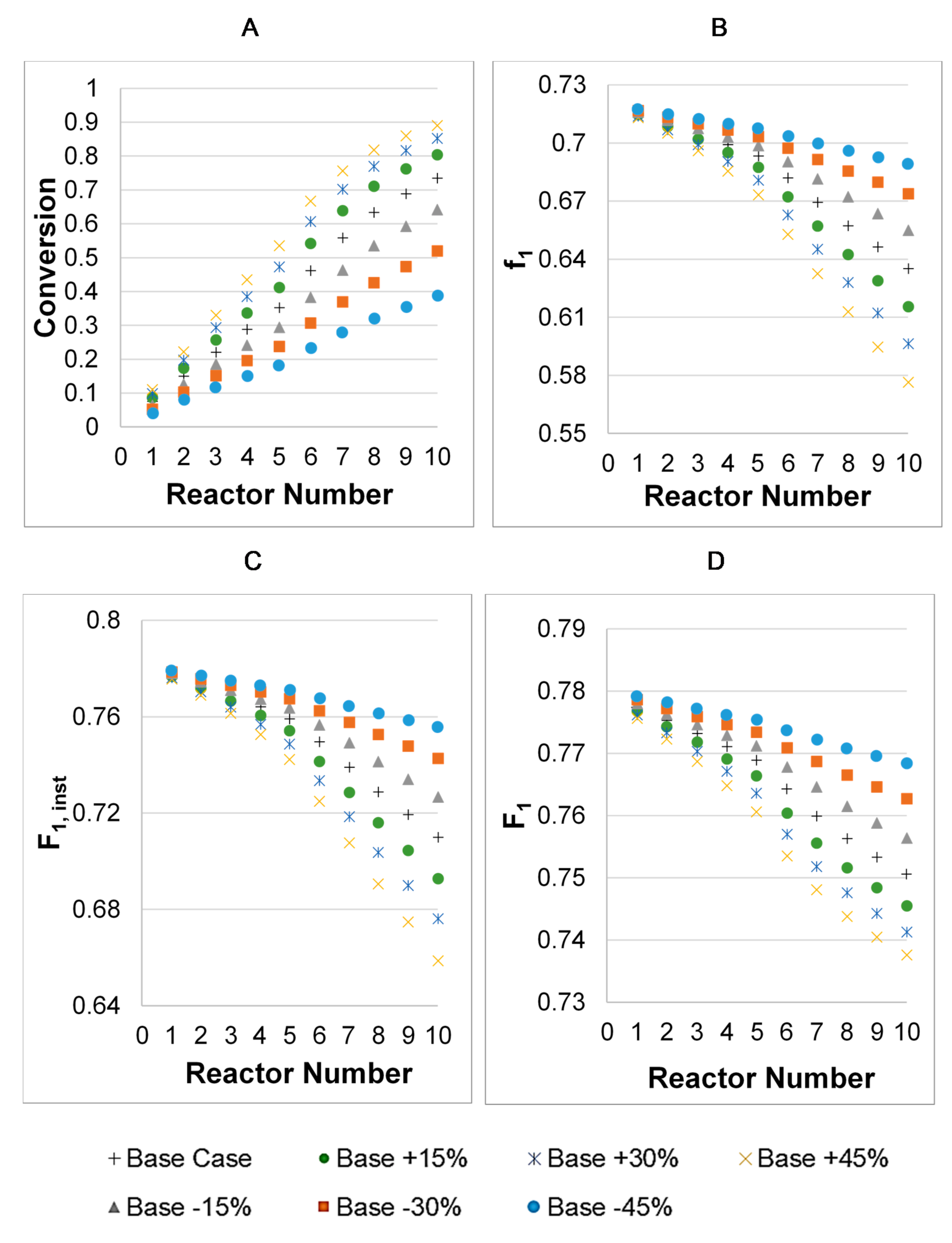
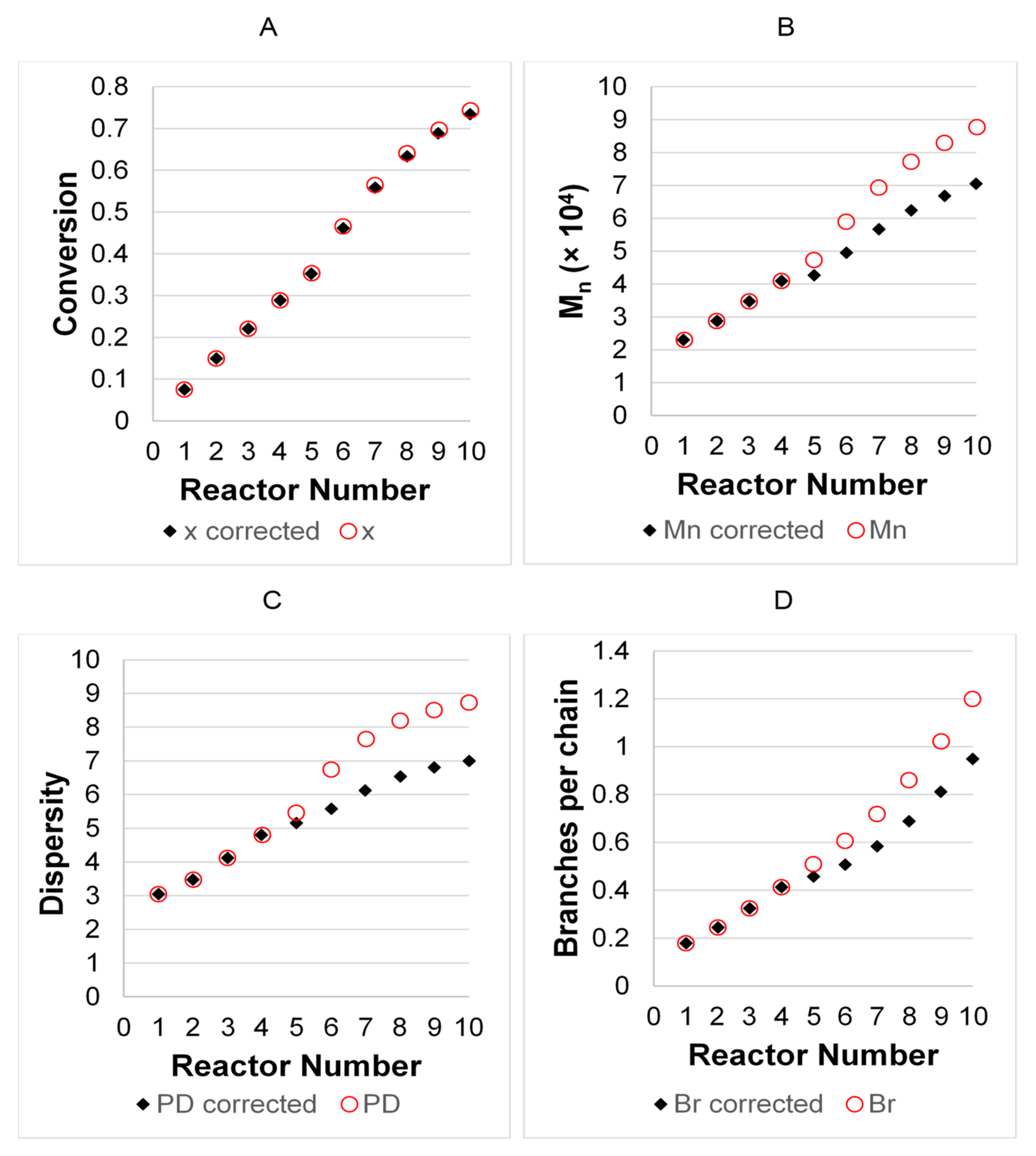
3.2. Model Validation
4. Conclusions
- -
- The radical compartmentalization was considered in more detail, both in the particle kinetics description and in the calculation of the moments of the MWD, and for the first time, a 0-1-2 model was used to describe these two important aspects in these processes. It was found that at least a 0-1 model should be used for an accurate description of the compartmentalization effects, especially for the first reactors in the train. A similar conclusion was recently put forward by Marien et al. [52] using stochastic simulation, although for a general miniemulsion system.
- -
- The monomer partitioning model was as simple as possible, and its parameters could be estimated a priori based on published physicochemical data. As mentioned by Madhuranthakam and Penlidis, [30] the use of more sophisticated thermodynamic models for monomer partitioning seems to be unnecessary for this kind of system.
- -
- A single modeling framework was presented in a unified form for the SBR and NBR systems. Though previous models have been applied with adaptations to both systems in some series of papers, it is believed that by presenting them together, it is easier to appreciate the similarities and differences in both systems.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Production Capacity of Styrene-Butadiene-Rubber Worldwide in 2018 and 2023. Available online: https://www.statista.com/statistics/1063647/styrene-butadiene-rubber-production-capacity-globally/ (accessed on 8 January 2020).
- The Global Nitrile Butadiene Rubber (NBR) Market to 2023: Global Production of NBR is Likely to Reach ~1,562 Thousand Tons. Available online: https://www.businesswire.com/news/home/20181126005386/en/Global-Nitrile-Butadiene-Rubber-NBR-Market-2023 (accessed on 8 January 2020).
- Kanetakis, J.; Wong, F.Y.C.; Hamielec, A.E.; MacGregor, J.F. Steady State Modeling of a Latex Reactor Train for the Production of Styrene-Butadiene Rubber. Chem. Eng. Commun. 1985, 35, 123–140. [Google Scholar] [CrossRef]
- Broadhead, T.O.; Hamielec, A.E. Dynamic modelling of the batch, semi-batch and continuous production of styrene/butadiene copolymers by emulsion polymerization. Die Makromol. Chem. 1985, 10, 105–128. [Google Scholar] [CrossRef]
- Penlidis, A.; MacGregor, J.F.; Hamielec, A.E. Dynamic modelling of emulsion polymerization reactors. AIChE J. 1985, 31, 881–889. [Google Scholar] [CrossRef]
- Smith, W.V.; Ewart, R.H. Kinetics of Emulsion Polymerization. J. Chem. Phys. 1948, 16, 592–599. [Google Scholar] [CrossRef]
- Storti, G.; Carrá, S.; Morbidelli, M.; Vita, G. Kinetics of multimonomer emulsion polymerization. The pseudo-homopolymerization approach. J. Appl. Polym. Sci. 1989, 37, 2443–2467. [Google Scholar] [CrossRef]
- Tobita, H.; Hamielec, A.E. Kinetics of free-radical copolymerization: The pseudo-kinetic rate constant method. Polymer 1991, 32, 2641–2647. [Google Scholar] [CrossRef]
- Saldívar, E.; Dafniotis, P.; Ray, W.H. Mathematical Modeling of Emulsion Copolymerization Reactors. I. Model Formulation and Application to Reactors Operating with Micellar Nucleation. J. Macromol. Sci. Part C Polym. Rev. 1998, 38, 207–325. [Google Scholar] [CrossRef]
- Ugelstad, J.; Mork, P.C.; Aasen, J.E. Kinetics of emulsion polymerization. J. Polym. Sci. 1967, A5, 2281–2288. [Google Scholar] [CrossRef]
- Yabuki, Y.; MacGregor, J.F. Product Quality Control in Semibatch Reactors Using Midcourse Correction Policies. Ind. Eng. Chem. Res. 1997, 36, 1268–1275. [Google Scholar] [CrossRef]
- Gugliotta, L.M.; Brandolini, M.C.; Vega, J.R.; Iturralde, E.O.; Azum, J.L.; Meira, G.R. Dynamic Model of a Continuous Emulsion Copolymerization of Styrene and Butadiene. Polym. React. Eng. 1995, 3, 201–233. [Google Scholar]
- Flory, P.J. Principles of Polymer Chemistry; Cornell University Press: Ithaca, NY, USA, 1953. [Google Scholar]
- Morton, M.; Kaizerman, S.; Altier, M.W. Swelling of latex particles. J. Colloid Sci. 1954, 9, 300–312. [Google Scholar] [CrossRef]
- Vega, J.R.; Gugliotta, L.M.; Brandolini, M.C.; Meira, G.R. Steady-State Optimization in a Continuous Emulsion Copolymerization of Styrene and Butadiene. Lat. Am. Appl. Res. 1995, 25, 207–214. [Google Scholar]
- Vega, J.R.; Gugliotta, L.M.; Meira, G.R. Continuous Emulsion Polymerization of Styrene and Butadiene. Reduction of Off-Spec Product between Steady-State. Lat. Am. Appl. Res. 1995, 25, 77–82. [Google Scholar]
- Minari, R.J.; Vega, J.R.; Gugliotta, L.M.; Meira, G.R. Continuous Emulsion Styrene-Butadiene Rubber (SBR) Process: Computer Simulation Study for Increasing Production and for Reducing Transients between Steady States. Ind. Eng. Chem. Res. 2006, 45, 245–257. [Google Scholar] [CrossRef]
- Godoy, J.L.; Minari, R.J.; Vega, J.R.; Marchetii, J.L. Multivariate statistical monitoring of an industrial SBR process. Soft-sensor for production and rubber quality. Chemom. Intell. Lab. Syst. 2011, 107, 258–268. [Google Scholar] [CrossRef]
- Zubov, A.; Pokorny, J.; Kosek, J. Styrene–butadiene rubber (SBR) production by emulsion polymerization: Dynamic modeling and intensification of the process. Chem. Eng. J. 2012, 207, 414–420. [Google Scholar] [CrossRef]
- Mustafina, S.; Miftakhov, E.; Mikhailova, T. Mathematical Study of Copolymer Composition and Compositional Heterogeneity during the Synthesis of Emulsion-Type Butadiene-Styrene Rubber. Int. J. Chem. Sci. 2014, 12, 1135–1144. [Google Scholar]
- Dubé, M.A.; Penlidis, A.; Mutha, R.K.; Cluett, W.R. Mathematical Modeling of Emulsion Copolymerization of Acrylonitrile/Butadiene. Ind. Eng. Chem. Res. 1996, 46, 4434–4448. [Google Scholar] [CrossRef]
- Vega, J.R.; Gugliotta, L.M.; Bielsa, R.O.; Brandolini, M.C.; Meira, G.R. Emulsion Copolymerization of Acrylonitrile and Butadiene. Mathematical Model of an Industrial Reactor. Ind. Eng. Chem. Res. 1997, 36, 1238–1246. [Google Scholar] [CrossRef]
- Rodríguez, V.I.; Estenoz, D.A.; Gugliotta, L.M.; Meira, G.R. Emulsion Copolymerization of Acrylonitrile and Butadiene. Calculation of the Detailed Macromolecular Structure. Int. J. Polym. Mater. 2002, 51, 511–529. [Google Scholar] [CrossRef]
- Vega, J.R.; Gugliotta, L.M.; Meira, G.R. Emulsion Copolymerization of Acrylonitrile and Butadiene. Semibatch Strategies for Controlling Molecular Structure on the Basis of Calorimetric Measurements. Polym. React. Eng. 2002, 10, 59–82. [Google Scholar] [CrossRef]
- Minari, R.J.; Gugliotta, L.M.; Vega, J.R.; Meira, G.R. Continuous emulsion copolymerization of acrylonitrile and butadiene: Computer simulation study for improving the rubber quality and increasing production. Comput. Chem. Eng. 2007, 31, 1073–1080. [Google Scholar] [CrossRef]
- Minari, R.J.; Gugliotta, L.M.; Vega, J.R.; Meira, G.R. Continuous emulsion copolymerization of acrylonitrile and butadiene: Simulation study for reducing transients during changes of grade. Ind. Eng. Chem. Res. 2007, 46, 7677–7683. [Google Scholar] [CrossRef]
- Clementi, L.A.; Suvire, R.B.; Rossomando, F.G.; Vega, J.R. A Closed-Loop Control Strategy for Producing Nitrile Rubber of Uniform Chemical Composition in a Semibatch Reactor: A Simulation Study. Macromol. React. Eng. 2018, 12, 1700054. [Google Scholar] [CrossRef]
- Washington, I.D. Dynamic Modelling of Emulsion Polymerization for the Continuous Production of Nitrile Rubber. Master’s Thesis, University of Waterloo, Waterloo, ON, Canada, 2008. [Google Scholar]
- Washington, I.D.; Duever, T.A.; Penlidis, A. Mathematical Modeling of Acrylonitrile-Butadiene Emulsion Copolymerization: Model Development and Validation. J. Macromol. Sci. Part A Pure Appl. Chem. 2010, 47, 747–769. [Google Scholar] [CrossRef]
- Madhuranthakam, C.M.R.; Penlidis, A. Modeling Uses and Analysis of Production for Acrylonitrile-Butadiene (NBR) Emulsions. Polym. Eng. Sci. 2011, 51, 1909–1918. [Google Scholar] [CrossRef]
- Madhuranthakam, C.M.R.; Penlidis, A. Improved Operating Scenarios for the Production of Acrylonitrile-Butadiene Emulsions. Polym. Eng. Sci. 2012, 53, 9–20. [Google Scholar] [CrossRef]
- Scott, A.J.; Nabifar, A.; Madhuranthakam, C.M.R.; Penlidis, A. Bayesian Design of Experiments Applied to a Complex Polymerization System: Nitrile Butadiene Rubber Production in a Train of CSTRs. Macromol. Theory Simul. 2015, 24, 13–27. [Google Scholar] [CrossRef]
- Vale, H.M.; McKenna, T.F. Synthesis of bimodal PVC latexes by emulsion polymerization: An experimental and modeling study. Macromol. Symp. 2006, 243, 261–267. [Google Scholar] [CrossRef]
- Dubé, M.A.; Saldívar-Guerra, E.; Zapata-González, I. Copolymerization. In Handbook of Polymer Synthesis, Characterization and Processing; Saldívar-Guerra, E., Vivaldo-Lima, E., Eds.; John Wiley and Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Blackley, D.C. Emulsion Polymerization; Applied Science Publishers: London, UK, 1975. [Google Scholar]
- Friis, N.; Nyhagen, L. A Kinetic Study of the Emulsion Polymerization of Vinyl Acetate. J. Appl. Polym. Sci. 1973, 17, 2311–2327. [Google Scholar] [CrossRef]
- Nomura, M. Desorption and Reabsorption of Free Radicals in Emulsion Polymerization. In Emulsion Polymerization; Piirma, I., Ed.; Academic Press: New York, NY, USA, 1982; pp. 191–219. [Google Scholar]
- Dougherty, E. The SCOPE Dynamic Model for Emulsion Polymerization. I. Theory. J. Appl. Polym. Sci. 1986, 32, 3051–3078. [Google Scholar] [CrossRef]
- Gardon, J.L. Emulsion polymerization. VI. Concentration of monomers in latex particles. J. Polym. Sci. Part A-1 Polym. Chem. 1968, 6, 2859–2879. [Google Scholar] [CrossRef]
- Ascher, U.M.; Petzold, L.R. Computer Methods for Ordinary Differential Equations and Differential-Algebraic Equations; Society for Industrial and Applied Mathematics: Philadelphia, PA, USA, 1998. [Google Scholar]
- Buback, M.; Gilbert, R.G.; Hutchinson, R.A.; Klumperman, B.; Kuchta, F.-D.; Manders, B.G.; O’Driscoll, K.F.; Russell, G.T.; Schweer, J. Critically evaluated rate coefficients for free-radical polymerization, 1. Propagation rate coefficients for styrene. Macromol. Chem. Phys. 1995, 196, 3267–3280. [Google Scholar] [CrossRef]
- Junkers, T.; Koo, S.P.S.; Barner-Kowollik, C. Determination of the propagation rate coefficient of acrylonitrile. Polym. Chem. 2010, 1, 438–441. [Google Scholar] [CrossRef]
- Saldívar, E.; Araujo, O.; Giudici, R.; López-Barrón, C. Modeling and Experimental Studies of Emulsion Copolymerization Systems. II. Styrenics, J. Appl. Polym. Sci. 2001, 79, 2380–2397. 2001, 79, 2380–2397. [Google Scholar]
- Deibert, S.; Bandermann, F.; Schweer, J.; Sarnecki, J. Propagation rate coefficient of free-radical polymerization of 1,3-butadiene. Makromol. Chem. Rapid Commun. 1992, 13, 351–355. [Google Scholar] [CrossRef]
- Bachmann, R.; Pallaske, U.; Schmidt, A.; Muller, H.G. DECHEMA Monographs; VCH: Weinham, Germany, 1995; Volume 131, pp. 65–74. [Google Scholar]
- Deibert, S.; Bandermann, F. Rate coefficients of the free-radical polymerization of 1,3-butadiene. Die Makromol. Chem. 1993, 194, 3287–3299. [Google Scholar] [CrossRef]
- Brandrup, J.; Immergut, E.H.; Grulke, E.A. (Eds.) Polymer Handbook, 4th ed.; John Wiley and Sons: New York, NY, USA, 1994. [Google Scholar]
- Mukerjee, P.; Mysels, K.J. Critical Micelle Concentrations of Aqueous Surfactant Systems; NSRDS-NBS 36; National Bureau of Standards: Washington, DC, USA, 1971. [Google Scholar]
- Rawlings, J.B.; Ray, W.H. The modeling of batch and continuous emulsion polymerization reactors. Part II: Comparison with experimental data from continuous stirred tank reactors. Polym. Eng. Sci. 1988, 28, 257–274. [Google Scholar] [CrossRef]
- Arve, D.S.C.; Manley, D.B.; Poling, B.E. Relative volatilities from mixture liquid densities for the n-butane/1,3-butadiene system. Fluid Phase Equilib. 1987, 35, 117–126. [Google Scholar] [CrossRef]
- Guyot, A.; Guillot, J.; Graillat, C.; Llauro, M.F. Controlled composition in emulsion copolymerization application to butadiene-acrylonitrile copolymers. J. Macromol. Sci.-Chem. 1984, A21, 683–699. [Google Scholar] [CrossRef]
- Marien, Y.W.; Van Steenberge, P.H.M.; D’hooge, D.; Marin, G.B. Particle by Particle Kinetic Monte Carlo Tracking of Reaction and Mass Transfer Events in Miniemulsion Free Radical Polymerization. Macromolecules 2019, 52, 1408–1423. [Google Scholar] [CrossRef]
- Marien, Y.W.; Van Steenberge, P.H.M.; Pich, A.; D’hooge, D. Coupled stochastic simulation of the chain length and particle size distribution in miniemulsion radical copolymerization of styrene and N-vinylcaprolactam. React. Chem. Eng. 2019, 4, 1935–1947. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction | Kinetics |
|---|---|
| Aqueous phase | |
| Redox initiation | |
| Particle phase, detailed terminal model 1 | |
| Propagation and cross-propagation (i,j = 1,2) | |
| Chain transfer to monomer (i,j = 1,2) | |
| Chain transfer to chain transfer agent T (i,j = 1,2) | |
| Particle phase, molecular weight distribution (MWD) pseudo-homopolymer form 2 | |
| Propagation (r = 1, …, ∞) | |
| Chain transfer to monomer (r = 1, …, ∞) | |
| Chain transfer to chain transfer agent (r = 1, …, ∞) | |
| Termination by combination and disproportionation (r,q = 1, …, ∞) 2 | |
| Chain transfer to polymer (q,r = 1,..,∞) 3 | |
| Internal or terminal double bond polymerization (q,r = 1, …, ∞) 3 | |
| Variable | Equation(s) | Number of Equations |
|---|---|---|
| Ordinary differential equations (ODEs) (mass or population balance equations) | ||
| Monomers, | 1 | 2 |
| Monomer bound in polymer, | 2 | 2 |
| Initiator, | 12 | 1 |
| Reducing agent 1, | 13 | 1 |
| Reducing agent 2, | 14 | 1 |
| Surfactant, | 15 | 1 |
| Chain transfer agent, | 18 | 1 |
| Water, | 20 | 1 |
| Number of particles con n radicals, | 7–9 | 3 |
| Live polymer moments | Supplementary Material Equations (S3f)–(S3i) with extensions Supplementary Material Equation (S3u), Supplementary Material Equation (S3v), Supplementary Material Equation (S3x), and Supplementary Material Equation (S3y) | 4 |
| Dead polymer moments | Supplementary Material Equations (S3j)–(S3r) with extensions Supplementary Material Equations (S3aa)–(S3af) | 9 |
| TOTAL ODEs | 26 | |
| Main auxiliary (algebraic) equations | ||
| Radicals in the aqueous phase, | 21 | 1 |
| Number of micelles per L of water, | 16 or 17 | 1 |
| Entry coefficient of radicals to particles, | Supplementary Material Equation (S1a) | 1 |
| Entry coefficient of radicals to micelles, | Supplementary Material Equation (S1b) | 1 |
| Desorption coefficient, | Supplementary Material Equation (S1c) | 1 |
| Chain transfer agent (CTA) concentration in particles, | Supplementary Material Equation (S1aa) | 1 |
| Monomer concentration in particles, | 23 or 25 | 1 |
| Total mass, | 29 | 1 |
| Number of branches per chain, | 36 | 1 |
| Closure expression for 3rd moments | Supplementary Material Equation (S3ag) | 1 |
| Total number of algebraic equations | 10 | |
| Parameter | Value | Units | Source/Comments |
|---|---|---|---|
| SBR System: monomer 1 butadiene, monomer 2 styrene | |||
| r1 | 1.4–1.58 | 1.4 from [46] (@ 50 C) | |
| r2 | 0.54–0.58 | 0.58 from [46] (@ 50 C) | |
| 1E, | 8540 | cal mol−1 | [47] |
| 1A, | 1.12 × 108 | L mol−1 s−1 | [47] |
| 2E, | 6220 | cal mol−1 | [46] |
| 2A, | 4.50 × 106 | L mol−1 s−1 | [46] |
| E, | 7770 | cal mol−1 | [41] Pulse laser polymerization (PLP)-determined |
| A, | 4.27 × 107 | L mol−1 s−1 | [41] PLP-determined |
| 100–250 | L mol−1 s−1 | Estimated | |
| 50–200 | L mol−1 s−1 | Estimated | |
| @ 10 C | 9.16 × 108 | L mol−1 s−1 | [45] PLP-determined |
| @ 10 C | 9.2 × 106 | L mol−1 s−1 | [46] |
| 9 × 10−5 | - | Estimated from [46] | |
| 9 × 10−5 | - | Assumed equal to | |
| 2 | - | Estimated | |
| 2 | - | Estimated | |
| 5 × 10−9 | m | Estimated | |
| 9.8 × 10−4 | mol L−1 | [48] | |
| 2.2 × 10−19 | m2 | Estimated | |
| b | 2000 | L mol−1 | [9] |
| 3.5 × 10−6 | mol m−2 | [9] | |
| 0.55 | - | Estimated | |
| f | 0.5 | - | Estimated |
| This work | |||
| This work | |||
| 3.55 × 10−15 | m2 s−1 | [49] | |
| 3.55 × 10−15 | m2 s−1 | [49] | |
| 2 × 10−15 | m2 s−1 | Estimated | |
| 2 × 10−15 | m2 s−1 | Estimated | |
| 20 | Estimated | ||
| 100 | Estimated | ||
| 200 | Estimated | ||
| 0.005–0.05 | L mol−1 s−1 | This work | |
| 618 | g L−1 | Estimated from [50] | |
| 906 | g L−1 | [49] | |
| 910 | g L−1 | Estimated from MSDS Sigma Aldrich St. Louis MOpolybutadiene Mw = 200,000 | |
| 1040 | g L−1 | [46] | |
| Density surfactant | 918 | g L−1 | Estimated |
| NBR System: monomer 1 butadiene, monomer 2 acrylonitrile 3NBR System: monomer 1 butadiene, monomer 2 acrylonitrile 3 | |||
| r1 | 0.30 | [51] | |
| r2 | 0.04 | [51] | |
| 2 | 6633 | L mol−1 s−1 | [24] |
| E, | 3690 | cal mol−1 | [42] PLP-determined |
| A, | 1.83 × 106 | L mol−1 s−1 | [42] PLP-determined |
| @ 10 C | 3.4 × 107 | L mol−1 s−1 | [28] |
| 6.5 × 10−4 | mol L−1 | [48] | |
| 3.8 × 10−19 | m2 | Estimated | |
| 10 | Estimated | ||
| 822 | g L−1 | [28] @ 10 C | |
| 1167 | g L−1 | [28] @ 10 C | |
| 3.5 | Estimated | ||
| Component | Parts per Hundred Monomer (pphm) | ||||
|---|---|---|---|---|---|
| Butadiene (M1) | 72 | ||||
| Styrene (M2) | 28 | ||||
| Initiator | 0.1–0.2 | ||||
| Reducing agent 1 1 | 0.02–0.05 | ||||
| Reducing agent 2 2 | 0.04–0.10 | ||||
| Chain transfer agent | 0.10–0.17 | ||||
| Surfactant | 3.0–5.0 | ||||
| Caustic potash | 0.5–0.7 | ||||
| Electrolyte (sodium carbonate, potassium chloride) | 0.2–0.4 | ||||
| Reactor No | R1–R5 | R6 | R7 | R8 | R9–R10 |
| Temperature | baseT (Tb) | Tb + 10 C | Tb + 7 C | Tb + 6 C | Tb + 2 C |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saldívar-Guerra, E.; Infante-Martínez, R.; Islas-Manzur, J.M. Mathematical Modeling of the Production of Elastomers by Emulsion Polymerization in Trains of Continuous Reactors. Processes 2020, 8, 1508. https://doi.org/10.3390/pr8111508
Saldívar-Guerra E, Infante-Martínez R, Islas-Manzur JM. Mathematical Modeling of the Production of Elastomers by Emulsion Polymerization in Trains of Continuous Reactors. Processes. 2020; 8(11):1508. https://doi.org/10.3390/pr8111508
Chicago/Turabian StyleSaldívar-Guerra, Enrique, Ramiro Infante-Martínez, and José María Islas-Manzur. 2020. "Mathematical Modeling of the Production of Elastomers by Emulsion Polymerization in Trains of Continuous Reactors" Processes 8, no. 11: 1508. https://doi.org/10.3390/pr8111508
APA StyleSaldívar-Guerra, E., Infante-Martínez, R., & Islas-Manzur, J. M. (2020). Mathematical Modeling of the Production of Elastomers by Emulsion Polymerization in Trains of Continuous Reactors. Processes, 8(11), 1508. https://doi.org/10.3390/pr8111508