Abstract
Plants produce a diverse portfolio of sesquiterpenes that are important in their response to herbivores and the interaction with other plants. Their biosynthesis from farnesyl diphosphate depends on the sesquiterpene synthases that admit different cyclizations and rearrangements to yield a blend of sesquiterpenes. Here, we investigate to what extent sesquiterpene biosynthesis metabolic pathways can be reconstructed just from the knowledge of the final product and the reaction mechanisms catalyzed by sesquiterpene synthases. We use the software package MedØlDatschgerl (MØD) to generate chemical networks and to elucidate pathways contained in them. As examples, we successfully consider the reachability of the important plant sesquiterpenes -caryophyllene, -humulene, and -farnesene. We also introduce a graph database to integrate the simulation results with experimental biological evidence for the selected predicted sesquiterpenes biosynthesis.
1. Introduction
Terpenes form a large and diverse class of natural products appearing particularly in the essential oils of many plants. They have commercial uses in medicine and as fragrances in perfumery. Synthetic derivatives of natural terpenes are also used as aromas and foodadditives [1]. Ecologically, they perform key functions both in direct plant defense and in indirect mechanisms involving herbivores and their natural enemies [2].
Terpenes are produced throughout the tree of life from the compounds isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP). Leopold Ružička [3] formulated the “biogenetic isoprene rule” according to which terpenes are the result of concatenating isoprene units in a “head-to-tail” fashion to form chains, from which then rings are formed. Prenyltransferases condense IPP and DMAPP to geranyl pyrophosphate (GPP), farnesyl pyrophosphate (FPP), and geranylgeranyl pyrophosphate (GGPP) as the entry points to the world of monoterpenes (), sesquiterpenes (), and diterpenes( [4]. FPP and GGPP can then be condensed further to form squalene () and even largermolecules [5].
These intermediates are substrates for a class of enzymes known collectively as terpene synthases (TPSs) to produce a wide variety of compounds. There are two major classes of TPS, which are distinguished by essential amino acid motifs [6,7]. Class I TPS, which are of interest in this contribution, convert linear, all-trans, isoprenoids, geranyl ()-, farnesyl ()-, or geranylgeranyl ()-diphosphate into numerous monoterpenes, sesquiterpenes, and diterpenes. They bind their substrates by the coordination of a divalent metal ion catalytic site consisting of a central cavity formed by mostly antiparallel -helices. This catalytic site has an aspartate-rich motif and often another motif in the C-terminal portion [8,9,10].
The mechanisms of sesquiterpenes synthesis involve the formation of C–C bonds, cationic intermediates, Wagner-Meerwein rearrangements, carbocation capture by water and hydride, as well as methyl- and allyl-shifts caused by conformational changes of intermediate cations [11,12,13]. A combinatorial reaction cascade inside the TPS combining different cyclization/rearrangement reactions yields a highly diverse set of sesquiterpenes from just a handful of simple acyclic precursors molecules. Nevertheless, there are only four dominating types of cyclization reactions initiating the complex reaction cascade: –, –, –, and – [14]. Even though the electrophilic chemical reaction mechanisms primarily determine the diversity of the products [11], many other factors influence multiproduct sesquiterpene enzymes and their cyclization cascades, such as , the metal cofactor [4], evolutionary forces for the functional divergence [15], plant tissue [16], and plant-plant interactions [17]. Throughout this contribution, we will refer to such a collection of factors as a Scenario.
The complexity of the portfolio of sesquiterpenes and their synthesis pathways calls for computational models that explain the observed diversity and can be used to predict biosynthetic pathways for particular terpene compounds. Within the field of systems biology, the reconstruction and exploration of metabolic networks is grounded in determining the metabolic capabilities from the enzymes encoded in the genome and the extensive knowledge on enzymes themselves collected, e.g., in the BRENDA database [18].
Several software tools have been developed to predict pathways from annotated genome sequences and to analyze and explore them. Pathway Tools [19], for instance, is a system designed for the BioCyc database collection [20]. Complementarily, versatile tools have become available to predict the metabolism of xenobiotics, in particular, aiming at drugs. Pattern recognition, machine learning techniques, and knowledge-based rules are used to identify a “site of metabolism” for a set of chemical products from biotransformation starting with a known parent molecule. Many of these tools are restricted to specific metabolic processes, although recently much more generally applicable tools such as Biotransformer [21] have become available. RetroRules [22] is a database of >400,000 highly specific reaction rules intended for metabolic engineering and, in particular, the prediction of novel and alternative products from de novo reactions of promiscuous enzymes. Possible reactions can, in principle, also be obtained with methods such as AFIR (Artificial Force Induced Reaction) [23] from Potential Energy Surfaces, however, the computational cost makes it difficult to use them for exploring large chemical spaces.
Here, we take a somewhat different approach in that we do not start from an extensive base of detailed chemical and biological knowledge but from a very generic representation of the reaction mechanisms underlying sesquiterpene synthesis. The molecules are represented as labeled graphs, abstracting away details of the spatial embedding of the molecules. The reactions are considered at the level of graph transformations and only use local context information to determine whether they are applicable to a given molecule or not. The representation of molecules as graphs and of reactions as graph transformations enables the efficient expansion of large networks of logically possible reactions. The integer flows in these networks correspond to feasible synthesis pathways, which can be identified efficiently as solutions of Integer Linear Programs (ILP). In Section 4, we describe in detail this approach and its computational efficiency as a key advantage, as well as adhering to FAIR Guiding Principles [24]. The software package MØD [25,26] makes it possible to explore the diversity of potential sesquiterpene synthesis pathways leading up to a user-defined set of reaction mechanisms (here, the types of rearrangements catalyzed by TPSs). Along with this, using a database of Scenarios taken from the literature, it is possible to filter from the universe of generated potential pathways those that are most plausibly expected in a given scenario on the basis of the available evidence. We have shown that such an explorative approach is indeed feasible and yields reasonable results without the need first to mine an extensive knowledge base. As examples, we consider the biosynthesis common plant sesquiterpenes as -caryophyllene, -humulene, and -farnesene.
2. Results
In order to propose a potential biosynthesis pathway, we have solved the corresponding formal reachability problem: Given known starting material(s), a target molecule of which the biosynthesis is unknown, and a set of reaction rules modeling the class(es) of enzymes suspected to be involved in the potential pathway, we ask whether the target is reachable from the starting material. The graph grammar rules implemented here accurately emulate the natural chemical mechanisms for multiproduct sesquiterpene enzymes and their cyclization cascades.
It has been shown by a reduction to the word problem for (semi) groups [27] that this type of reachability question for chemical transformation systems is Turing undecidable. Hence, in practice, we only solve a restricted problem: Is the target reachable from the starting material within a bounded number of steps. This question can be solved efficiently within the MØD framework by expanding a network of potential reactions. Within this network, the reachability question reduces to the existence of a hyperpath connecting prescribed sources and targets — a problem that is solved efficiently by ILP. The reachability question for molecules of interest can be investigated under varying conditions by modulating the sets of reactions and/or additional molecules, e.g., nucleophiles, present during a reaction networks expansion and pathway search.
Terpene cyclization involves highly reactive cationic intermediates vulnerable to a nucleophilic attack or elimination reactions. This characteristic allows us to tie the changing mixture of reachable sesquiterpene products to variations in the external constraints which can, in many cases, be mapped to changes in the environmental conditions. A particular set of external conditions previously described in the literature, will, in the following, be called a Scenario. We also consider how some selected generated pathways fit into a Scenario, acknowledging their advantages and limitations.
2.1. Protonation-Dependent Diphosphate Cleavage
Natural class I terpene synthases precisely pre-orient their acyclic diphosphate substrates in their active site pockets in a reaction-ready conformation. Then, the complex reaction cascade is initiated by cleaving off the diphosphate group from the pre-oriented substrate molecule, a -dependent [4] process that requires divalent metal ions as cofactor [28,29]. It has been shown that this ionization step of the allylic diphosphate is the rate-limiting step in a class I terpene synthetase catalysis [30]. Then, the enzyme guides the resulting highly reactive carbocation via rearrangements of the carbon skeleton towards various polycyclic product molecules. The highly reactive carbocation intermediates can scavenge nucleophiles, present in the enzyme’s active site, or can saturate the positive charge by deprotonation, terminating the rearrangement cascade in an early stage. A fair example of a rearrangement cascade is the isomerization of FPP to Nerolidyl Diphosphate (NPP) [31], where the diphosphate anion terminates the reaction cascade by reattaching to the carbocationic intermediate immediately after an allylic rearrangement has occurred. The reachability of NPP from FPP is trivial to ascertain in a constraint-independent simulation (Figure 1) using only the cleavage of the diphosphate group and the addition of a nucleophile to a cation in the reaction set (Figure 7).
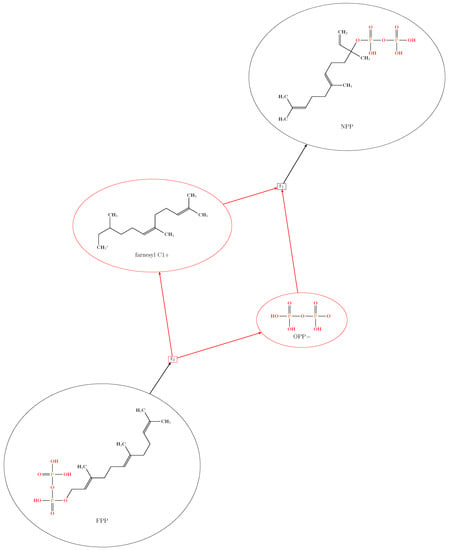
Figure 1.
The plotted result of Simulation 01 for OPP (Diphosphate) cleavage from FPP (Farnesyl Diphosphate).
2.2. Synthesis of -Caryophyllene, -Humulene, -Farnesene, and their Side Products
(E)--caryophyllene is produced from FPP and is emitted by different plant tissues, often in response to a herbivore attack [16,32,33]. There is ample evidence that TPS catalysis produces a mixture of sesquiterpenes rather than a single sesquiterpene product [2,34,35]. The same mechanism that potentially produces -caryophyllene, -humulene, and -farnesene also can produce additional compounds. Simulation 02 shows the computational reachability of these compounds (Figure 2) together with as an intermediate compound, and the predicted side compounds and . For this simulation, the applied set of rules are presented in Figure 8.
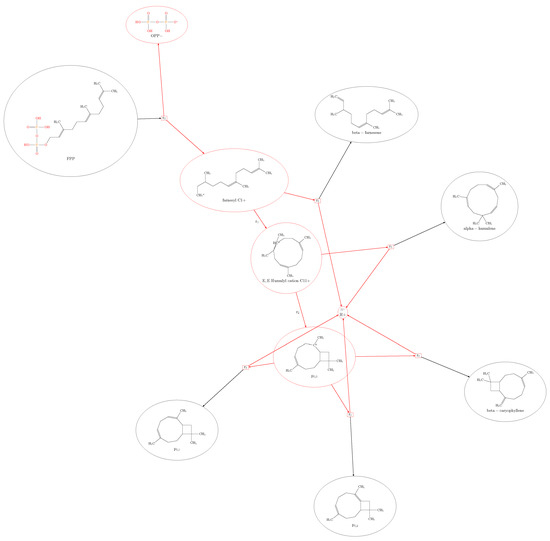
Figure 2.
The plotted result of Simulation 02 for -caryophyllene, -humulene, -farnesene, and the side-predicted compounds from FPP.
2.3. Large-Scale Exploration of Terpene Space
Simulation 03 uses a more explorative derivation graph strategy. In this example, a vast diversity of the feasible compounds is generated starting with an FPP and a water molecule, an extended set of rules (Figure 9) iteratively applied seven times (Figure 3).
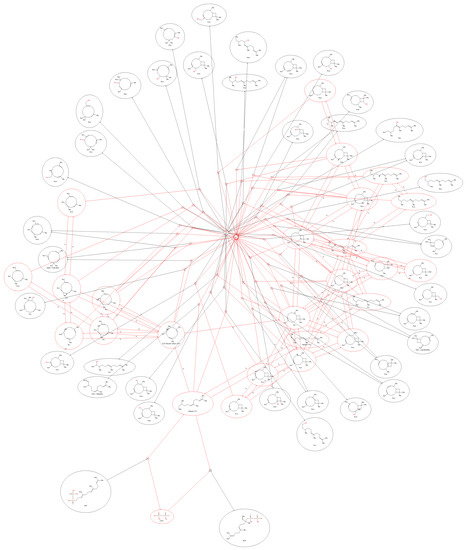
Figure 3.
The exploratory strategy yielded 55 predicted molecules: Stable, neutral molecules are outlined in black, while cations and anions are outlined in red.
2.4. 2Path-Sesquiterpenes Database
A reasonable question dealing with the predicted data is how to explain them. Another matter is how to express them properly. Graph databases are a suitable tool for this purpose that has been demonstrated to be an efficient and convenient way to store and explore metabolic networks [36,37]. Here, we used a proposed graph database (2Path-Sesquiterpenes) to store and enrich the simulation data with experimental evidence related to the predicted sesquiterpenes, the Scenarios. As an example of this use, any simulation using the provided set of rules can be stored using the complementary code Processes_Store, also available on GitHub.
Once stored, the generated chemical mechanisms level metabolic network can be traversed and handled through graph database query languages such as Cypher (https://neo4j.com/developer/cypher-query-language) or Gremlin (https://tinkerpop.apache.org/gremlin.html) and visualized using tools such as Cytoscape [38]. Figure 4 shows a selected example of a query result for a stored simulation into Neo4J graph database (https://neo4j.com). This example shows how a particular generated pathway can be retrieved using Cypher queries, as well as its integration with Scenarios.
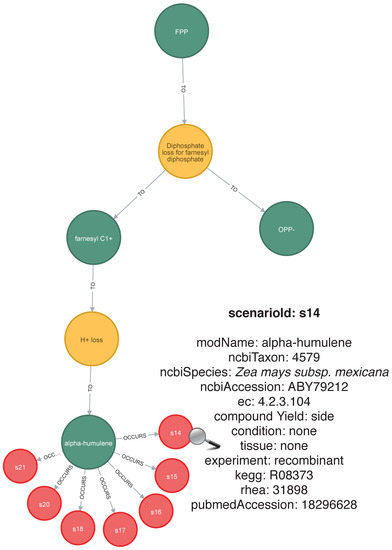
Figure 4.
An example of a generated pathway stored in the Neo4J graph database. In this example, yellow nodes (label “Rules”) denote the rules “diphosphate loss” and “H+ loss”. The green nodes (label “Compound”) denote compounds from FPP to -humulene and other generated compounds. The red nodes (label “Scenario”) denote the Scenarios for the -humulene biosynthesis. The details inside each Scenario node, as the experimental conditions, the plant tissue, EC (Enzyme Commission) numbers for the reactions, and cross-references, can be reached using the web interface, which is native for the Neo4J graph database. In this example, we expose these details for the particular Scenario S14.
3. Discussion
The metabolic networks have been abstracted by various data structures, including substrate graphs, bipartite graphs, directed hypergraphs, reaction graphs, stoichiometric matrix, and Petri-net [39,40,41]. Directed hypergraphs can overcome the conceptual limitations of the graph modeling of biological processes such as multilateral relationships, which are not compatible with graph edges [39]. These hypergraphs properties allow for multilateral relationships between the nodes resulting in a suitable description of biological processes [39]. For example, in a metabolic reaction such as (), a hypergraph allows the edges to connect more than two nodes.
A directed hypergraph is the result of a rule-based graph grammar that transforms compounds (abstracted as undirected graphs). Thus, the graph transformation framework MedØlDatschgerl granted a very intuitive way to explore the sesquiterpene biosynthesis through simulations of its cascade rearrangements. In this work, the proposed graph grammar rules emulate a set of natural cascade rearrangements. The resulting chemical network varies depending on the combination of rules, iteration steps, derivation strategy, and initial compounds. By exploring the possible chemical mechanisms of these reactions, it is reasonable to establish connections with the experimental results to draw from this universe of possibilities, those that can occur naturally or synthetically under certain circumstances.
Constraint-based models (CBM) have enormous potential in enhancing the understanding of reconstructed/predicted metabolic networks and predictive computational models by integrating biological evidence [42,43,44]. Omics studies can address important questions, as biosynthetic pathways, a selection of plants of interest, the environmental influence on the gene expression and the metabolome profile, and many further questions. 2Path-Sesquiterpenes is an optional feature of this work. It helps to address such questions focusing on plant sesquiterpene biosynthesis by aggregating the putative biological meaning to the predicted chemical network.
There are some related works as AFIR [23]/GRRM, RetroRules [22], and Biotransformer [21]. Isegawa et al. [45] had made computational simulations predicting pathways for terpene formation from a humulyl cation and other intermediate molecules using AFIR [23]/GRRM of which the chemical results align with ours, which allowed for a cross-confirmation. Compared to this work, the AFIR [23]/GRRM approach is a fundamentally distinct approach because it uses both a different data structure to design molecules and applies artificial forces between two or more reacting molecules. Both the Biotransformer and RetroRules [22] uses chemical reaction descriptions and rules encoded by SMARTS [46] and SMIRKS [47], and they are quite distinct from this work regarding the method. Different from these three related works, in 2Path-Sesquiterpenes, the geometry of chemical bonds are indistinguishable due to the data structure (undirected graphs) that abstracts the compound molecules.
Another matter is making the simulation results findable, accessible, interoperable, and reusable (FAIR). For this purpose, 2Path-Sesquiterpenes offers the option of storing the simulation results in a graph database. Metabolic network databases have been constructed since 1989 [48] through distinct methods, and many of them have been made available over time mostly due both to advances in metabolic network reconstruction methods and to the expansion of omic data. Despite their extensive range, such as KEGG (Kyoto Encyclopedia of Genes and Genomes) [49], Metacyc [50], and Reactome [51], to name a few, most of them do not provide information at the level of chemical mechanisms and intermediate compounds of a reaction, except for MACiE [52], which still offers rare data on biosynthetic sesquiterpene reactions. Also, graph databases can bring significant query performance improvements for selected problems including metabolic networks [51], confirming their suitability for this purpose. Table 1 shows an overview of some features of the 2Path-Sesquiterpenes and its related works.

Table 1.
A summary of some features of the 2Path-Sesquiterpenes and its related works.
4. Methods
4.1. Molecules as Undirected Graphs, Graph Transformation, Hypergraphs, and Integer Hyperflows
Labeled graphs are commonly used in the chemical literature to represent molecules. Undirected graphs are a convenient and natural way to model chemical molecules where the vertices denote atom types and the number of edges between the vertices denotes bond types. An undirected graph G can be defined as an ordered pair consisting of a nonempty set of vertices V and a set of edges E disjoint of V [53]. Figure 5 shows an example of this abstraction for representing molecules as undirected graphs where Figure 5a shows the set of vertices V composed of and and Figure 5b shows the set of vertices V composed by and .
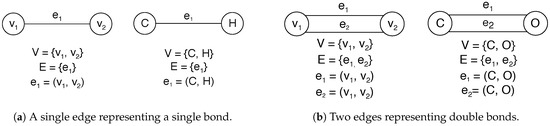
Figure 5.
The general concepts of undirected graphs and their abstraction for representing molecules as undirected graphs using labels as atoms and number of edges as bonds.
Graph grammars are formal systems describing rule-based graph transformation that generalize the much more commonly used term-rewriting systems [54]. In this context, the rules of a graph grammar transform undirected graphs (molecules) in other undirected graphs (molecules) through rules of a given graph grammar formalism. Thus, chemical reactions correspond to the transformations of graphs with particular features:
- (i)
- Reactions may change the number of molecules; hence, both input (substrate) and output (product) graphs are not necessarily connected.
- (ii)
- All atoms are preserved, i.e., a chemical reaction defines a bijection between the vertex sets of input and output graphs.
- (iii)
- Electrons are preserved as well, implying restrictive conditions on the way edges (bonds) can change, corresponding to chemical reaction mechanisms.
We favor the so-called double pushout (DPO) formalism [55] as a model of chemistry because it guarantees the structural reversibility of reactions [56] and it conveniently exposes the representation of the chemical transition state as part of the rule. In DPO graph rewriting, a transformation rule is of the form where L, R, and K are the left graph, right graph, and context graph, respectively. These three graphs are connected by graph morphisms and , describing the embedding of the context into the L and R. The application of the rule p to a graph G requires that L “matches” a part of G. The existence of another graph morphism captures this, the matching morphism . Together, the rule p and the matching morphism m uniquely define the transformation of the substrate G to the product H by requiring that all morphisms in the following commutative diagram exist:
In the context of modeling chemistry, we consider only injective graph morphisms, and we require that the restrictions of r and l to the vertex sets are bijective, ensuring a preservation of the atoms. Since each chemical reaction transforms a set of substrate molecules into a set of product molecules, chemical networks are directed hypergraphs, with molecules as vertices and concrete reactions as hyperedges. The iterated application of reaction rules to a set of starting molecules generates the network (directed hypergraph) of reachable molecules, i.e., the chemical space defined by the given starting molecules and reaction rules. Figure 6 shows a concrete example of a rule denoting the OPP loss and its application as a matching morphism in a molecule of FPP according to Diagram (1).
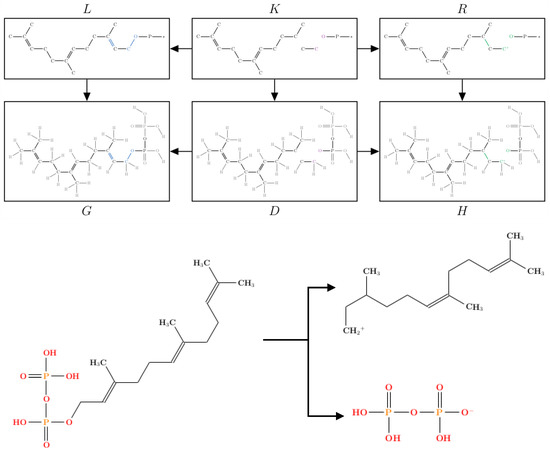
Figure 6.
An example of a graph grammar rule for the OPP loss applied to the FPP molecule. The resulting pathway is a directed hypergraph with the reachable molecules.
Chemical reactions preserve mass, atom types, and charges. Chemical reaction pathways, therefore, form flows in the reaction hypergraph that connect a set of input molecules with a set of output molecules [39,57]. As an immediate consequence, reachability questions in chemical reaction networks translate into the existence of integer flows [26], which is efficiently evaluated by means of Integer Linear Programming (ILP).
4.2. Simulations
Simulations were performed using MedØlDatschgerl (MØD) [25], a software package that combines a DPO graph rewriting engine and an ILP solver to generate and analyze large-scale reaction networks. MØD provides a Python interface (the Python 3 module PyMØD comprising bindings to the underlying library libMØD) as well as a system of generic exploration strategies [58] to guide and restrict the generation process. Graph transformation rules are specified manually in GML format [59]; substrate molecules can be provided either in GML or SMILES [60] format.
We have designed DPO graph transformation rules representing chemical mechanisms involved in the production of the plant sesquiterpenes -caryophyllene, -humulene, and -farnesene from its precursor FPP, exploring the generation of distinct compounds through simulations with varying sets of rules. The presented simulations are available on GitHub. Figure 7 shows the rules employed in Simulation 01, Figure 8 shows the rules employed in Simulation 02, and Figure 9 shows the rules that, together with rules of Figure 7 and Figure 8, were employed in Simulation 03.
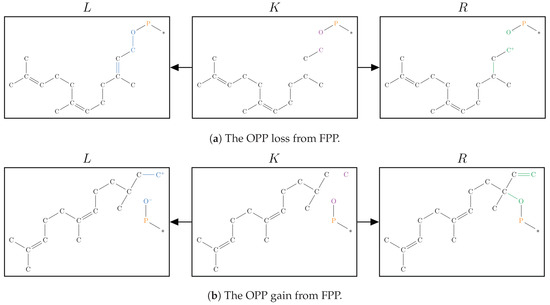
Figure 7.
The graphical representation of the set of graph grammar rules used in Simulation 01.
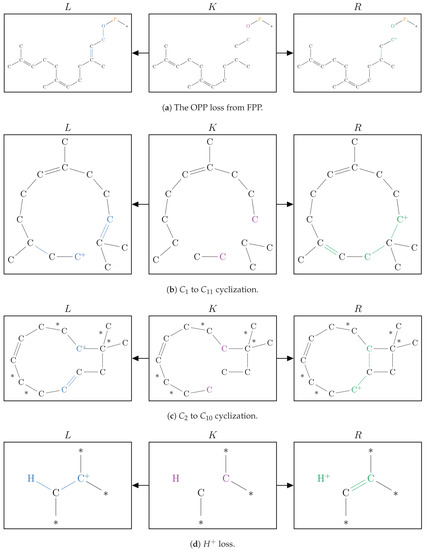
Figure 8.
The graphical representation of the set of graph grammar rules used in Simulation 02.
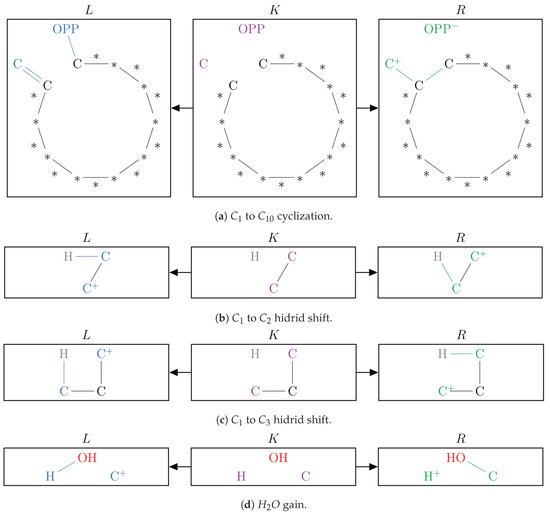
Figure 9.
The graphical representation of the set of graph grammar rules that, together with Rules of Figure 7 and Figure 8, were used in Simulation 03.
4.3. Database Storage
NoSQL Graph Databases are Database Management Systems (DBMS) that can store graphs natively. They have been used in research with biological data, especially in cases where data integration is a determining factor [37]. In this work, we have used the Neo4J graph database to optionally store both the generated sesquiterpenes biosynthesis pathways and the previous data knowledge from Scenarios. The database was named 2Path-Sesquiterpenes, and its schema was modeled using the graphical diagram description called GRAPHED [61]. The modeled schema showed in Figure 10 minimizes the transition from the generated hypergraph and links it to the data from Scenarios. It makes it possible to associate the metadata to the predicted pathways to establish a relationship between the predictions and biological evidence.
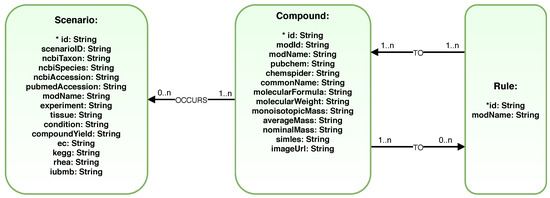
Figure 10.
A 2Path-Sesquiterpenes database scheme: Each Compound can be related to zero or more Rules. Each Rule needs to be related to one or more Compound. Each Compound can be related to zero or more Scenarios.
In the 2Path-Sesquiterpenes database, the nodes labeled as Compound represent the compounds generated during the simulation. The nodes labeled as Reaction represent the directed hyperedges, i.e., a chemical mechanism meant by an application of a graph grammar rule. All relationships between a Compound and a Reaction are directed and labeled as TO. Figure 10 shows all data inside each Node.
The 2Path-Sesquiterpenes database nodes labeled as Scenarios provide a kernel of manually curated biological experimental evidence about constraints under which the compound are produced. The Scenarios provide an NCBI accession number for the enzymes; a PUBMED accession number for the associated publication with the experimental results; the experimental conditions; the plant tissue using EMBL-EBI Plant Ontology (https://www.ebi.ac.uk/ols/ontologies/po); the compound yield; the EC numbers for the reactions; and the cross references to KEGG [49], Rhea [62] and ExploEnz (IUBMB) [63] in a taxonomic range of species. A relationship labeled as OCCURS is created between a node Scenarios and a node Compound when there is a biological scenario supporting the biosynthesis of this predicted compound.
5. Conclusions
Rule-based generative transformation systems, such as the graph grammars used here, provide a mathematically sound way to answer reachability questions in combinatorial, potentially infinite, search spaces. Here, we have used DPO graph transformations, as implemented by MedØlDatschgerl [25] to model the specific combination of cyclizations reactions catalyzed by plant sesquiterpene synthases (STPS) using a small number of transformation rules. The results help to explain the diversity of sesquiterpenes, including common plant sesquiterpenes as -caryophyllene,-humulene, and -farnesene, through combinations of specific cyclization reactions. The generative approach produces a local view of the metabolic or chemical network that is naturally represented as a hypergraph. Reachability then translates to the existence of pathways, which can be decided by integer linear programming. The networks and pathways are exported to a PDF report and optionally can be stored and traversed in a graph database adhering to FAIR Guiding Principles [24]. This makes it possible to integrate into the database the predicted results by simulations with Scenarios, which are experimental evidence on the biosynthesis reactions from the literature. Computational de novo pathway discovery is of particular interest for reactions catalyzed by multi-product enzymes as in the case of STPS because the combinatorial complexity of products in a multistep synthesis quickly exceeds the limits of manual analysis. It also enables a systematic analysis of the synthetic and heterologous biology with potential applications, e.g., in sustainable bioeconomy. The work presented here is intended as a proof of concept. In future work, it will be expanded in several directions. Additional graph grammar rules can be included to extend the space of reachable sesquiterpenes and/or to include other classes of terpenes. Functionalization to a broad array of terpenoids can also be modeled by the generative approach discussed here. On the other hand, we plan to expand the collection of experimental scenarios and to develop a user-friendly interface to facilitate the integration of experimental knowledge and computational predictions.
Author Contributions
Conceptualization, W.M.C.d.S. and C.F.; methodology C.F., J.L.A., M.T.H., M.E.M.T.W., and P.F.S.; software, C.F. and J.L.A.; simulations, W.M.C.d.S.; data curation, J.L.A. and C.F; interpretation of results, C.F., J.L.A., M.M.B., and P.F.S.; writing—first draft W.M.C.d.S.; all authors contributed to writing and editing.
Funding
This research was funded in part by CAPES through a sandwich scholarship to W.M.C.d.S. It was additionally supported in part by the Independent Research Fund Denmark, Natural Sciences, grant DFF-7014-00041.
Acknowledgments
W.M.C.d.S. thanks CAPES for the scholarship and gratefully acknowledges the hospitality of Leipzig and Vienna Universities.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of the data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
The following abbreviations are used in this manuscript:
| AAM | Atom to Atom Mapping |
| AFIR | Artificial Force-Induced Reaction |
| CBM | Constraint-based models |
| DBMS | Database Management Systems |
| DMAPP | Dimethylallyl Pyrophosphate |
| DPO | Double pushout graph rewriting |
| FAIR | Findability, Accessibility, Interoperability, and Reuse of digital assets |
| FBA | Flux Balanced Analysis |
| FPP | Farnesyl Diphosphate |
| GGPP | Geranylgeranyl Diphosphate |
| GML | Graph Modeling Language |
| GRRM | Global Reaction Route Mapping |
| GPP | Geranyl Diphosphate |
| ILP | Integer Linear Programming |
| IPP | Isopentenyl Pyrophosphate |
| IUBMB | International Union of Biochemistry and Molecular Biology |
| MEP | Methylerythritol phosphate pathway |
| MVA | Mevalonate pathway |
| NPP | Nerolidyl Diphosphate |
| NoSQL | Not Only Structured Query Language |
| OPP | Diphosphate |
| Portable Document Format | |
| PGDB | Pathway/Genome database |
| STPS | Sesquiterpene Synthases |
| TPS | Terpene Synthase |
References
- Breitmaier, E. Terpenes: Flavors, Fragrances, Pharmaca, Pheromones; Wiley-VCH: Hoboken, NJ, USA, 2006. [Google Scholar] [CrossRef]
- Cheng, A.X.; Xiang, C.Y.; Li, J.X.; Yang, C.Q.; Hu, W.L.; Wang, L.J.; Lou, Y.G.; Chen, X.Y. The rice (E)-β-caryophyllene synthase (OsTPS3) accounts for the major inducible volatile sesquiterpenes. Phytochemistry 2007, 68, 1632–1641. [Google Scholar] [CrossRef]
- Ružička, L. The isoprene rule and the Biogenesis of terpenic compounds. Cell. Mol. Life Sci. 1953, 9, 357–367. [Google Scholar] [CrossRef]
- Vattekkatte, A.; Garms, S.; Brandt, W.; Boland, W. Enhanced structural diversity in terpenoid biosynthesis: Enzymes, substrates and cofactors. Org. Biomol. Chem. 2018, 16, 348–362. [Google Scholar] [CrossRef]
- Wink, M. Biochemistry of Plant Secondary Metabolism; John Willey & Sons Inc.: Hoboken, NJ, USA, 2010; Volume 40. [Google Scholar]
- Chen, F.; Tholl, D.; Bohlmann, J.; Pichersky, E. The family of terpene synthases in plants: A mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 2011, 66, 212–229. [Google Scholar] [CrossRef]
- Liu, W.; Feng, X.; Zheng, Y.; Huang, C.H.; Nakano, C.; Hoshino, T.; Bogue, S.; Ko, T.P.; Chen, C.-C.; Cui, Y.; et al. Structure, function and inhibition of ent-kaurene synthase from Bradyrhizobium japonicum. Sci. Rep. 2014, 4, 6214. [Google Scholar] [CrossRef]
- Lesburg, C.A. Crystal Structure of Pentalenene Synthase: Mechanistic Insights on Terpenoid Cyclization Reactions in Biology. Science 1997, 277, 1820–1824. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, E.; Lin, F.Y. Terpene biosynthesis: Modularity rules. Angew. Chem. Int. Ed. 2012, 51, 1124–1137. [Google Scholar] [CrossRef] [PubMed]
- Kempinski, C.; Jiang, Z.; Bell, S.; Chappell, J. Metabolic engineering of higher plants and algae for isoprenoid production. Adv. Biochem. Eng. Biotechnol. 2015, 148, 161–199. [Google Scholar]
- Degenhardt, J.; Köllner, T.G.; Gershenzon, J. Monoterpene and sesquiterpene synthases and the origin of terpene skeletal diversity in plants. Phytochemistry 2009, 70, 1621–1637. [Google Scholar] [CrossRef] [PubMed]
- Schifrin, A.; Khatri, Y.; Kirsch, P.; Thiel, V.; Schulz, S.; Bernhardt, R. A single terpene synthase is responsible for a wide variety of sesquiterpenes in Sorangium cellulosum Soce56. Org. Biomol. Chem. 2016, 14, 3385–3393. [Google Scholar] [CrossRef]
- Tholl, D. Terpene synthases and the regulation, diversity and biological roles of terpene metabolism. Curr. Opin. Plant Biol. 2006, 9, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Christianson, D.W. Structural and Chemical Biology of Terpenoid Cyclases. Chem. Rev. 2017, 117, 11570–11648. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Li, G.; Köllner, T.G.; Jia, Q.; Gershenzon, J.; Chen, F. Positive Darwinian selection is a driving force for the diversification of terpenoid biosynthesis in the genus Oryza. BMC Plant Biol. 2014, 14, 239. [Google Scholar] [CrossRef]
- Tholl, D.; Chen, F.; Petri, J.; Gershenzon, J.; Pichersky, E. Two sesquiterpene synthases are responsible for the complex mixture of sesquiterpenes emitted from Arabidopsis flowers. Plant J. 2005, 42, 757–771. [Google Scholar] [CrossRef] [PubMed]
- Kigathi, R.N.; Weisser, W.W.; Reichelt, M.; Gershenzon, J.; Unsicker, S.B. Plant volatile emission depends on the species composition of the neighboring plant community. BMC Plant Biol. 2019, 19, 58. [Google Scholar] [CrossRef] [PubMed]
- Jeske, L.; Placzek, S.; Schomburg, I.; Chang, A.; Schomburg, D. BRENDA in 2019: A European ELIXIR core data resource. Nucleic Acids Res. 2018, 47, D542–D549. [Google Scholar] [CrossRef] [PubMed]
- Karp, P.D.; Latendresse, M.; Caspi, R. The pathway tools pathway prediction algorithm. Stand. Genom. Sci. 2011, 5, 424. [Google Scholar] [CrossRef]
- Karp, P.D.; Billington, R.; Caspi, R.; Fulcher, C.A.; Latendresse, M.; Kothari, A.; Keseler, I.M.; Krummenacker, M.; Midford, P.E.; Ong, Q.; et al. The BioCyc collection of microbial genomes and metabolic pathways. Briefings Bioinf. 2017. [Google Scholar] [CrossRef]
- Djoumbou-Feunang, Y.; Fiamoncini, J.; Gil-de-la Fuente, A.; Greiner, R.; Manach, C.; Wishart, D.S. BioTransformer: A comprehensive computational tool for small molecule metabolism prediction and metabolite identification. J. Cheminform. 2019, 11, 2. [Google Scholar] [CrossRef]
- Duigou, T.; du Lac, M.; Carbonell, P.; Faulon, J.L. RetroRules: A database of reaction rules for engineering biology. Nucleic Acids Res. 2018, 47, D1229–D1235. [Google Scholar] [CrossRef]
- Maeda, S.; Harabuchi, Y.; Takagi, M.; Taketsugu, T.; Morokuma, K. Artificial Force Induced Reaction (AFIR) Method for Exploring Quantum Chemical Potential Energy Surfaces. Chem. Rec. 2016, 16, 2232–2248. [Google Scholar] [CrossRef]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.W.; da Silva Santos, L.B.; Bourne, P.E.; et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef]
- Andersen, J.L.; Flamm, C.; Merkle, D.; Stadler, P.F.; Andersen, J.L.; Flamm, C.; Merkle, D.; Stadler, P.F. A software package for chemically inspired graph transformation. In International Conference on Graph Transformation; Springer: Cham, Switzerland, 2016; pp. 73–88. [Google Scholar]
- Andersen, J.L.; Flamm, C.; Merkle, D.; Stadler, P.F. Chemical Transformation Motifs—Modelling Pathways as Integer Hyperflows. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017, 5963, 510–523. [Google Scholar] [CrossRef]
- Smith, W.D. Computational Complexity of Synthetic Chemistry—Basic Facts; Technical Report; NECI: Princeton, NJ, USA, 1997. [Google Scholar]
- Picaud, S.; Olsson, M.E.; Brodelius, M.; Brodelius, P.E. Cloning, expression, purification and characterization of recombinant (+)-germacrene D synthase from Zingiber officinale. Arch. Biochem. Biophys. 2006, 452, 17–28. [Google Scholar] [CrossRef]
- Farzadfar, S.; Zarinkamar, F.; Behmanesh, M.; Hojati, M. Magnesium and manganese interactively modulate parthenolide accumulation and the antioxidant defense system in the leaves of Tanacetum parthenium. J. Plant Physiol. 2016, 202, 10–20. [Google Scholar] [CrossRef]
- Zhang, F.; Chen, N.; Zhou, J.; Wu, R. Protonation-dependent diphosphate cleavage in FPP cyclases and synthases. ACS Catal. 2016, 6, 6918–6929. [Google Scholar] [CrossRef]
- Cane, D.E.; Iyengar, R. The enzymic conversion of farnesyl to nerolidyl pyrophosphate: Role of the pyrophosphate moiety. J. Am. Chem. Soc. 1979, 101, 3385–3388. [Google Scholar] [CrossRef]
- Kollner, T.G.; Held, M.; Lenk, C.; Hiltpold, I.; Turlings, T.C.; Gershenzon, J.; Degenhardt, J. A Maize (E)-beta-Caryophyllene Synthase Implicated in Indirect Defense Responses against Herbivores Is Not Expressed in Most American Maize Varieties. Plant Cell Online 2008, 20, 482–494. [Google Scholar] [CrossRef]
- Irmisch, S.; Krause, S.T.; Kunert, G.; Gershenzon, J.; Degenhardt, J.; Köllner, T.G. The organ-specific expression of terpene synthase genes contributes to the terpene hydrocarbon composition of chamomile essential oils. BMC Plant Biol. 2012, 12, 84. [Google Scholar] [CrossRef]
- Chen, F. Biosynthesis and Emission of Terpenoid Volatiles from Arabidopsis Flowers. Plant Cell Online 2003, 15, 481–494. [Google Scholar] [CrossRef]
- Yu, F.; Okamto, S.; Nakasone, K.; Adachi, K.; Matsuda, S.; Harada, H.; Misawa, N.; Utsumi, R. Molecular cloning and functional characterization of α-humulene synthase, a possible key enzyme of zerumbone biosynthesis in shampoo ginger (Zingiber zerumbet Smith). Planta 2008, 227, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Brandizi, M.; Singh, A.; Rawlings, C.; Hassani-Pak, K. Towards FAIRer Biological Knowledge Networks Using a Hybrid Linked Data and Graph Database Approach. J. Integr. Bioinform. 2018, 15. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, W.M.; Wercelens, P.; Walter, M.E.M.; Holanda, M.; Brígido, M. Graph Databases in Molecular Biology. In Brazilian Symposium on Bioinformatics; Springer: Cham, Switzerland, 2018; pp. 50–57. [Google Scholar]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2010, 27, 431–432. [Google Scholar] [CrossRef] [PubMed]
- Klamt, S.; Haus, U.U.; Theis, F. Hypergraphs and cellular networks. PLoS Comput. Biol. 2009, 5, e1000385. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Dash, S.; Ng, C.Y.; Maranas, C.D. A review of computational tools for design and reconstruction of metabolic pathways. Synth. Syst. Biotechnol. 2017, 2, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Cherdal, S.; Mouline, S. Modelling and Simulation of Biochemical Processes Using Petri Nets. Processes 2018, 6, 97. [Google Scholar] [CrossRef]
- Blazier, A.S.; Papin, J.A. Integration of expression data in genome-scale metabolic network reconstructions. Front. Physiol. 2012, 3 AUG, 299. [Google Scholar] [CrossRef]
- Øyås, O.; Stelling, J. Genome-scale metabolic networks in time and space. Curr. Opin. Syst. Biol. 2018, 8, 51–58. [Google Scholar] [CrossRef]
- Fang, C.; Fernie, A.R.; Luo, J. Exploring the Diversity of Plant Metabolism. Trends Plant Sci. 2018, 24, 83–98. [Google Scholar] [CrossRef]
- Isegawa, M.; Maeda, S.; Tantillo, D.J.; Morokuma, K. Predicting pathways for terpene formation from first principles–routes to known and new sesquiterpenes. Chem. Sci. 2014, 5, 1555–1560. [Google Scholar] [CrossRef]
- Systems, D.C.I. SMARTS—A Language for Describing Molecular Patterns. 2008. Available online: http://www.daylight.com/dayhtml/doc/theory/theory.smarts.html (accessed on 30 January 2019).
- Systems, D.C.I. A Reaction Transform Language. Available online: http://daylight.com/dayhtml/doc/theory/theory.smirks.html (accessed on 30 January 2019).
- Selkov, E.E.; Goryanin, I.I.; Kaimatchnikov, N.P.; Shevelev, E.L.; Yunus, I.A. Factographic data bank on enzymes and metabolic pathways. Stud. Biophys. 1989, 129, 155–164. [Google Scholar]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Caspi, R.; Billington, R.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Midford, P.E.; Ong, Q.; Ong, W.K.; et al. The MetaCyc Database of metabolic pathways. Nucleic Acids Res. 2014, 42, 471–480. [Google Scholar] [CrossRef]
- Fabregat, A.; Korninger, F.; Viteri, G.; Sidiropoulos, K.; Marin-Garcia, P.; Ping, P.; Wu, G.; Stein, L.; D’Eustachio, P.; Hermjakob, H. Reactome graph database: Efficient access to complex pathway data. PLoS Comput. Biol. 2018, 14, 1–13. [Google Scholar] [CrossRef]
- Holliday, G.L.; Andreini, C.; Fischer, J.D.; Rahman, S.A.; Almonacid, D.E.; Williams, S.T.; Pearson, W.R. MACiE: Exploring the diversity of biochemical reactions. Nucleic Acids Res. 2012, 40, D783–D789. [Google Scholar] [CrossRef]
- Bondy, J.A.; Murty, U.S.R. Graph Theory with Applications; Elsevier Science Publishing Co., Inc.: New York, NY, USA, 1976; Volume 290. [Google Scholar]
- Ehrig, H.; Ehrig, K.; Prange, U.; Taenthzer, G. Fundamentals of Algebraic Graph Transformation; Springer: Berlin, Germany, 2006. [Google Scholar] [CrossRef]
- Löwe, M. Algebraic approach to single-pushout graph transformation. Theory Comput. Sci. 1993, 109, 181–224. [Google Scholar] [CrossRef]
- Andersen, J.L.; Flamm, C.; Merkle, D.; Stadler, P.F. Inferring chemical reaction patterns using rule composition in graph grammars. J. Syst. Chem. 2013, 4, 4. [Google Scholar] [CrossRef]
- Zeigarnik, A.V. On Hypercycles and Hypercircuits in Hypergraphs. In Discrete Mathematical Chemistry; DIMACS Series in Discrete Mathematics and Theoretical Computer Science; Hansen, P., Fowler, P.W., Zheng, M., Eds.; American Mathematical Society: Providence, RI, USA, 2000; Volume 51, pp. 377–383. [Google Scholar]
- Andersen, J.L.; Flamm, C.; Merkle, D.; Stadler, P.F. Generic Strategies for Chemical Space Exploration. Int. J. Comput. Biol. Drug Des. 2014, 7, 225–258. [Google Scholar] [CrossRef]
- Himsolt, M. GML: A Portable Graph File Format; Universität Passau: Passau, Germany, 1997. [Google Scholar]
- Minkiewicz, P.; Iwaniak, A.; Darewicz, M. Annotation of peptide structures using SMILES and other chemical codes-practical solutions. Molecules 2017, 22, 2075. [Google Scholar] [CrossRef]
- Van Erven, G.; Silva, W.; Carvalho, R.; Holanda, M. GRAPHED: A graph description diagram for graph databases. In World Conference on Information Systems and Technologies; Springer: Cham, Switzerland, 2018; pp. 1141–1151. [Google Scholar]
- Alcántara, R.; Axelsen, K.B.; Morgat, A.; Belda, E.; Coudert, E.; Bridge, A.; Cao, H.; De Matos, P.; Ennis, M.; Turner, S.; et al. Rhea—A manually curated resource of biochemical reactions. Nucleic Acids Res. 2011, 40, D754–D760. [Google Scholar] [CrossRef]
- McDonald, A.G.; Boyce, S.; Tipton, K.F. ExplorEnz: The primary source of the IUBMB enzyme list. Nucleic Acids Res. 2008, 37, D593–D597. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).