1. Introduction
Hyperbranched (HB) polymers are specialty polymeric materials, possessing compact architecture, a vast number of end groups that can be functionalized, and specific space inside the molecule. A wide variety of potential applications have been and are being developed [
1]. The HB polymers are macromolecules in between deterministic linear chains and dendrimer structures [
2], and their properties are influenced significantly by their detailed branched architecture. The prediction and control of HB architecture is essential to produce higher quality polymers, which opens up a challenging field for the chemical engineers to develop novel production processes.
Basic chemical reaction engineering textbooks emphasize the importance in clarifying the fundamental chemical behavior in three representative reactor types; batch reactor, plug flow reactor (PFR), and continuous stirred-tank reactor (CSTR) [
3]. Ideally, a PFR is equivalent to a batch reactor by changing the reaction time to the residence time. In this article, the differences in branched architecture formed in a batch reactor and a CSTR are considered.
For the synthesis of HB polymers consisting of tri-branched monomeric units, the two major chemical methods used are step growth polymerization of AB
2-type monomer and self-condensing vinyl polymerization (SCVP). The former is a classical synthetic route originally considered by Flory [
4], but recent development in polymer chemistry has made it possible to change the chemical reactivity of the second B group freely [
5], and has established the chemical control method for the branching frequency.
Figure 1 shows the reaction scheme of the step growth polymerization of AB
2 type monomer, considered in this article. In the figure, T is the terminal unit with both B’s being unreacted, L is the linearly incorporated unit with one of two B’s being reacted, and D is the dendritic unit in which both B’s have reacted. The reaction rate constant,
kT is for the reaction between an A group and a B group in T, while
kL is for the reaction between A and B in L. The reactivity of the second B group is represented by the reactivity ratio,
r defined by:
The magnitude of
r can be changed from 0 to infinity at will [
5] by using appropriate chemical systems. For instance, it is possible to produce HB polymers with 100% degree of branching (DB, the exact definition will be given shortly), but the quasi-linear polymer can also be produced even with
DB = 1, as illustrated earlier [
5]. Process control in combination with chemical control is needed to synthesize well-designed HB polymers.
In this theoretical study, the branched molecular architecture is investigated by using a Monte Carlo (MC) simulation method proposed earlier for a batch reactor [
6,
7] and for a CSTR [
8,
9]. In the MC simulation, the structure of each HB polymer can be investigated, and any desired structural information could be extracted.
Figure 2 shows an example of HB polymer generated in the present MC simulation for a CSTR. In
Figure 2, FP means the focal point unit that possesses an unreacted A group. Note that in the ring-free model employed in this article, there is only one unit in a molecule that bears unreacted A group.
This article is aimed at establishing the most fundamental characteristics of HB polymers formed based on the ideal chemical kinetics, and the non-idealities, such as cyclization and shielding [
10] are not considered. Both cyclization and shielding depend heavily on the 3D architecture, and it is of prime importance to establish the ideal architecture first. As for the cyclization, only one ring per molecule is allowed. Because smaller rings have a better chance of being formed [
11], the effect on the global architecture of large polymers, which is of major interest in this article, may not be significant. On the other hand, the shielding effect depends on the crowding of the 3D architecture, and inferences on crowding could be obtained from the present type of structural investigation.
One of the most simple and fundamental information of the HB architecture is the degree of branching (DB). The DB of an HB polymer was originally defined by [
12]:
where
P is the degree of polymerization (total number of units in a polymer molecule), i.e.,
P =
T +
D +
L. Note that every time the L-type unit is converted to D, the number of T units increases by one, and therefore, the relationship,
T =
D + 1 always holds true for any HB architecture, as long as the ring formation through the intramolecular reaction between the focal point A group and the unreacted B group in the same molecule is neglected.
When there are no L units, such as for the perfect dendron,
DB = 1. With the definition given by Equation (2), however, DB cannot go down to 0 because linear polymer structure always possesses one T unit at its tail. To avoid this problem, at the same time, to make balanced comparison with the HB polymers synthesized via SCVP in which the focal point is always the L-type, the following definition for the DB was proposed [
6].
where FP means the focal point. In this article, DB defined by Equation (3) is used. For the case of the HB polymer shown in
Figure 2,
P = 27,
D = 10, and the FP is the D-type, the DB is calculated to be
DB = (2)(9)/(24) = 0.75. Obviously, the DB of large polymers, i.e., with
P >> 1, is given by
, in either type of definition.
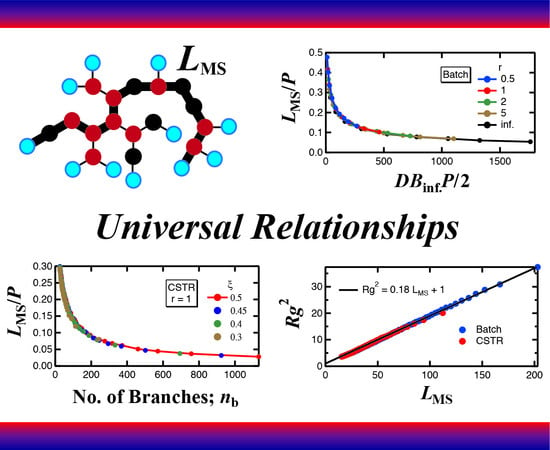
Figure 3 shows the relationship between the values of
DB and
P for batch polymerization with
r = 1 when the conversion of A group is
xA = 0.9. The figure was prepared using the unpublished data obtained in the investigation reported earlier [
6]. In the figure, each red dot shows a pair of values,
DB and
P, for each polymer molecule generated in the MC simulation. The circular symbols with a blue line show the average DB within each intervals of
P, which shows the expected DB for the given
P-value,
. The DB-value converges to
DBinf, as the degree of polymerization
P increases, i.e., for large polymers. On the other hand, the black broken line shows the magnitude of average DB of the whole reaction system. It is clearly shown that the values of DB are distributed around
DBinf, rather than the average DB of the whole system, and
DBinf is larger than the average DB. These characteristics hold true irrespective of the magnitude of reactivity ratio
r, not only for a batch reactor [
7] but also for a CSTR [
8,
9]. In this article, the HB architecture of large polymers, for which
has reached a constant value
DBinf, is investigated in detail, both for a batch reactor and a CSTR.
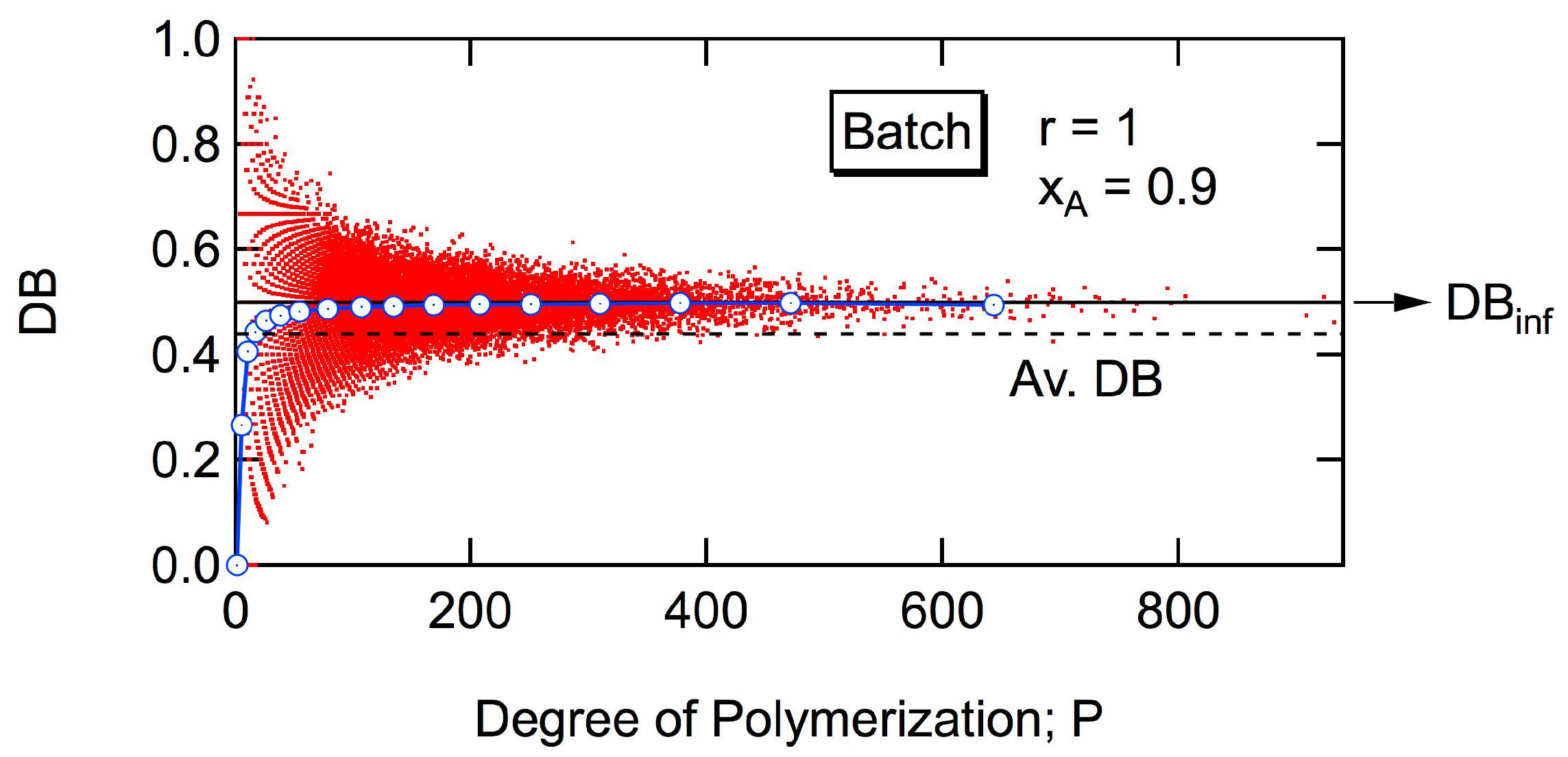
An interesting characteristic of
DBinf is that the magnitude of
DBinf is essentially kept constant, irrespective of the conversion level for a given reactivity ratio
r, both for a batch reactor [
6,
7] and for a CSTR [
8,
9]. Note that the average DB of the whole reaction system increases with conversion, but
DBinf does not change. The value of
DBinf can be estimated by the reactivity ratio
r, as shown in
Figure 4. Obviously,
DBinf = 1 for the cases with
r = ∞, both for batch and CSTR, but the value of
DBinf for a CSTR is always larger than that for a batch reactor, as long as
r is finite. Incidentally, analytic relationship between
DBinf and
r was established previously [
6,
7], and a smooth curve was drawn for batch polymerization in
Figure 4. On the other hand, a general equation for a CSTR has not been reported, and five data points reported in the earlier publication [
9] were plotted and connected.
Based on the structural information shown in
Figure 2, it is possible to determine the 3D size, represented by the mean-square radius of gyration under the unperturbed condition,
. One of the methods used to determine the value of
is the Wiener index (WI) [
13]. The WI is related with
through the following relationship [
14],
where
l is the random walk segment length, and
N is the number of such segments in the polymer molecule.
N is related with
P through
N =
P/
u, where
u is the number of monomeric units in a segment. In the previous investigation [
6,
7,
8,
9], as well as the present article,
u = 1 is used. Define
Rg2 by the following equation,
where
WIu = 1 is the value of WI when
u = 1.
Rg2 is the value of
normalized by the squared monomer-unit length, and is proportional to
, at least for large polymers,
P >> 1. Note that
Rg2 is equal to the value of
u/
l2, which is unchanged irrespective of the magnitude of
u, as long as the number
N of steps is large enough.
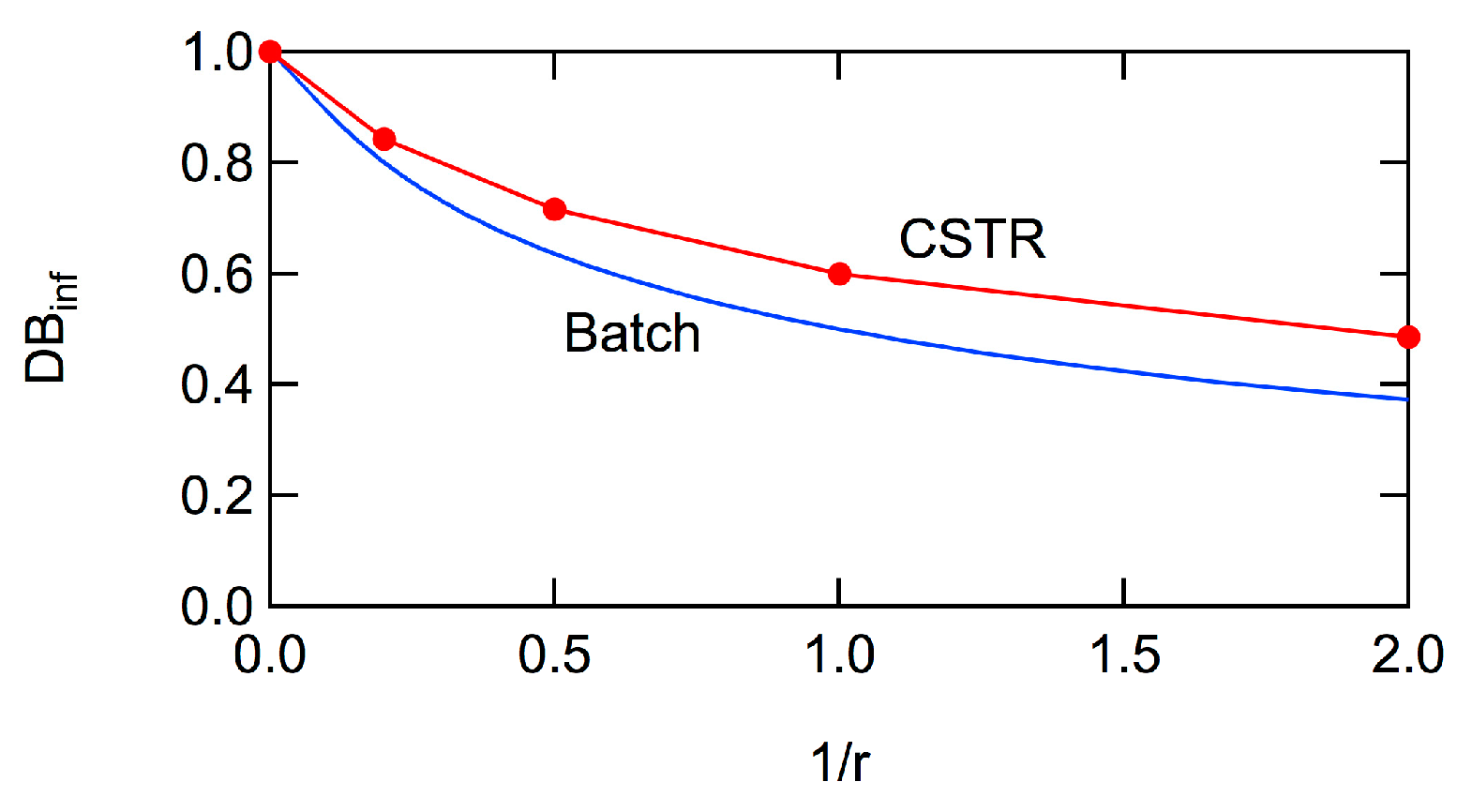
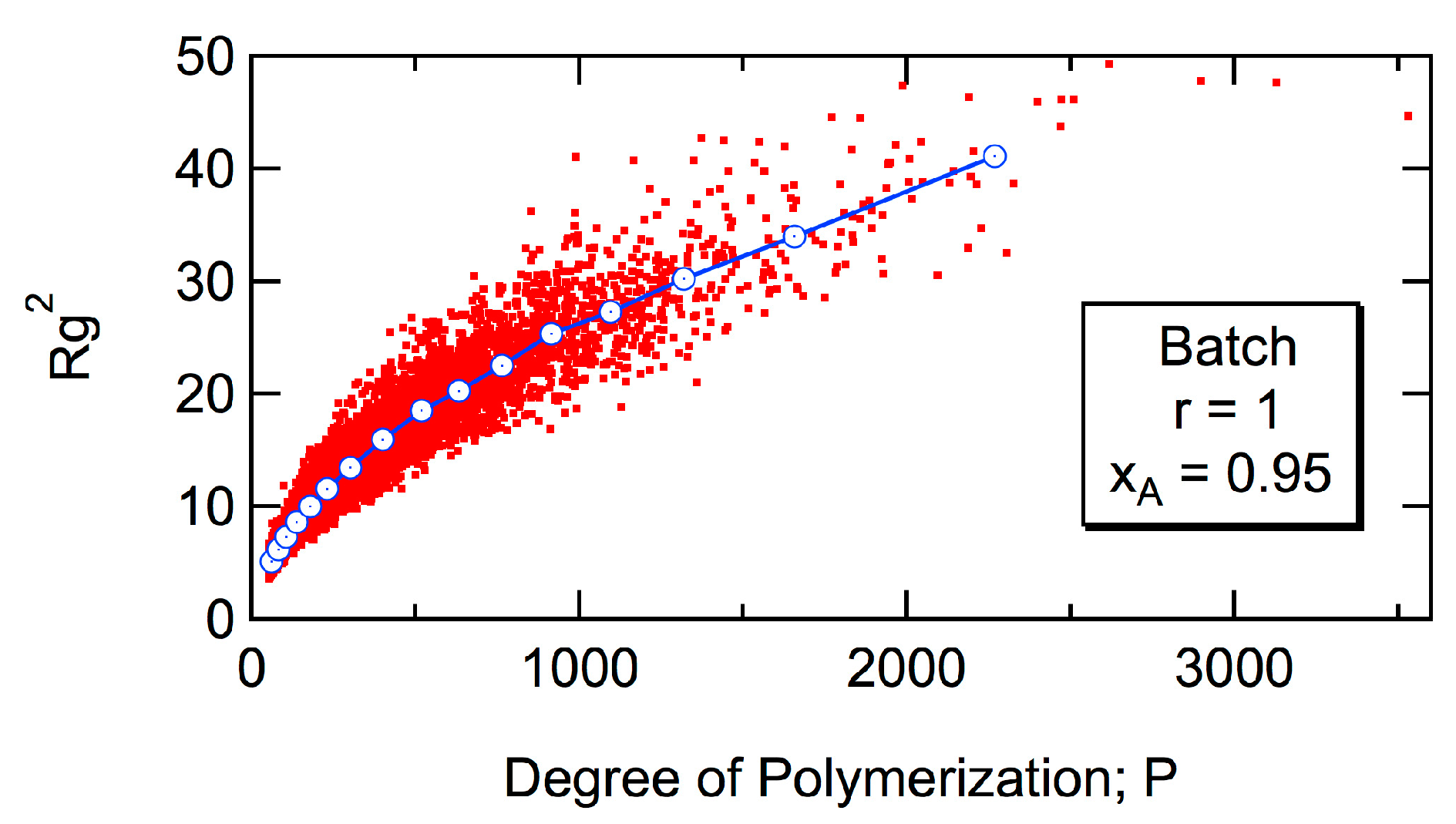
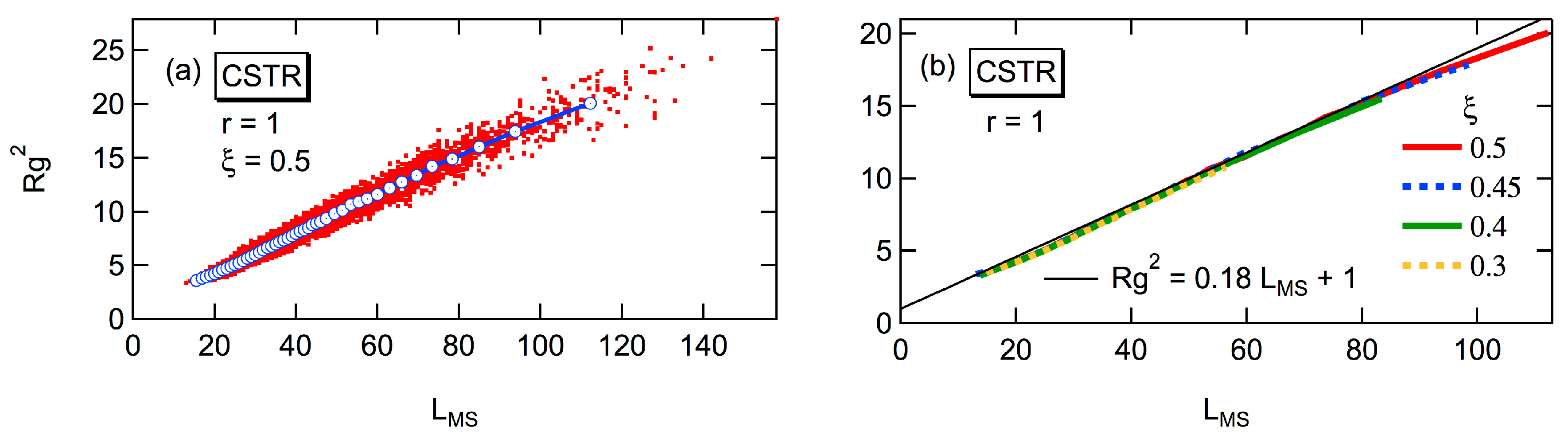
Figure 5 shows the expected
Rg2 of the polymer molecule whose degree of polymerization is
P for batch polymerization with the reactivity ratio,
r = 1. As shown in the figure, the relationship between
Rg2 and
P does not change with the progress of conversion,
xA [
6,
7]. The curve moves to smaller
Rg2, as the reactivity ratio
r is increased [
7]. However, even with
r = ∞ for which
DB = 1,
Rg2 is much larger than that for the perfect dendron [
7], which is shown by the black curve in
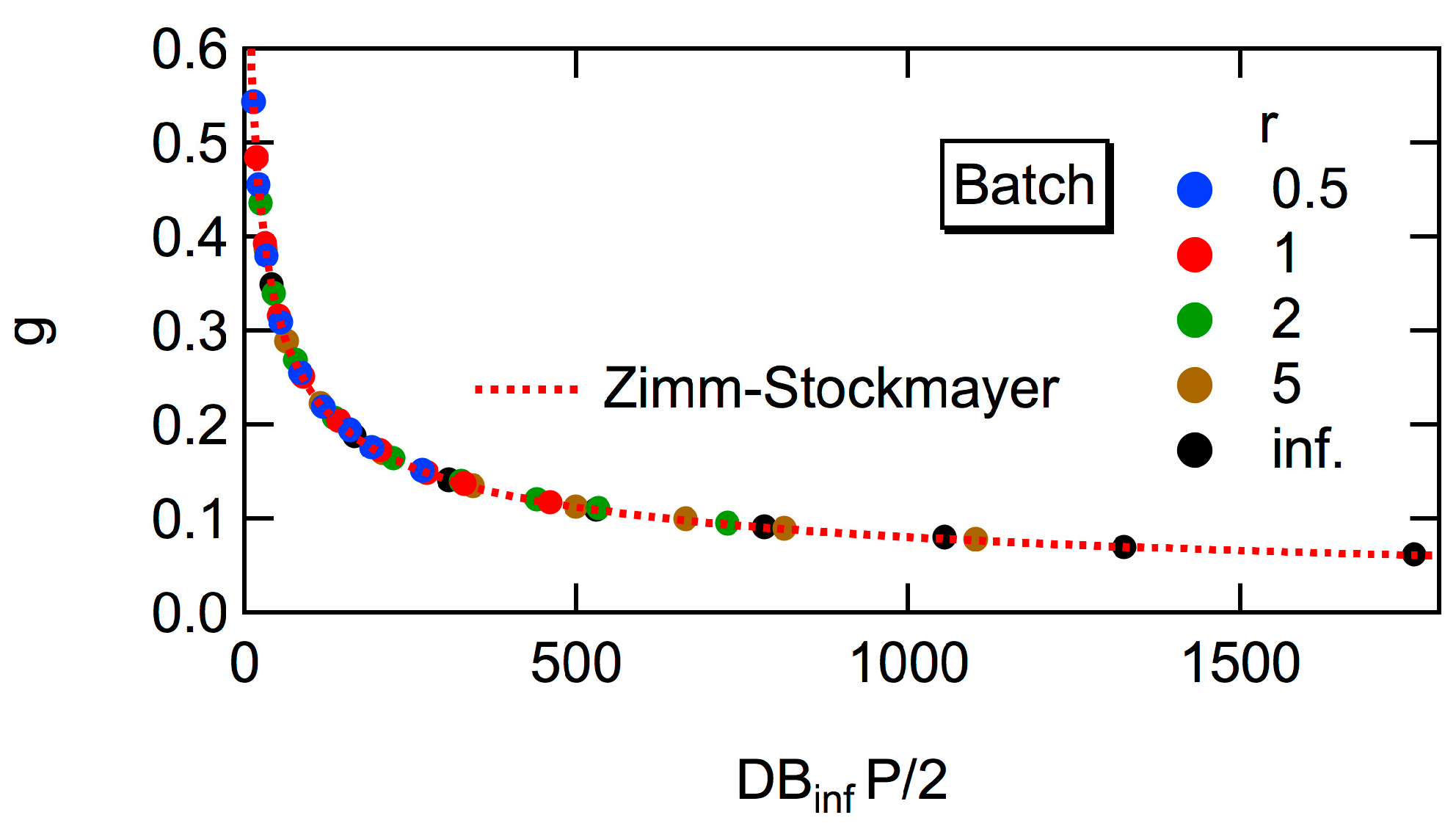
Figure 4. For large polymers, the power law
Rg2 ~
P0.5 is valid, irrespective of the magnitude of
r [
6,
7]. The power exponent, 0.5 is the same as for the random branched polymers, represented by the Zimm-Stockmayer equation [
15]. In this article, the universal relationship concerning
Rg2, that is independent of
r, will be reported, and the relationship with the Zimm-Stockmayer equation will be discussed quantitatively.
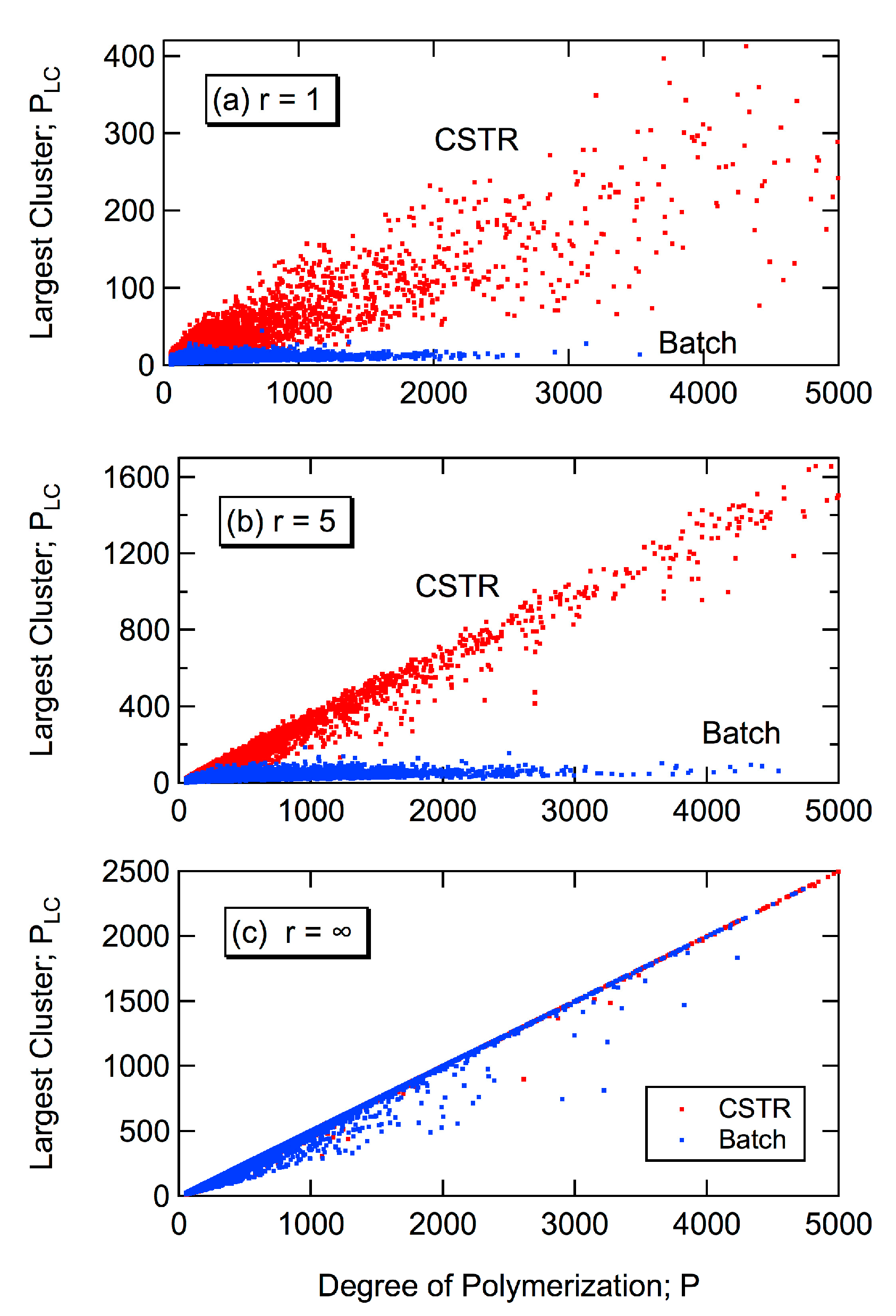
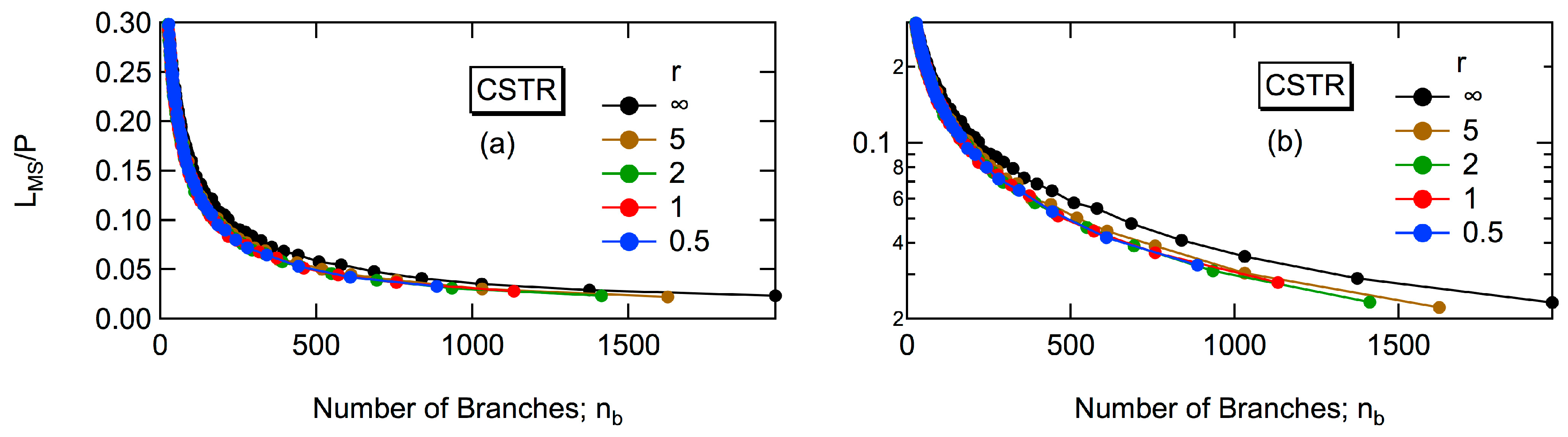
For a CSTR, the variance of
Rg2 for large polymers is quite large. It is not perfectly clear, but the relationship between
Rg2 and
P does not change significantly, even when the steady state conversion level is changed [
8,
9]. It was clearly demonstrated that a CSTR produces polymers with much smaller
Rg2 compared with batch polymerization, even when the value of
DBinf is deliberately set to be the same for both types of reactors [
8]. In this article, the reason for obtaining much more compact architecture in a CSTR is explored by considering the properties of the largest tri-branched cluster in a polymer molecule. The tri-branched clusters are shown by the group of D units surrounded by the red closed frames in
Figure 2, and the largest cluster for this example consists of six units. Note that although the focal point unit is the D type, it is connected to only two other units and it is not considered as a tri-branched unit. A universal relationship concerning
Rg2, independent of the steady state conversion level, will also be reported for a CSTR.
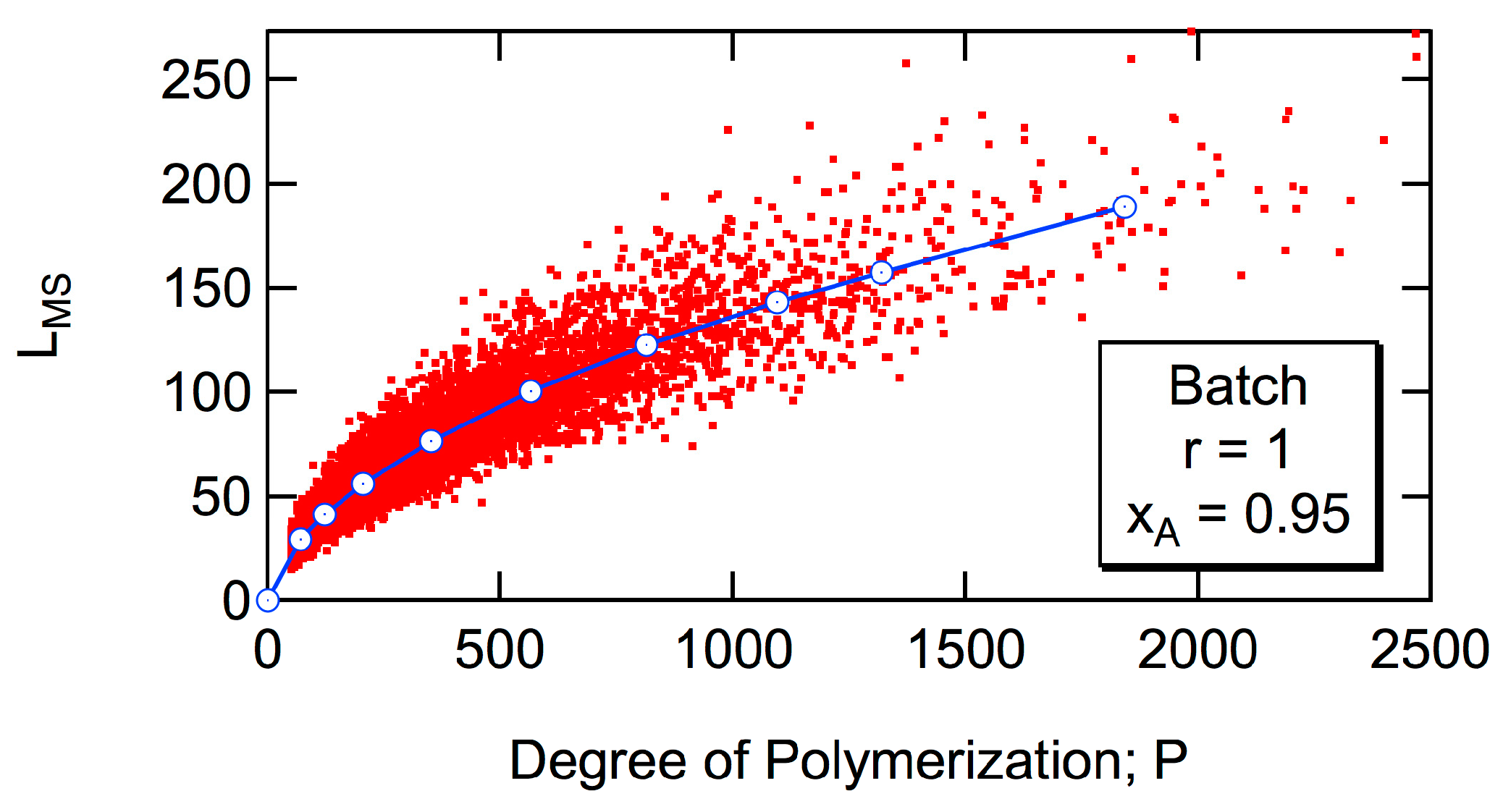
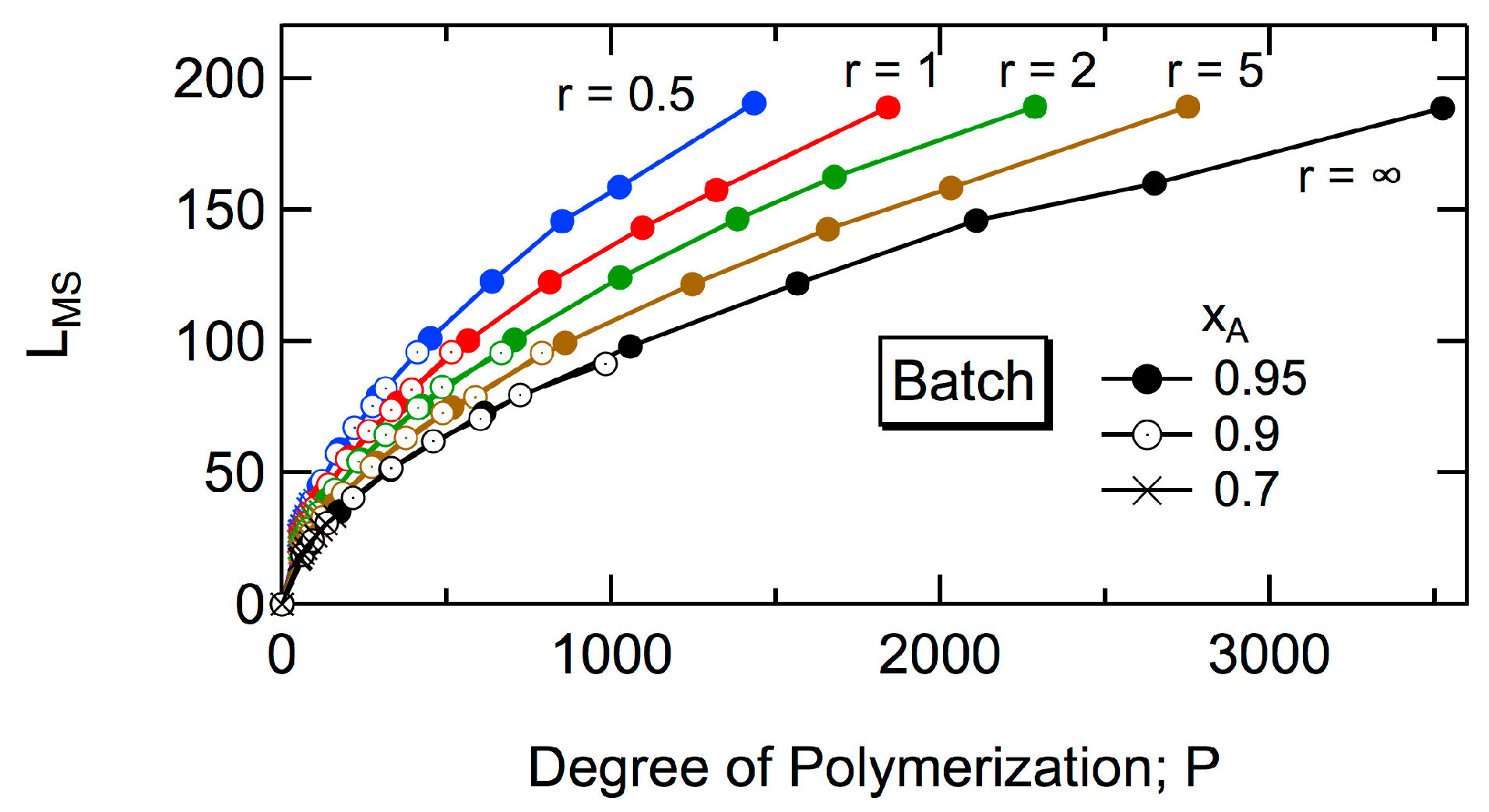
Another structural information investigated in this article is the maximum span length,
LMS. Here, the span length refers to the distance in the monomeric units [
16], and
LMS is equivalent to the longest end-to-end distance [
17]. In the case of HB polymer shown in
Figure 2,
LMS = 12, which is the distance between the units with a star. There are two routes having
LMS = 12, starting from the unit with a star to the unit with star 1 or 2. Interesting universal relationships will be reported for the magnitude of
LMS.
In this article, the HB architecture formed in a batch and a CSTR is compared and discussed, by investigating the properties of the largest tri-branched cluster, and the magnitudes of Rg2 and LMS. For Rg2 and LMS, the universal relationships are sought. Note that the universal relationships reported so far for Rg2 are with respect to the conversion level, and they change with the reactivity ratio r. In this article, further unification is explored.
2. Methods
The MC simulation method proposed earlier for batch polymerization [
6,
7], and that for a CSTR [
8,
9] were used to determine the branched architecture, as shown in
Figure 2. This MC simulation method is based on the random sampling technique [
18,
19], and the polymer molecules were selected from the final product on a weight basis. The whole molecular architecture of each selected molecule was reconstructed by strictly following the history of branched structure formation. By generating a large number of polymer molecules in the simulation, the statistical properties were determined effectively. In the present investigation, the structural properties of large sized polymer molecules are highlighted, and 10
4 polymer molecules with
P > 50 were collected to determine statistically valid estimates. The reactivity ratios investigated were
r = 0.5, 1, 2, 5, and ∞.
For a CSTR, the polymerization behavior at steady state is fully described by the reactivity ratio
r and the dimensionless number
ξ, sometimes referred to as the Damköhler number for the second order reaction, defined by:
where [A]
0 is the initial concentration of A group, or equivalently, the initial monomer concentration, and
is the mean residence time [
3]. The HB polymer shown in
Figure 2 was generated for a CSTR with the condition,
r = 2 and
ξ = 0.35. The conversion of A group,
xA increases with
ξ. To set the value of
ξ corresponds to fixing the steady state conversion level,
xA.
For a CSTR, it was found that the weight-average molecular weight cannot reach the steady state for large
cases [
8,
9], and similar behavior was also reported for the SCVP [
20,
21] which is another route to synthesize HB polymers. The upper limit
ξ-values above which the weight-average molecular weight cannot reach the steady state for a given reactivity ratio
r was shown graphically in the earlier publication [
9]. For example, the upper limit value of
ξ is
ξUL = 0.5 for
r = 1, and
ξUL = 0.25 for
r = ∞. In the present investigation, the MC simulations were conducted for the cases with
ξ ≤
ξUL.
The WI was calculated for each polymer molecule generated in the MC simulation, by setting up the distance matrix, {
dij}, where
dij is the distance in the number of monomeric units between the
ith and
jth unit. The WI when
u = 1 is given by [
13,
14]:
The maximum span length,
LMS was determined by finding the largest value of
dij in the distance matrix.
The statistical properties of various types of clusters in HB polymers were determined from the structural information, shown in
Figure 2.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
























