Metabolic Modeling of Clostridium difficile Associated Dysbiosis of the Gut Microbiota

Abstract
1. Introduction
2. Results
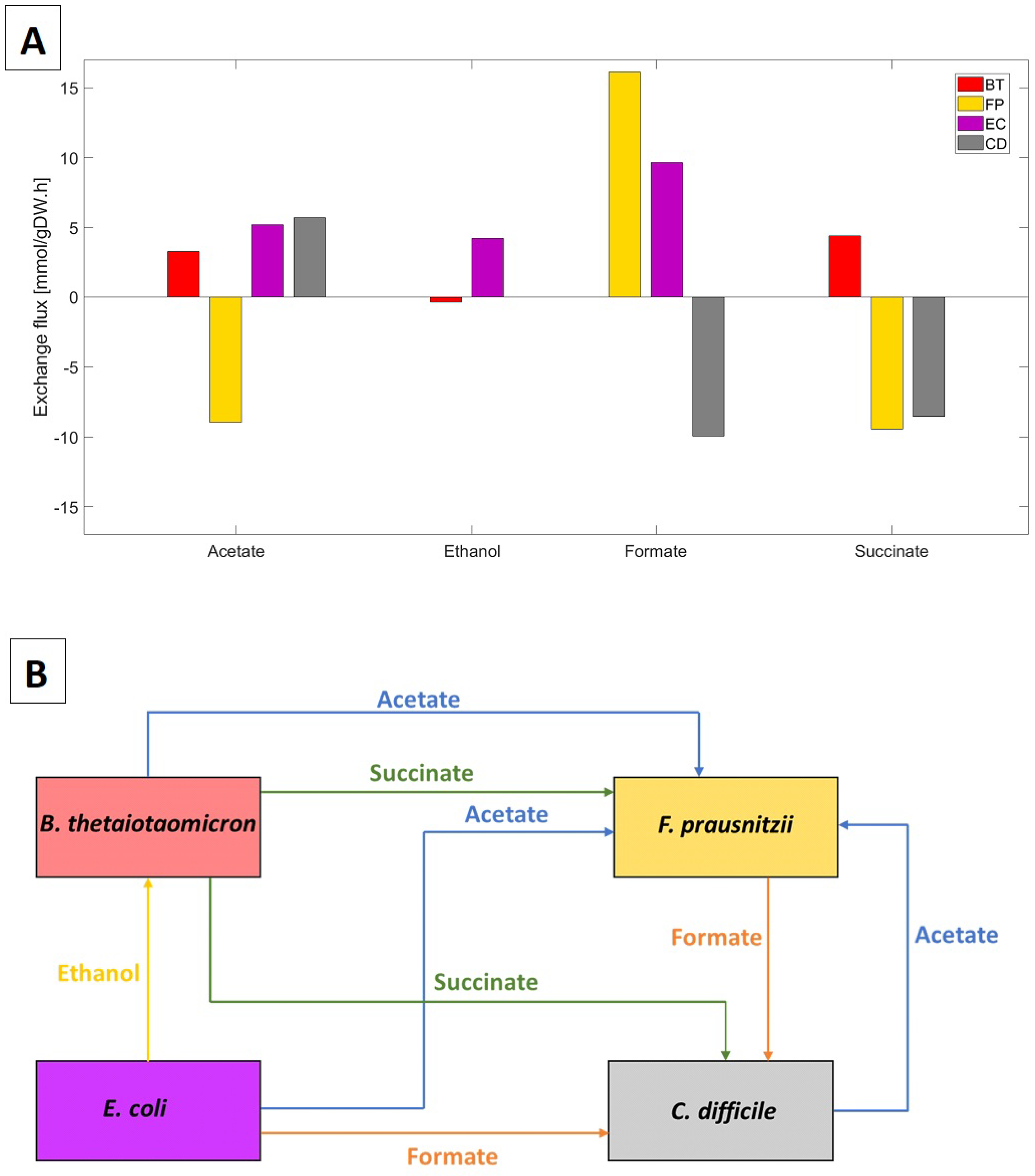
2.1. Discovery of Putative Byproduct Cross-Feeding Relationships
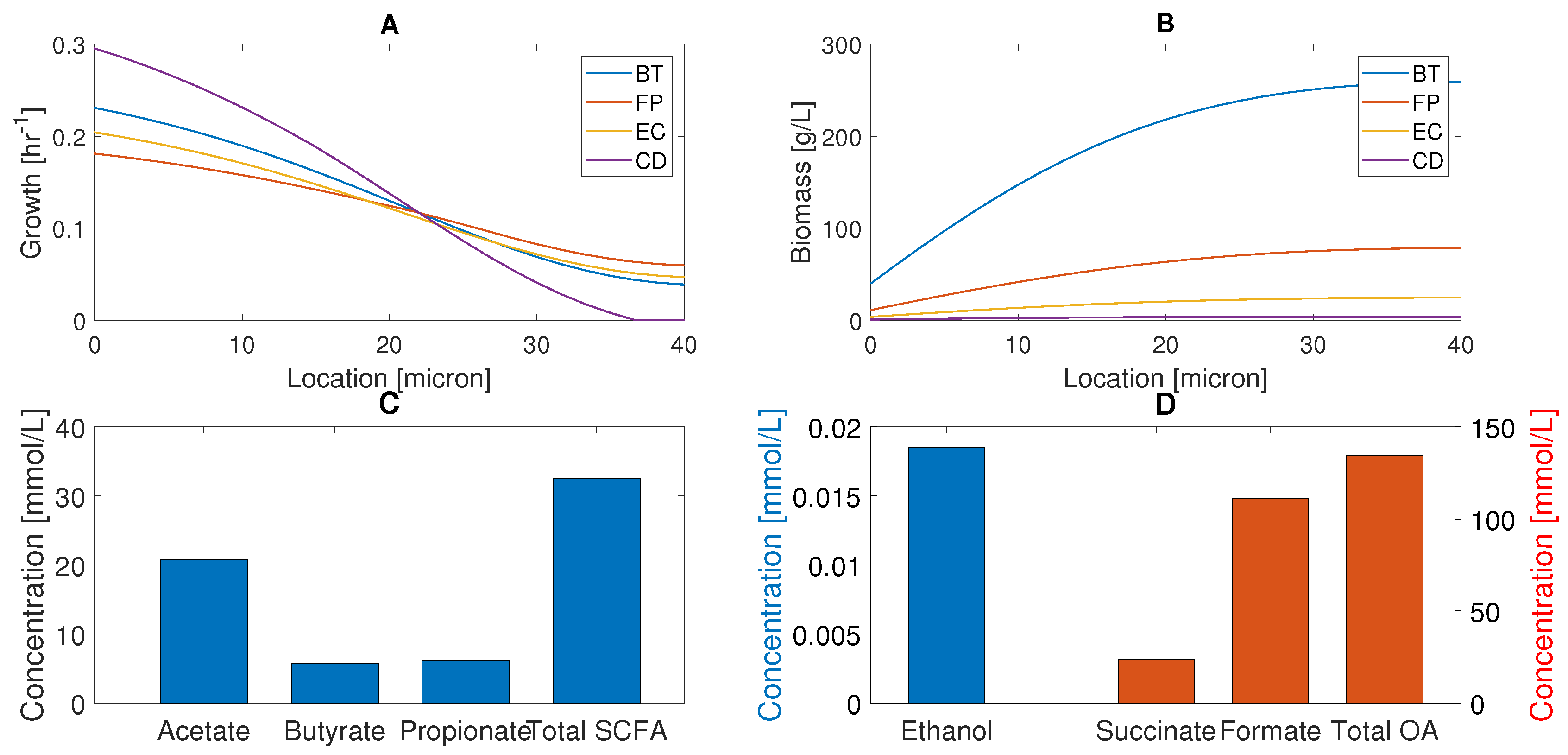
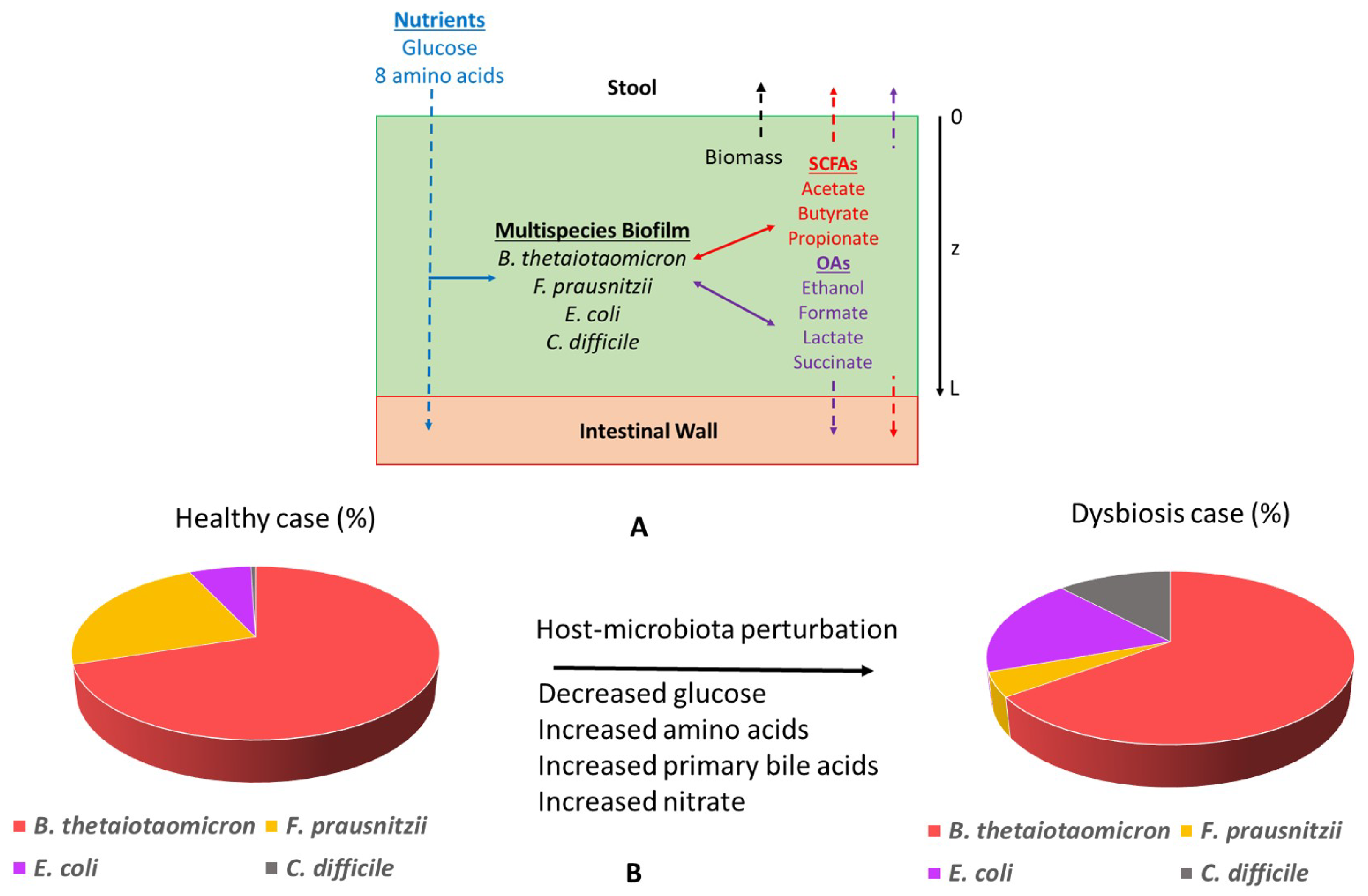
2.2. Characterization of Healthy Gut Microbiota
2.3. Glucose and Amino Acid Perturbations
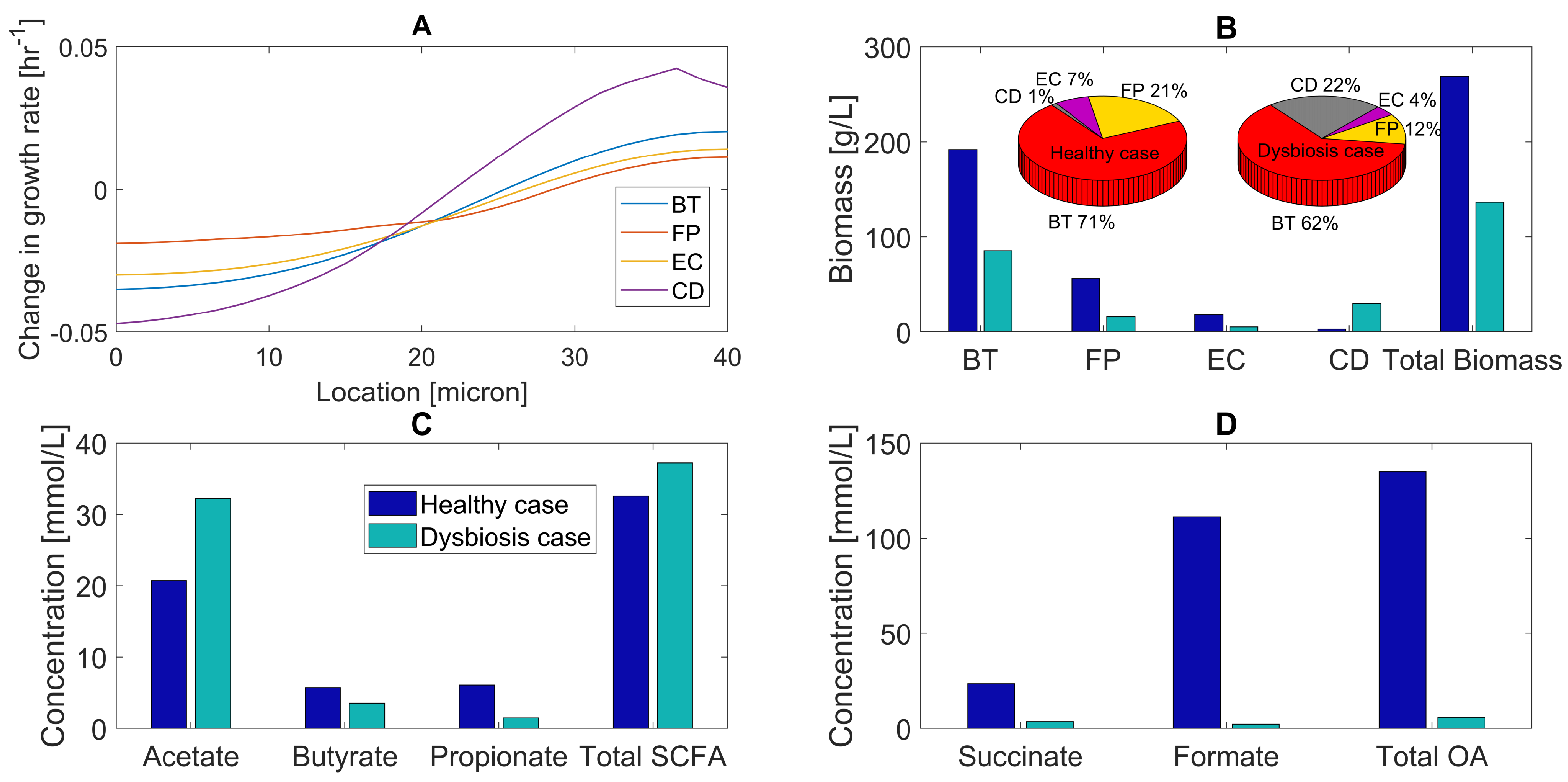
2.4. Primary Bile Acid Perturbations
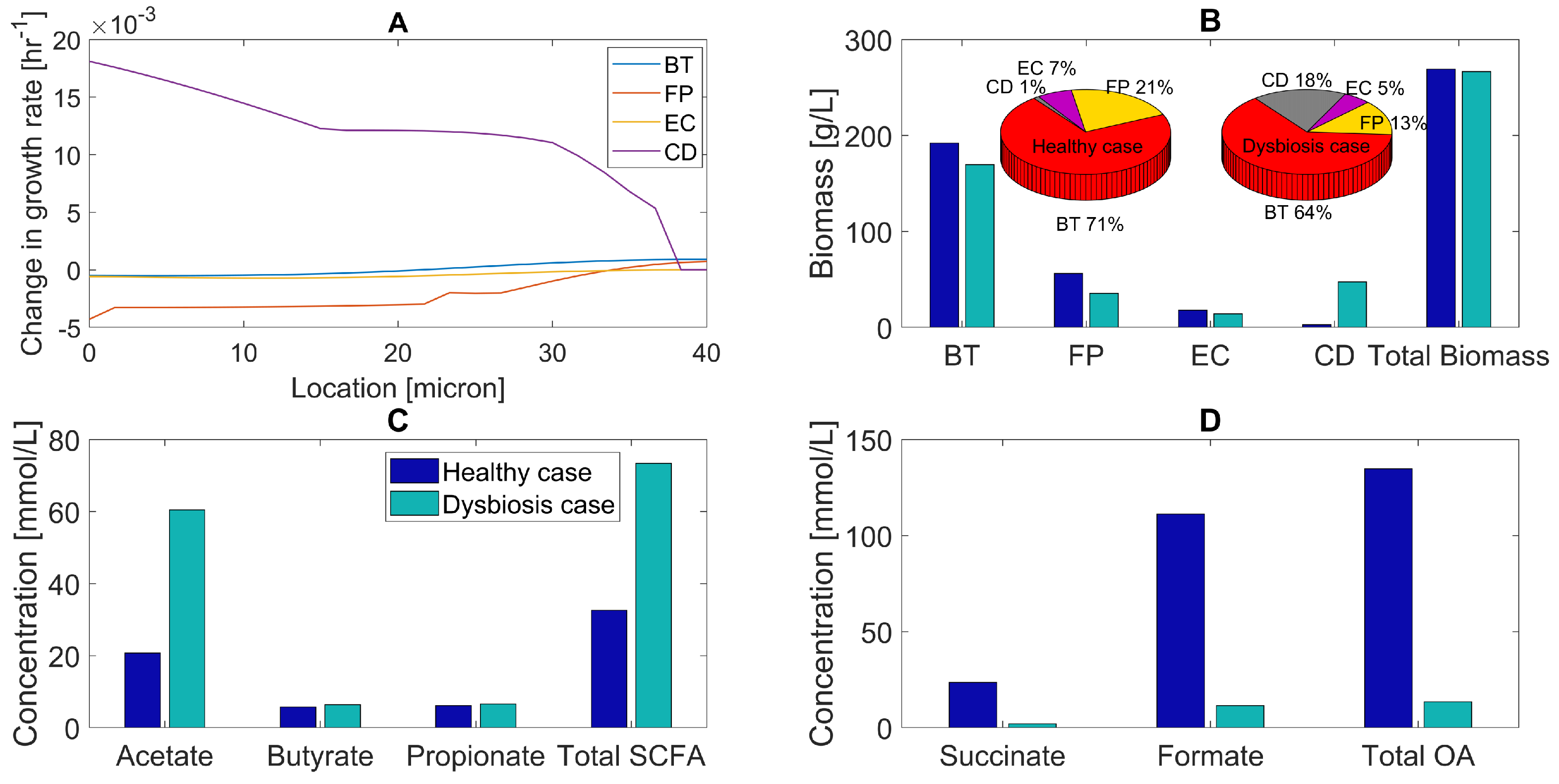
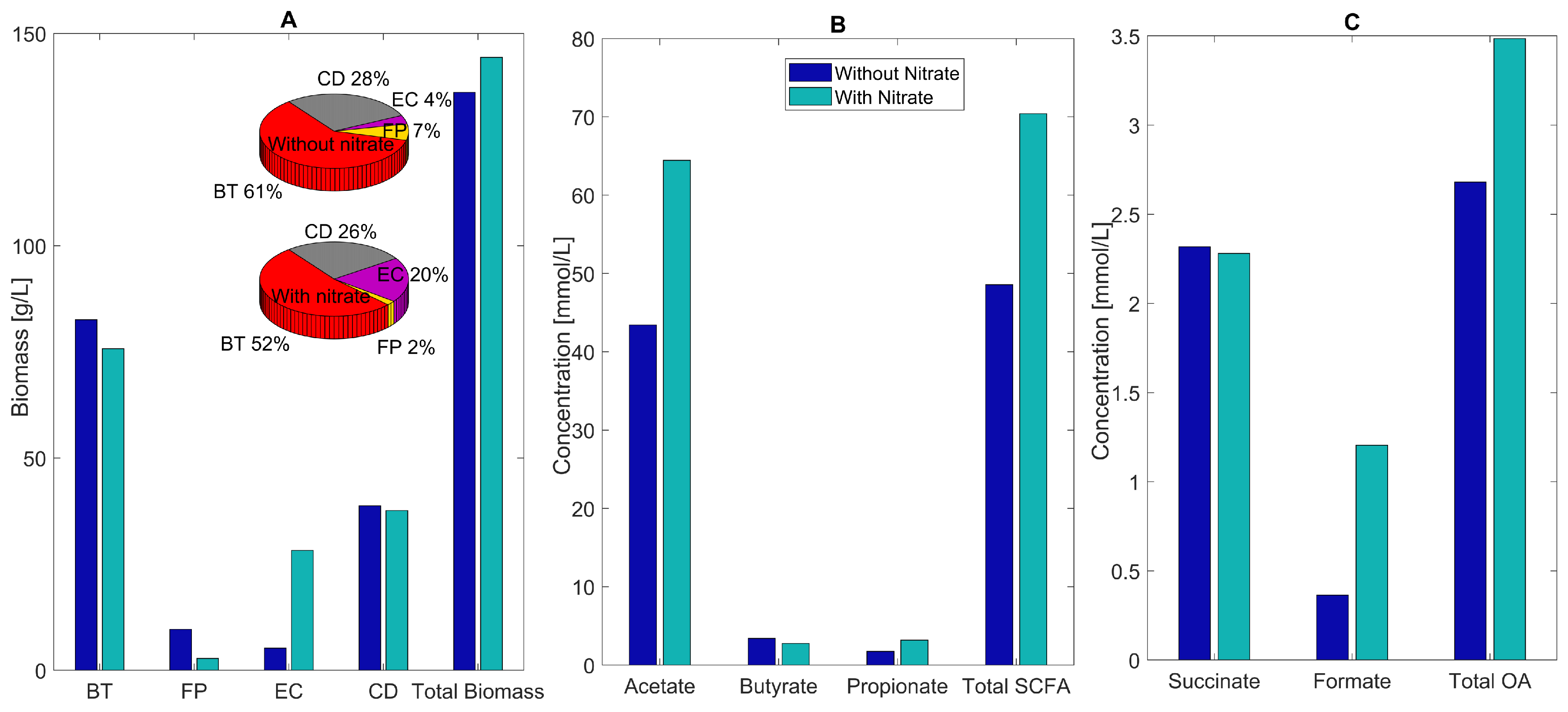
2.5. Host-Derived Nitrate Perturbations
3. Discussion
4. Materials and Methods
4.1. Biofilm Model Formulation and Solution
4.2. Biofilm Model Parameterization and Tuning
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ATPM | ATP maintenance |
| BT | Bacteroides thetaiotaomicron |
| CD | Clostridium difficile |
| CDI | Clostridium difficile infection |
| DFBAlab | Dynamic flux balance analysis laboratory |
| EC | Escherichia coli |
| FBA | Flux balance analysis |
| FP | Faecalibacterium prausnitzii |
| IBD | Inflammatory bowel diseases |
| LP | Linear program |
| MDM | Minimal defined media |
| OA | Organic acid |
| ODE | Ordinary differential equation |
| PDE | Partial differential equation |
| SCFA | Short chain fatty acid |
References
- Gill, S.R.; Pop, M.; DeBoy, R.T.; Eckburg, P.B.; Turnbaugh, P.J.; Samuel, B.S.; Gordon, J.I.; Relman, D.A.; Fraser-Liggett, C.M.; Nelson, K.E. Metagenomic analysis of the human distal gut microbiome. Science 2006, 312, 1355–1359. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-bacterial mutualism in the human intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, A.J.; Harris, N.L. Interactions between commensal intestinal bacteria and the immune system. Nat. Rev. Immunol. 2004, 4, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Bäumler, A.J.; Sperandio, V. Interactions between the microbiota and pathogenic bacteria in the gut. Nature 2016, 535, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, S.; Steed, H.; Macfarlane, G.T. Intestinal bacteria and inflammatory bowel disease. Crit. Rev. Clin. Lab. Sci. 2009, 46, 25–54. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Rajilić-Stojanović, M.; Smidt, H.; De Vos, W.M. Diversity of the human gastrointestinal tract microbiota revisited. Environ. Microbiol. 2007, 9, 2125–2136. [Google Scholar] [CrossRef]
- Zoetendal, E.; Rajilić-Stojanović, M.; De Vos, W. High-throughput diversity and functionality analysis of the gastrointestinal tract microbiota. Gut 2008, 57, 1605–1615. [Google Scholar] [CrossRef]
- Gerritsen, J.; Smidt, H.; Rijkers, G.T.; Vos, W.M. Intestinal microbiota in human health and disease: The impact of probiotics. Genes Nutr. 2011, 6, 209. [Google Scholar] [CrossRef]
- Young, V.B.; Schmidt, T.M. Antibiotic-associated diarrhea accompanied by large-scale alterations in the composition of the fecal microbiota. J. Clin. Microbiol. 2004, 42, 1203–1206. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.; DeCoffe, D.; Molcan, E.; Gibson, D.L. Diet-induced dysbiosis of the intestinal microbiota and the effects on immunity and disease. Nutrients 2012, 4, 1095–1119. [Google Scholar] [CrossRef] [PubMed]
- Anitha, M.; Reichardt, F.; Tabatabavakili, S.; Nezami, B.G.; Chassaing, B.; Mwangi, S.; Vijay-Kumar, M.; Gewirtz, A.; Srinivasan, S. Intestinal dysbiosis contributes to the delayed gastrointestinal transit in high-fat diet fed mice. CMGH Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Alou, M.T.; Lagier, J.C.; Raoult, D. Diet influence on the gut microbiota and dysbiosis related to nutritional disorders. Hum. Microb. J. 2016, 1, 3–11. [Google Scholar] [CrossRef]
- Agus, A.; Denizot, J.; Thévenot, J.; Martinez-Medina, M.; Massier, S.; Sauvanet, P.; Bernalier-Donadille, A.; Denis, S.; Hofman, P.; Bonnet, R.; et al. Western diet induces a shift in microbiota composition enhancing susceptibility to Adherent-Invasive E. coli infection and intestinal inflammation. Sci. Rep. 2016, 6, 19032. [Google Scholar] [CrossRef] [PubMed]
- Theriot, C.M.; Bowman, A.A.; Young, V.B. Antibiotic-induced alterations of the gut microbiota alter secondary bile acid production and allow for Clostridium difficile spore germination and outgrowth in the large intestine. MSphere 2016, 1, e00045-15. [Google Scholar] [CrossRef]
- Winter, S.E.; Winter, M.G.; Xavier, M.N.; Thiennimitr, P.; Poon, V.; Keestra, A.M.; Laughlin, R.C.; Gomez, G.; Wu, J.; Lawhon, S.D.; et al. Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science 2013, 339, 708–711. [Google Scholar] [CrossRef]
- Frank, D.N.; Amand, A.L.S.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef] [PubMed]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015, 26, 26191. [Google Scholar] [CrossRef]
- Surawicz, C.M.; Brandt, L.J.; Binion, D.G.; Ananthakrishnan, A.N.; Curry, S.R.; Gilligan, P.H.; McFarland, L.V.; Mellow, M.; Zuckerbraun, B.S. Guidelines for diagnosis, treatment, and prevention of Clostridium difficile infections. Am. J. Gastroenterol. 2013, 108, 478. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.K.; Johnson, S.; Samore, M.H.; Bliss, D.Z.; Gerding, D.N. Primary symptomless colonisation by Clostridium difficile and decreased risk of subsequent diarrhoea. Lancet 1998, 351, 633–636. [Google Scholar] [CrossRef]
- Kyne, L.; Warny, M.; Qamar, A.; Kelly, C.P. Asymptomatic carriage of Clostridium difficile and serum levels of IgG antibody against toxin A. N. Engl. J. Med. 2000, 342, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Barbut, F.; Petit, J.C. Epidemiology of Clostridium difficile-associated infections. Clin. Microbiol. Infect. 2001, 7, 405–410. [Google Scholar] [CrossRef]
- Kato, H.; Kita, H.; Karasawa, T.; Maegawa, T.; Koino, Y.; Takakuwa, H.; Saikai, T.; Kobayashi, K.; Yamagishi, T.; Nakamura, S. Colonisation and transmission of Clostridium difficile in healthy individuals examined by PCR ribotyping and pulsed-field gel electrophoresis. J. Med. Microbiol. 2001, 50, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, E.; Kato, H.; Kita, H.; Karasawa, T.; Maegawa, T.; Koino, Y.; Matsumoto, K.; Takada, T.; Nomoto, K.; Tanaka, R.; et al. Clostridium difficile colonization in healthy adults: transient colonization and correlation with enterococcal colonization. J. Med. Microbiol. 2004, 53, 167–172. [Google Scholar] [CrossRef]
- Poutanen, S.M.; Simor, A.E. Clostridium difficile-associated diarrhea in adults. Can. Med. Assoc. J. 2004, 171, 51–58. [Google Scholar] [CrossRef]
- Furuya-Kanamori, L.; Marquess, J.; Yakob, L.; Riley, T.V.; Paterson, D.L.; Foster, N.F.; Huber, C.A.; Clements, A.C. Asymptomatic Clostridium difficile colonization: epidemiology and clinical implications. BMC Infect. Dis. 2015, 15, 516. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.Y.; Antonopoulos, D.A.; Kalra, A.; Tonelli, A.; Khalife, W.T.; Schmidt, T.M.; Young, V.B. Decreased diversity of the fecal microbiome in recurrent Clostridium difficile-associated diarrhea. J. Infect. Dis. 2008, 197, 435–438. [Google Scholar] [CrossRef]
- Bartlett, J.G. Clostridium difficile: History of its role as an enteric pathogen and the current state of knowledge about the organism. Clin. Infect. Dis. 1994, 18, S265–S272. [Google Scholar] [CrossRef]
- Pérez-Cobas, A.E.; Moya, A.; Gosalbes, M.J.; Latorre, A. Colonization resistance of the gut microbiota against Clostridium difficile. Antibiotics 2015, 4, 337–357. [Google Scholar] [CrossRef] [PubMed]
- Pothoulakis, C. Pathogenesis of Clostridium difficile-associated diarrhoea. Eur. J. Gastroenterol. Hepatol. 1996, 8, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Theriot, C.M.; Young, V.B. Interactions between the gastrointestinal microbiome and Clostridium difficile. Annu. Rev. Microbiol. 2015, 69, 445–461. [Google Scholar] [CrossRef] [PubMed]
- Mylonakis, E.; Ryan, E.T.; Calderwood, S.B. Clostridium difficile-associated diarrhea: A review. Arch. Intern. Med. 2001, 161, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Jarrad, A.M.; Karoli, T.; Blaskovich, M.A.; Lyras, D.; Cooper, M.A. Clostridium difficile drug pipeline: Challenges in discovery and development of new agents. J. Med. Chem. 2015, 58, 5164–5185. [Google Scholar] [CrossRef] [PubMed]
- Lessa, F.C.; Mu, Y.; Bamberg, W.M.; Beldavs, Z.G.; Dumyati, G.K.; Dunn, J.R.; Farley, M.M.; Holzbauer, S.M.; Meek, J.I.; Phipps, E.C.; et al. Burden of Clostridium difficile infection in the United States. N. Engl. J. Med. 2015, 372, 825–834. [Google Scholar] [CrossRef]
- Dubberke, E.R.; Olsen, M.A. Burden of Clostridium difficile on the healthcare system. Clin. Infect. Dis. 2012, 55, S88–S92. [Google Scholar] [CrossRef]
- Dawson, L.F.; Valiente, E.; Faulds-Pain, A.; Donahue, E.H.; Wren, B.W. Characterisation of Clostridium difficile biofilm formation, a role for Spo0A. PLoS ONE 2012, 7, e50527. [Google Scholar] [CrossRef]
- Dhapa, T.; Leuzzi, R.; Ng, Y.K.; Baban, S.T.; Adamo, R.; Kuehne, S.A.; Scarselli, M.; Minton, N.P.; Serruto, D.; Unnikrishnan, M. Multiple factors modulate biofilm formation by the anaerobic pathogen Clostridium difficile. J. Bacteriol. 2013, 195, 545–555. [Google Scholar]
- Donelli, G.; Vuotto, C.; Cardines, R.; Mastrantonio, P. Biofilm-growing intestinal anaerobic bacteria. FEMS Immunol. Med. Microbiol. 2012, 65, 318–325. [Google Scholar] [CrossRef]
- Semenyuk, E.G.; Laning, M.L.; Foley, J.; Johnston, P.F.; Knight, K.L.; Gerding, D.N.; Driks, A. Spore formation and toxin production in Clostridium difficile biofilms. PLoS ONE 2014, 9, e87757. [Google Scholar] [CrossRef] [PubMed]
- Swidsinski, A.; Weber, J.; Loening-Baucke, V.; Hale, L.P.; Lochs, H. Spatial organization and composition of the mucosal flora in patients with inflammatory bowel disease. J. Clin. Microbiol. 2005, 43, 3380–3389. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, S.; Dillon, J. Microbial biofilms in the human gastrointestinal tract. J. Appl. Microbiol. 2007, 102, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Costerton, J.W.; Stewart, P.S.; Greenberg, E.P. Bacterial biofilms: A common cause of persistent infections. Science 1999, 284, 1318–1322. [Google Scholar] [CrossRef] [PubMed]
- Anderl, J.N.; Franklin, M.J.; Stewart, P.S. Role of antibiotic penetration limitation in Klebsiella pneumoniae biofilm resistance to ampicillin and ciprofloxacin. Antimicrob. Agents Chemother. 2000, 44, 1818–1824. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.S.; Costerton, J.W. Antibiotic resistance of bacteria in biofilms. Lancet 2001, 358, 135–138. [Google Scholar] [CrossRef]
- Stewart, P.S. Mechanisms of antibiotic resistance in bacterial biofilms. Int. J. Med. Microbiol. 2002, 292, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Zuroff, T.R.; Bernstein, H.; Lloyd-Randolfi, J.; Jimenez-Taracido, L.; Stewart, P.S.; Carlson, R.P. Robustness analysis of culturing perturbations on Escherichia coli colony biofilm beta-lactam and aminoglycoside antibiotic tolerance. BMC Microbiol. 2010, 10, 185. [Google Scholar] [CrossRef] [PubMed]
- Shreiner, A.B.; Kao, J.Y.; Young, V.B. The gut microbiome in health and in disease. Curr. Opin. Gastroenterol. 2015, 31, 69. [Google Scholar] [CrossRef] [PubMed]
- Henson, M.A.; Phalak, P. Byproduct Cross Feeding and Community Stability in an In Silico Biofilm Model of the Gut Microbiome. Processes 2017, 5, 13. [Google Scholar] [CrossRef]
- Henson, M.A.; Phalak, P. Microbiota dysbiosis in inflammatory bowel diseases: in silico investigation of the oxygen hypothesis. BMC Syst. Biol. 2017, 11, 145. [Google Scholar] [CrossRef] [PubMed]
- Rivière, A.; Selak, M.; Lantin, D.; Leroy, F.; De Vuyst, L. Bifidobacteria and butyrate-producing colon bacteria: Importance and strategies for their stimulation in the human gut. Front. Microbiol. 2016, 7, 979. [Google Scholar] [CrossRef] [PubMed]
- Ríos-Covián, D.; Ruas-Madiedo, P.; Margolles, A.; Gueimonde, M.; de los Reyes-Gavilán, C.G.; Salazar, N. Intestinal short chain fatty acids and their link with diet and human health. Front. Microbiol. 2016, 7, 185. [Google Scholar] [CrossRef] [PubMed]
- Jakobsdottir, G.; Xu, J.; Molin, G.; Ahrne, S.; Nyman, M. High-fat diet reduces the formation of butyrate, but increases succinate, inflammation, liver fat and cholesterol in rats, while dietary fibre counteracts these effects. PLoS ONE 2013, 8, e80476. [Google Scholar] [CrossRef] [PubMed]
- Ferreyra, J.A.; Wu, K.J.; Hryckowian, A.J.; Bouley, D.M.; Weimer, B.C.; Sonnenburg, J.L. Gut microbiota- produced succinate promotes C. difficile infection after antibiotic treatment or motility disturbance. Cell Host Microbe 2014, 16, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Köpke, M.; Straub, M.; Dürre, P. Clostridium difficile is an autotrophic bacterial pathogen. PLoS ONE 2013, 8, e62157. [Google Scholar] [CrossRef] [PubMed]
- Spor, A.; Koren, O.; Ley, R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat. Rev. Microbiol. 2011, 9, 279–290. [Google Scholar] [CrossRef]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef]
- Byrne, C.; Chambers, E.; Morrison, D.; Frost, G. The role of short chain fatty acids in appetite regulation and energy homeostasis. Int. J. Obes. 2015, 39, 1331–1338. [Google Scholar] [CrossRef]
- Campbell, J.M.; Fahey, G.C.; Wolf, B.W. Selected indigestible oligosaccharides affect large bowel mass, cecal and fecal short-chain fatty acids, pH and microflora in rats. J. Nutr. 1997, 127, 130–136. [Google Scholar] [CrossRef]
- Theriot, C.M.; Koenigsknecht, M.J.; Carlson Jr, P.E.; Hatton, G.E.; Nelson, A.M.; Li, B.; Huffnagle, G.B.; Li, J.Z.; Young, V.B. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat. Commun. 2014, 5, 3114. [Google Scholar] [CrossRef] [PubMed]
- Utzschneider, K.M.; Kratz, M.; Damman, C.J.; Hullarg, M. Mechanisms linking the gut microbiome and glucose metabolism. J. Clin. Endocrinol. Metab. 2016, 101, 1445–1454. [Google Scholar] [CrossRef] [PubMed]
- Carmody, R.N.; Gerber, G.K.; Luevano, J.M.; Gatti, D.M.; Somes, L.; Svenson, K.L.; Turnbaugh, P.J. Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe 2015, 17, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shen, D.; Fang, Z.; Jie, Z.; Qiu, X.; Zhang, C.; Chen, Y.; Ji, L. Human gut microbiota changes reveal the progression of glucose intolerance. PLoS ONE 2013, 8, e71108. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.; Calos, M.; Myers, A.; Self, W.T. Analysis of proline reduction in the nosocomial pathogen Clostridium difficile. J. Bacteriol. 2006, 188, 8487–8495. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, S.; Burman, L.G.; Åkerlund, T. Suppression of toxin production in Clostridium difficile VPI 10463 by amino acids. Microbiology 1999, 145, 1683–1693. [Google Scholar] [CrossRef] [PubMed]
- Neumann-Schaal, M.; Hofmann, J.D.; Will, S.E.; Schomburg, D. Time-resolved amino acid uptake of Clostridium difficile 630Δerm and concomitant fermentation product and toxin formation. BMC Microbiol. 2015, 15, 281. [Google Scholar] [CrossRef] [PubMed]
- Antonopoulos, D.A.; Huse, S.M.; Morrison, H.G.; Schmidt, T.M.; Sogin, M.L.; Young, V.B. Reproducible community dynamics of the gastrointestinal microbiota following antibiotic perturbation. Infect. Immunity 2009, 77, 2367–2375. [Google Scholar] [CrossRef] [PubMed]
- Antharam, V.C.; Li, E.C.; Ishmael, A.; Sharma, A.; Mai, V.; Rand, K.H.; Wang, G.P. Intestinal dysbiosis and depletion of butyrogenic bacteria in Clostridium difficile infection and nosocomial diarrhea. J. Clin. Microbiol. 2013, 51, 2884–2892. [Google Scholar] [CrossRef]
- Vincent, C.; Manges, A. Antimicrobial use, human gut microbiota and Clostridium difficile colonization and infection. Antibiotics 2015, 4, 230–253. [Google Scholar] [CrossRef]
- Schippa, S.; Conte, M.P. Dysbiotic events in gut microbiota: Impact on human health. Nutrients 2014, 6, 5786–5805. [Google Scholar] [CrossRef]
- Tamboli, C.; Neut, C.; Desreumaux, P.; Colombel, J. Dysbiosis in inflammatory bowel disease. Gut 2004, 53, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Littman, D.R. The microbiome in infectious disease and inflammation. Annu. Rev. Immunol. 2012, 30, 759–795. [Google Scholar] [CrossRef] [PubMed]
- Ling, Z.; Liu, X.; Jia, X.; Cheng, Y.; Luo, Y.; Yuan, L.; Wang, Y.; Zhao, C.; Guo, S.; Li, L.; et al. Impacts of infection with different toxigenic Clostridium difficile strains on faecal microbiota in children. Sci. Rep. 2014, 4, 7485. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef]
- Setchell, K.; Lawson, A.; Tanida, N.; Sjövall, J. General methods for the analysis of metabolic profiles of bile acids and related compounds in feces. J. Lipid Res. 1983, 24, 1085–1100. [Google Scholar] [PubMed]
- Winston, J.A.; Theriot, C.M. Impact of microbial derived secondary bile acids on colonization resistance against Clostridium difficile in the gastrointestinal tract. Anaerobe 2016, 41, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Rea, M.C.; Dobson, A.; O’Sullivan, O.; Crispie, F.; Fouhy, F.; Cotter, P.D.; Shanahan, F.; Kiely, B.; Hill, C.; Ross, R.P. Effect of broad-and narrow-spectrum antimicrobials on Clostridium difficile and microbial diversity in a model of the distal colon. Proc. Natl. Acad. Sci. USA 2011, 108, 4639–4644. [Google Scholar] [CrossRef]
- Sorg, J.A.; Sonenshein, A.L. Bile salts and glycine as cogerminants for Clostridium difficile spores. J. Bacteriol. 2008, 190, 2505–2512. [Google Scholar] [CrossRef] [PubMed]
- Sorg, J.A.; Sonenshein, A.L. Inhibiting the initiation of Clostridium difficile spore germination using analogs of chenodeoxycholic acid, a bile acid. J. Bacteriol. 2010, 192, 4983–4990. [Google Scholar] [CrossRef] [PubMed]
- Allegretti, J.R.; Kearney, S.; Li, N.; Bogart, E.; Bullock, K.; Gerber, G.K.; Bry, L.; Clish, C.B.; Alm, E.; Korzenik, J. Recurrent Clostridium difficile infection associates with distinct bile acid and microbiome profiles. Aliment. Pharmacol. Ther. 2016, 43, 1142–1153. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.H.; Kennedy, M.J.; Fekety, F.R. Use of sodium taurocholate to enhance spore recovery on a medium selective for Clostridium difficile. J. Clin. Microbiol. 1982, 15, 443–446. [Google Scholar] [PubMed]
- Buggy, B.; Hawkins, C.; Fekety, R. Effect of adding sodium taurocholate to selective media on the recovery of Clostridium difficile from environmental surfaces. J. Clin. Microbiol. 1985, 21, 636–637. [Google Scholar] [PubMed]
- Wilson, K.H. Efficiency of various bile salt preparations for stimulation of Clostridium difficile spore germination. J. Clin. Microbiol. 1983, 18, 1017–1019. [Google Scholar] [PubMed]
- Seekatz, A.M.; Young, V.B. Clostridium difficile and the microbiota. J. Clin. Investig. 2014, 124, 4182–4189. [Google Scholar] [CrossRef]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef]
- Engevik, M.A.; Engevik, K.A.; Yacyshyn, M.B.; Wang, J.; Hassett, D.J.; Darien, B.; Yacyshyn, B.R.; Worrell, R.T. Human Clostridium difficile infection: Inhibition of NHE3 and microbiota profile. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G497–G509. [Google Scholar] [CrossRef]
- Dannheim, H.; Will, S.E.; Schomburg, D.; Neumann-Schaal, M. Clostridioides difficile 630Δerm in silico and in vivo–quantitative growth and extensive polysaccharide secretion. FEBS Open Biol. 2017, 7, 602–615. [Google Scholar] [CrossRef]
- Heinken, A.; Sahoo, S.; Fleming, R.M.; Thiele, I. Systems-level characterization of a host-microbe metabolic symbiosis in the mammalian gut. Gut Microbes 2013, 4, 28–40. [Google Scholar] [CrossRef]
- Heinken, A.; Khan, M.T.; Paglia, G.; Rodionov, D.A.; Harmsen, H.J.; Thiele, I. Functional metabolic map of Faecalibacterium prausnitzii, a beneficial human gut microbe. J. Bacteriol. 2014, 196, 3289–3302. [Google Scholar] [CrossRef]
- Baumler, D.J.; Peplinski, R.G.; Reed, J.L.; Glasner, J.D.; Perna, N.T. The evolution of metabolic networks of E. coli. BMC Syst. Biol. 2011, 5, 182. [Google Scholar] [CrossRef] [PubMed]
- Henson, M.A.; Phalak, P. Suboptimal community growth mediated through metabolite cross-feeding promotes species diversity in the gut microbiota. PLoS Comput. Biol. 2018, 14, e1006558. [Google Scholar] [CrossRef] [PubMed]
- Ouwerkerk, J.P.; de Vos, W.M.; Belzer, C. Glycobiome: Bacteria and mucus at the epithelial interface. Best Pract. Res. Clin. Gastroenterol. 2013, 27, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.L.; Spinler, J.K.; Savidge, T.C. Structural and functional changes within the gut microbiota and susceptibility to Clostridium difficile infection. Anaerobe 2016, 41, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Browne, H.P.; Forster, S.C.; Anonye, B.O.; Kumar, N.; Neville, B.A.; Stares, M.D.; Goulding, D.; Lawley, T.D. Culturing of ‘unculturable’human microbiota reveals novel taxa and extensive sporulation. Nature 2016, 533, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Staley, C.; Weingarden, A.R.; Khoruts, A.; Sadowsky, M.J. Interaction of gut microbiota with bile acid metabolism and its influence on disease states. Appl. Microbiol. Biotechnol. 2017, 101, 47–64. [Google Scholar] [CrossRef] [PubMed]
- Myers, S.P. The causes of intestinal dysbiosis: A review. Altern. Med. Rev. 2004, 9, 180–197. [Google Scholar]
- Singh, R.K.; Chang, H.W.; Yan, D.; Lee, K.M.; Ucmak, D.; Wong, K.; Abrouk, M.; Farahnik, B.; Nakamura, M.; Zhu, T.H.; et al. Influence of diet on the gut microbiome and implications for human health. J. Transl. Med. 2017, 15, 73. [Google Scholar] [CrossRef]
- Kamada, N.; Chen, G.Y.; Inohara, N.; Núñez, G. Control of pathogens and pathobionts by the gut microbiota. Nat. Immunol. 2013, 14, 685–690. [Google Scholar] [CrossRef]
- Weingarden, A.R.; Chen, C.; Bobr, A.; Yao, D.; Lu, Y.; Nelson, V.M.; Sadowsky, M.J.; Khoruts, A. Microbiota transplantation restores normal fecal bile acid composition in recurrent Clostridium difficile infection. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 306, G310–G319. [Google Scholar] [CrossRef]
- Heinken, A.; Thiele, I. Systematic prediction of health-relevant human-microbial co-metabolism through a computational framework. Gut Microbes 2015, 6, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Shoaie, S.; Nielsen, J. Elucidating the interactions between the human gut microbiota and its host through metabolic modeling. Front. Genet. 2014, 5, 86. [Google Scholar] [CrossRef]
- Thiele, I.; Heinken, A.; Fleming, R.M. A systems biology approach to studying the role of microbes in human health. Curr. Opin. Biotechnol. 2013, 24, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Glick-Bauer, M.; Yeh, M.C. The health advantage of a vegan diet: Exploring the gut microbiota connection. Nutrients 2014, 6, 4822–4838. [Google Scholar] [CrossRef] [PubMed]
- Swainston, N.; Mendes, P.; Kell, D.B. An analysis of a ‘community-driven’reconstruction of the human metabolic network. Metabolomics 2013, 9, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Thiele, I.; Swainston, N.; Fleming, R.M.; Hoppe, A.; Sahoo, S.; Aurich, M.K.; Haraldsdottir, H.; Mo, M.L.; Rolfsson, O.; Stobbe, M.D.; et al. A community-driven global reconstruction of human metabolism. Nat. Biotechnol. 2013, 31, 419. [Google Scholar] [CrossRef] [PubMed]
- Thiele, I.; Sahoo, S.; Heinken, A.; Heirendt, L.; Aurich, M.K.; Noronha, A.; Fleming, R.M. When metabolism meets physiology: Harvey and Harvetta. bioRxiv 2018. [Google Scholar] [CrossRef]
- Dai, Z.L.; Wu, G.; Zhu, W.Y. Amino acid metabolism in intestinal bacteria: Links between gut ecology and host health. Front. Biosci. 2011, 16, 1768–1786. [Google Scholar] [CrossRef]
- Horn, H.; Lackner, S. Modeling of biofilm systems: A review. In Productive Biofilms; Springer: Cham, Swizterland, 2014; pp. 53–76. [Google Scholar]
- Lawley, T.D.; Clare, S.; Walker, A.W.; Stares, M.D.; Connor, T.R.; Raisen, C.; Goulding, D.; Rad, R.; Schreiber, F.; Brandt, C.; et al. Targeted restoration of the intestinal microbiota with a simple, defined bacteriotherapy resolves relapsing Clostridium difficile disease in mice. PLoS Pathog. 2012, 8, e1002995. [Google Scholar] [CrossRef]
- Chen, J.; Gomez, J.A.; Höffner, K.; Phalak, P.; Barton, P.I.; Henson, M.A. Spatiotemporal modeling of microbial metabolism. BMC Syst. Biol. 2016, 10, 21. [Google Scholar] [CrossRef]
- Phalak, P.; Chen, J.; Carlson, R.P.; Henson, M.A. Metabolic modeling of a chronic wound biofilm consortium predicts spatial partitioning of bacterial species. BMC Syst. Biol. 2016, 10, 90. [Google Scholar] [CrossRef] [PubMed]
- Gomez, J.A.; Hoffner, K.; Barton, P.I. DFBAlab: A fast and reliable MATLAB code for dynamic flux balance analysis. BMC Bioinform. 2014, 15, 409. [Google Scholar] [CrossRef] [PubMed]
- Meadows, A.L.; Karnik, R.; Lam, H.; Forestell, S.; Snedecor, B. Application of dynamic flux balance analysis to an industrial Escherichia coli fermentation. Metab. Eng. 2010, 12, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.S. A review of experimental measurements of effective diffusive permeabilities and effective diffusion coefficients in biofilms. Biotechnol. Bioeng. 1998, 59, 261–272. [Google Scholar] [CrossRef]
- Stewart, P.S. Diffusion in biofilms. J. Bacteriol. 2003, 185, 1485–1491. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nutrient | Healthy | High Amino Acids, Low Glucose | High Primary Bile Acids | High Nitrate |
|---|---|---|---|---|
| Glucose | 8.0 | 4.0 | 8.0 | 4.0 |
| Cysteine | 0.5 | 1.0 | 0.5 | 1.0 |
| Isoleucine | 0.5 | 1.0 | 0.5 | 1.0 |
| Leucine | 0.5 | 1.0 | 0.5 | 1.0 |
| Methionine | 0.5 | 1.0 | 0.5 | 1.0 |
| Proline | 0.5 | 1.0 | 0.5 | 1.0 |
| Serine | 0.5 | 1.0 | 0.5 | 1.0 |
| Tryptophan | 0.5 | 1.0 | 0.5 | 1.0 |
| Valine | 0.5 | 1.0 | 0.5 | 1.0 |
| Nitrate | 0 | 0 | 0 | 0.4 |
| Taurocholate | 0 | 0 | 1.5 | 1.5 |
| Symbol | Parameter | Value | Units | Source |
|---|---|---|---|---|
| L | Biofilm thickness | 40 | m | [115] |
| Biomass bulk concentrations | 0 | g/L | [50] | |
| Byproduct bulk concentrations | 0 | mmol/L | [50] | |
| Diffusion coefficient | ||||
| Biomass | 2 × 10 | cm/s | [50] | |
| Glucose | 2.01 × 10 | cm/s | [116] | |
| Cysteine | 2.45 × 10 | cm/s | [116] | |
| Isoleucine | 2.19 × 10 | cm/s | [116] | |
| Leucine | 2.19 × 10 | cm/s | [116] | |
| Methionine | 2.21 × 10 | cm/s | [116] | |
| Proline | 2.51 × 10 | cm/s | [116] | |
| Serine | 2.64 × 10 | cm/s | [116] | |
| Tryptophan | 1.89 × 10 | cm/s | [116] | |
| Valine | 2.49 × 10 | cm/s | [116] | |
| Acetate | 3.03 × 10 | cm/s | [116] | |
| Butyrate | 1.74 × 10 | cm/s | [116] | |
| CO | 1.15 × 10 | cm/s | [116] | |
| Ethanol | 3.97 × 10 | cm/s | [116] | |
| Formate | 4.23 × 10 | cm/s | [116] | |
| Lactate | 3.1 × 10 | cm/s | [116] | |
| Propionate | 4.03 × 10 | cm/s | [116] | |
| Succinate | 2.82 × 10 | cm/s | [116] | |
| Nitrate | 1.29 × 10 | cm/s | [116] | |
| Taurocholate | 7.29 × 10 | cm/s | [116] | |
| Mass transfer coefficient | ||||
| Biomass | 6 × 10 | cm/s | [50] | |
| Glucose | 2 × 10 | cm/s | [50] | |
| Amino acid | 2 × 10 | cm/s | [50] | |
| Byproduct | 5 × 10 | cm/s | [50] | |
| Butyrate | 8.5 × 10 | cm/s | Tuned | |
| Propionate | 1.35 × 10 | cm/s | Tuned | |
| Nitrate | 1.5 × 10 | cm/s | Tuned | |
| Taurocholate | 2 × 10 | cm/s | Tuned | |
| Maximum uptake rate | ||||
| Glucose | 10 | mmol/gDW/h | [114] | |
| Amino acid | 1 | mmol/gDW/h | [114] | |
| Byproduct | 10 | mmol/gDW/h | [50] | |
| Michaelis–Menten constant | ||||
| Glucose | 0.5 | mmol/L | [114] | |
| Amino acids | 0.1 | mmol/L | [114] | |
| Byproduct | 0.5 | mmol/L | [50] | |
| ATP maintenance | ||||
| B. thetaiotaomicron | 4.25 | mmol/gDW/h | Tuned | |
| F. prausnitzii | 3.4 | mmol/gDW/h | Tuned | |
| E. coli | 2.75 | mmol/gDW/h | Tuned | |
| C. difficile | 8.43 | mmol/gDW/h | Tuned |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phalak, P.; Henson, M.A. Metabolic Modeling of Clostridium difficile Associated Dysbiosis of the Gut Microbiota. Processes 2019, 7, 97. https://doi.org/10.3390/pr7020097
Phalak P, Henson MA. Metabolic Modeling of Clostridium difficile Associated Dysbiosis of the Gut Microbiota. Processes. 2019; 7(2):97. https://doi.org/10.3390/pr7020097
Chicago/Turabian StylePhalak, Poonam, and Michael A. Henson. 2019. "Metabolic Modeling of Clostridium difficile Associated Dysbiosis of the Gut Microbiota" Processes 7, no. 2: 97. https://doi.org/10.3390/pr7020097
APA StylePhalak, P., & Henson, M. A. (2019). Metabolic Modeling of Clostridium difficile Associated Dysbiosis of the Gut Microbiota. Processes, 7(2), 97. https://doi.org/10.3390/pr7020097