1. Introduction
Adipose tissue and its derivatives are being increasingly used as autografts. Bioactive fractions or transformed products of fat include microfat, nanofat, stromal vascular fraction (SVF), and adipose-derived stem cells (ATD-MSC). Indications for the use of these autologous tissue derivatives are several and enlarging [
1], i.e., cosmetic surgery for creating, increasing, or reconstructing volumes, reconstructive surgery for revitalizing mortified areas [
2], and orthopedic surgery for anti-inflammatory effects in degenerating joints, ligaments, and tendons [
3,
4,
5]. The European Union and American regulations impose some specific requirements when processing harvested fat. As with any other tissue for immediate autografting, users must respect minimal manipulation requirements. Indeed, the enzymatic digestion of adipose tissue, which disrupts the extracellular matrix (ECM) with the release of SVF, is considered high manipulation and must be performed into cell factories. Notably, the FDA also considers that significant physical processing is more than minimal manipulation when the structural integrity of the tissue is altered.
An increasing number of systems have been developed to process or isolate adipose tissue elements overcoming donor variations, risk of infections, and unpredictability of the final product. The most common method is cell isolation based on enzymatic digestion to obtain SVF, but it has high costs with an impact on safety and efficacy [
6]. To date, many techniques, fully compliant with the minimal manipulation EU and US requirements, have been developed to extract these fractions. Non-enzymatic isolation methods use mechanical forces to separate the cells or cell aggregates from adipose tissue, with a sound efficacy even with a lower cell-yield compared to enzymatic-dependent techniques [
7,
8]. Indeed, the success of the treatment seems independent of ATD-MSC quantity and differentiation ability, but likely more dependent on MSCs’ ability to release growth factors and cytokines in an adaptive paracrine fashion [
9]. Our research group has developed a novel technique based on a semipermeable membrane commonly used for dialysis purposes. This method, while being completely adherent to the minimal manipulation concept, would appear to be delicate as well as extremely effective and efficient in isolating extracellular matrix components and discarding lipoaspiration byproducts with no recognized bioactive effects. In this study, we cytologically and histologically characterized the final product of the Lipocell filtration-based method and determined its efficiency in the clearance of residues.
2. Materials and Methods
2.1. Sample Collection and Processing
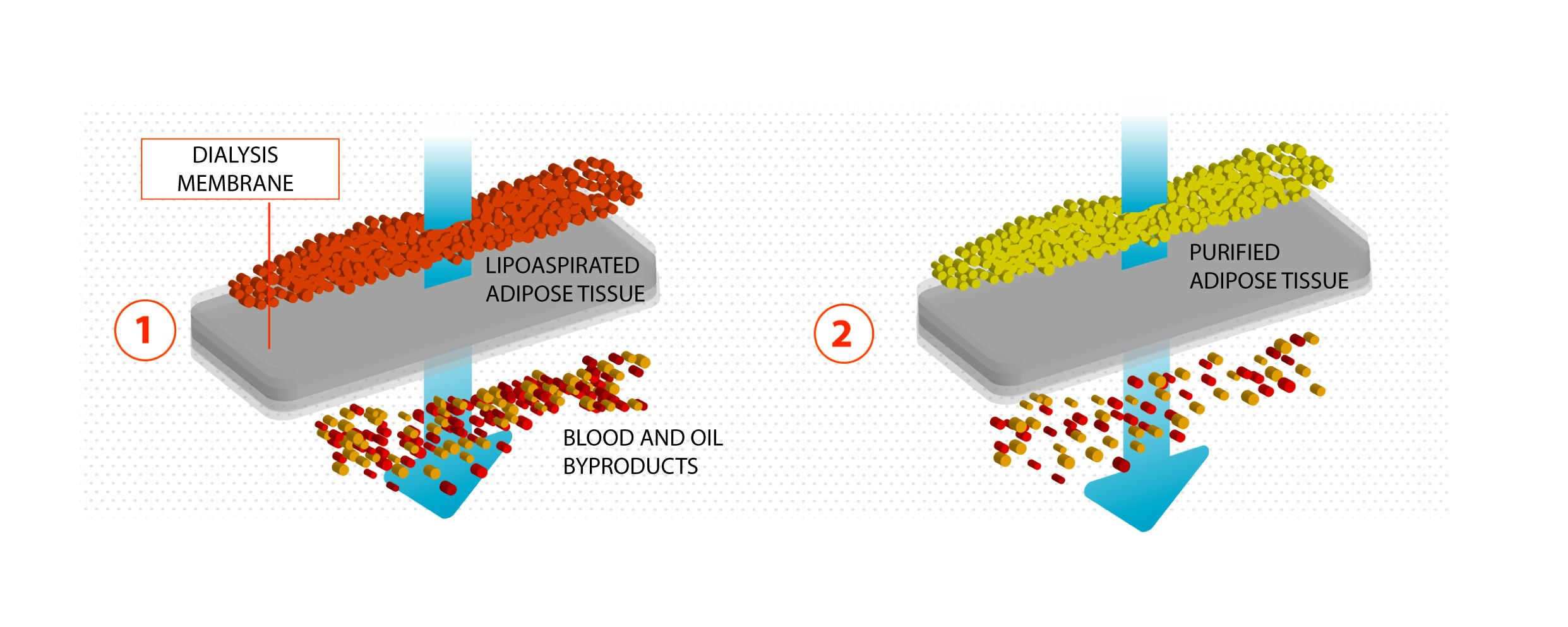
Lipoaspirate samples were collected from surplus aesthetic liposuction material obtained from patients, who signed written informed consent. Patients underwent liposuction after Klein solution infiltration (lidocaine 2% and adrenaline 1 mg/mL in 500 mL NaCl 0.9% solution). Klein solution infiltration and liposuction were performed with fenestrated blunt cannulae. The lipoaspirates were processed with Lipocell (Tiss’You, Domagnano, Republic of San Marino) according to instructions. Briefly, the lipoaspirate was inserted into the device where it was dialyzed with a filter (15 µm, 20 µm, or 50 µm) and washed with 300–500 mL of washing solution (NaCl 0.9%, phosphate buffered saline (PBS), or Ringer’s lactate (RL). Gentle brushing was done on the outside of the bag to facilitate the washing. The whole procedure lasted for an average of 5 min. At the end, the lipoaspirate appeared clear from blood, and the flowing washing solution was transparent; the purified lipoaspirate could be recovered with a 10 mL syringe from the output connection. The volume of the final product was dependent on the liposuction technique. Averagely, 50% up to 70% of harvested material could be recovered after the processing.
2.2. Isolation of SVF from Adipose Tissue
SVFs were isolated from fresh and Lipocell-processed adipose tissue by enzymatic digestion with Collagenase NB4 (Serva Electrophoresis, Heidelberg, Germany) 0.15 U/mL using a tube rotator for 40 min at 37 °C. The activity of the enzyme was neutralized by the addition of Dulbecco’s modified Eagle’s medium (DMEM) low glucose ±10% FBS, and then the samples were centrifuged at 1700 rpm for 10 min. The pellet was resuspended in medium, then passed through 100 µm and 70 µm cell strainers, washed by saline solution, collected, and counted. The phenotype of ATD-MSCs contained in the SVF was evaluated at T0, soon after SVF isolation. The SVF cells were seeded in T25 flasks and cultured in DMEM with 10% FBS, 2 mM glutamine, and 1% antibiotics (Thermo Fisher Scientific, Waltham, MA, USA), and the medium was replaced to eliminate nonadherent cells after 24 h. After 7 days of culture, the growth rate was calculated by counting the number of cells grown for mL of adipose tissue. Then, MSCs were cultured for two passages, then detached, and their phenotype was analyzed.
2.3. Phenotype of ATD-MSCs
Cell surface markers of ATD-MSCs were analyzed by flow cytometry on fresh SVF derived from adipose tissue. ATD-MSCs were identified as CD105+/CD73+/CD90+/CD271+/CD45− cells, pericytes were CD146+/CD31−/CD34− cells, endothelial cells were CD31+/CD34−/CD146− cells, stromal cells were CD34+/CD31−/CD146−/CD45− cells, monocytes were CD14±cells, and lymphocytes were CD3+ cells. Standard labelling protocol was performed with the following fluorochrome-conjugated antibodies and isotypic controls: human CD105 PE (Invitrogen, Carlsbad, CA, USA), CD73 FITC (kindly provided by Professor Malavasi, University of Turin, Turin, Italy), CD90 PerCP (Biolegend, San Diego, CA, USA) CD45 PerCP, CD3 PerCP, CD271 APC, IgG1 PE, IgG1 APC, and IgG2a PerCP (Miltenyi Biotec, Bergisch Gladbach, Germany), and IgG1 FITC-conjugated (Immunostep, Salamanca, Spain). About 105 events/sample were used for capture with CellQuest software. ATD-MSC percentages were previously normalized on the absolute number of viable cells. All data were analyzed with FlowLogic software (Miltenyi Biotec, Bergisch Gladbach, Germany).
2.4. Washing Efficiency Assay
250 mL of lipoaspirate from a unique donor was collected and split into three homogenous 50 mL syringes. Each sample was put in a different Lipocell device (15 µm, 20 µm, or 50 µm) and washed homogenously for 5 min. The procedures were carried out synchronously to assess the washing efficiency of the devices. Purified adipose tissues were then recovered in tubes and centrifuged at 300×
g for 10 min using untreated lipoaspirate as a control sample. Water phases were transferred in plastic cuvettes (optical path = 1 cm) and measured with a UV-Vis Spectrophotometer (Lambda 365, PerkinElmer, Waltham, MA, USA). Hemoglobin (Hb) concentrations were calculated accordingly to references [
10].
2.5. Histology and Staining
Adipose tissue purified with the Lipocell procedure and controls were fixed in 4% paraformaldehyde (PFA) then washed in PBS-1X and embedded in 1% agar. After the dehydration process (50% ethanol 30 min, 70% ethanol 30 min, 90% ethanol 45 min, 95% ethanol 30 min, 100% ethanol 90 min × 2, and xylene 60 min × 2) the adipose tissue was embedded in paraffin. The slices, obtained by the microtome cut, were rehydrated (xylene 5 min × 2, 100% ethanol 5 min × 2, 95% ethanol 3 min, 90% ethanol 3 min, and 70% ethanol 3 min), then stained in hematoxylin for 4 min and dehydrated for eosin staining (6 min). Finally, the sections were mounted with EUKITT mounting media (O. Kindler GmbH & CO, Freiburg im Breisgau, Germany).
2.6. ECM Extraction and Collagen Type II
ECM extraction procedure was performed at 40 °C to prevent freezing of the oil components of lipoaspirate samples. Then, 3 mL of each sample was diluted in distilled water (1:1) and homogenized for 5 min using a Polytron PT2500 with pulsation. The tissue suspension was then centrifuged at 1800× g for 5 min, and the upper layer of oil components was discarded; this process was repeated several times until the complete removal of oil components. The tissue was minced and covered with HNTG Lysis buffer (50 mM HEPES, 150 mM NaCl, 10% Glycerol, and 1% Triton X-100), homogenized 20 times and rotated at 4 °C for 30 min. The lysate was centrifuged at 18,000× g at 4 °C for 20 min. Type II collagen was quantified with COL 2 Elisa Kit (Cloud-Clone, Corp., Houston, TX, USA) according to the manufacturer’s instructions.
2.7. Glycosaminoglycans Quantification
GAG content was quantified with the Dimethylmethylene Blue Assay (DMMB) assay. Samples were freeze-dried to a constant weight, and 10 mg of samples were digested in papain buffer (125 mg/mL papain, 5 mM cysteine-, 50 HCl, and 5 mM disodium EDTA in PBS) at 60 °C for 24 h. Then, 50 µL of each sample was mixed with 250 µL of 1.9-dimethyl-methylene blue (Sigma-Aldrich, St. Louis, MO, USA) in a 96-well microtiter plate, and the absorbance was measured at 530 nm. The amount of GAG content was calculated by reference to a standard curve prepared using different concentrations of chondroitin sulfate sodium salt from shark cartilage (Sigma-Aldrich, St. Louis, MO, USA).
2.8. Collagen Quantification
10 µL of extracted ECM was mixed thoroughly with 100 µL of 37% hydrochloric acid. The homogenate was incubated at 120 °C for 3 h to ensure complete hydrolysis. After hydrolysis, 10 µL from each sample was transferred to a 96-well microplate (Greiner Cellstar 96 well flat bottom, Sigma-Aldrich, St. Louis, MO, USA) and left overnight at 60 °C. After evaporation, the hydroxyproline content was determined using a Hydroxyproline Assay Kit (Sigma-Aldrich, St. Louis, MO, USA). The absorbance at 560 nm was determined using a microplate reader (Infinite 2000Pro, Tecan, Männedorf, Switzerland) and converted to hydroxyproline content using a standard curve; total collagen concentration was obtained by multiplying by 8 the hydroxyproline content [
11].
4. Discussion
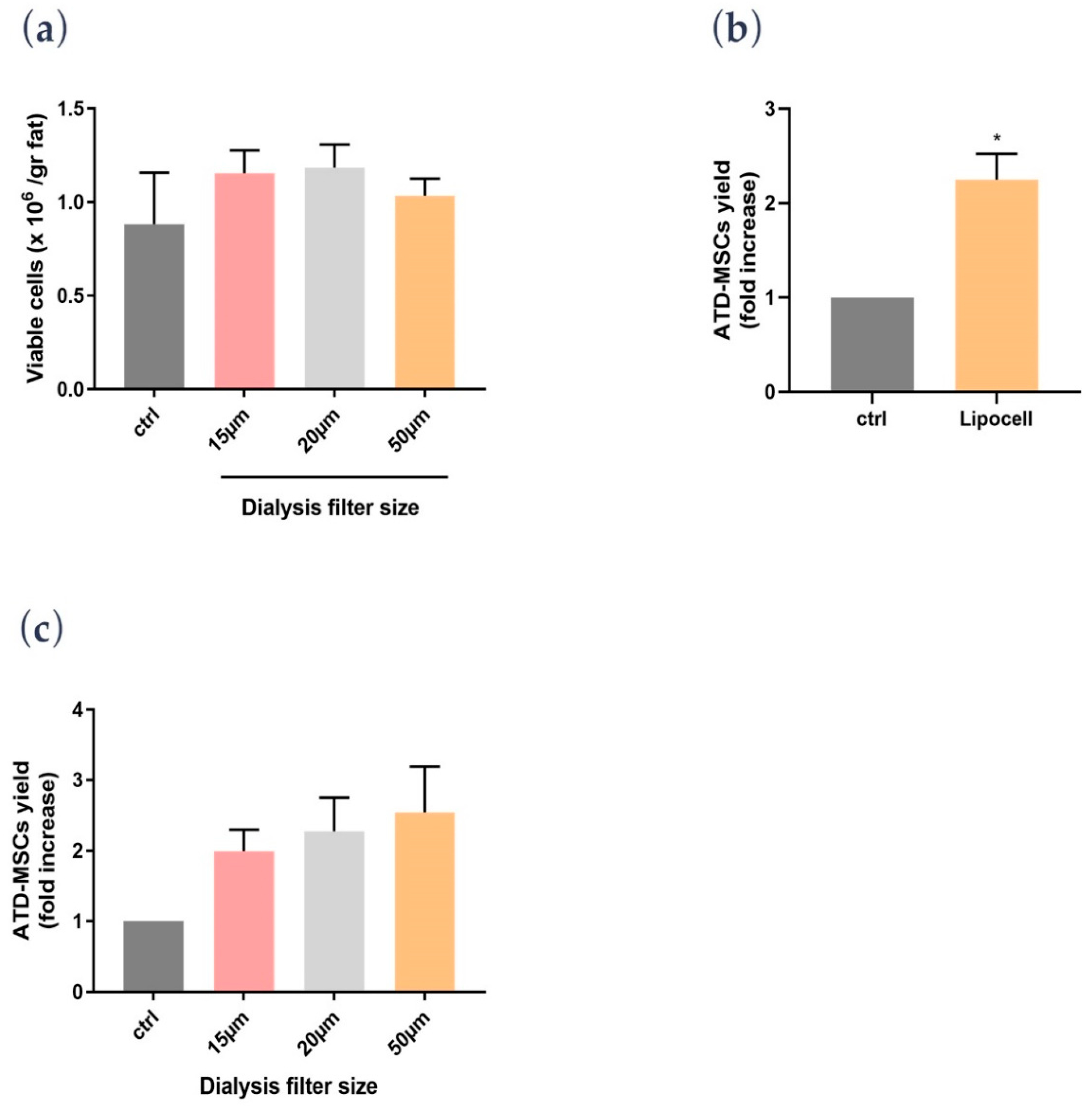
In the present study, we showed that Lipocell is an efficient method to purify adipose tissue from undesired byproducts, such as blood and oil, and it preserves ATD-MSC viability and ECM integrity. The whole procedure was done in a closed-loop system, and it is performable intra-operatively in a sterile environment, being compliant to EU and US regulations about minimal manipulation of cell and tissue therapies. Lipocell processing lasts for an average of 5 min; standard centrifugation usually requires up to 15 min. With this novel technique, the whole treatment (lipoaspiration, processing, and in-situ injection) can be done within 30 min. Even without cell isolation or purification, the procedure provided a considerable cell yield: over two times more ATD-MSCs than basal levels, likely due to purification from blood cells and other elements. This cell yield was not dependent on filter size, which only influenced oil and blood purification. Unlike traditional methods such as centrifugation or enzymatic treatment, ECM was not disrupted by the procedure, as demonstrated by H&E staining and biochemical assay. The maintenance of ECM integrity is crucial to preserve tissue volume, to provide temporary mechanical functions, and to guide the complex multicellular processes of tissue regeneration [
12,
13].
A novel finding was the effect of RL on ATD-MSCs; indeed, while cell counts were not different among saline, PBS, and RL after the procedure, the proliferation assay showed a better ATD-MSC proliferative potential with RL than saline and PBS. To explain this RL-mediated effect on proliferation, we hypothesized that cells are deprived of blood supply after the liposuction, and the sudden lack of oxygen can trigger anaerobic respiration; providing lactate ions can drive the cell respiration back to the aerobic pathway thus ameliorating cell survival [
14,
15]. Based on these preliminary data, RL may be considered as a standard for processing and washing adipose tissue to preserve ATD-MSC proliferation ability.
Subcutaneous adipose tissue is an abundant source of ATD-MSCs, thus it is a promising tool for several therapeutic applications in plastic and regenerative medicine. An increasing number of systems have been developed to process or isolate adipose tissue elements according to the low manipulation rules laid down by the EU and US. Non-enzymatic isolation methods use mechanical forces, such as centrifugation, to separate the cells or cell aggregates from adipose tissue, but ECM can be disrupted by centrifugal forces, and the procedure is usually performed away from the sterile field, increasing the risk of infections. Indeed, tissue regeneration is regulated by specific environmental stimuli, providing trophic signals and immune modulation, also dependent on the maintenance of micro-environment and ECM architecture [
9]. The gentle mechanical force applied by Lipocell preserves extracellular matrix and structural integrity, according to our analysis. Lipocell is distinguished from other devices by its simplicity, speed, and operator-independency. Moreover, the dialysis mechanism of Lipocell avoids the risk of fat obstruction. The Lipocell final product displays a preserved structural integrity in compliance with FDA requirements, avoiding excessive clustering that can alter the mechanical properties of fat. Moreover, Lipocell mechanical forces are less dependent on the strength of the operator [
16].
A limitation of this first study on the Lipocell device is the lack of ATD-MSC multi-lineage differentiation assays, but the focus of this work was to test ATD-MSC viability and ECM integrity. Further investigations will be performed to explore the activity of Lipocell-derived ATD-MSCs, even though it is known that the therapeutic effect of purified adipose tissue depends on a multitude of elements and not only on their ability to differentiate into different tissues [
1,
9].
In conclusion, Lipocell is a promising tool in the booming field of regenerative medicine. It is effective in optimizing adipose tissue recovery for subsequent autologous injection (i.e., subcutaneous, intra-articular, or intra-epithelial), according to the low manipulation requirements.

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}