Modeling and Observer-Based Monitoring of RAFT Homopolymerization Reactions


Abstract
1. Introduction
2. Model and Observer Design
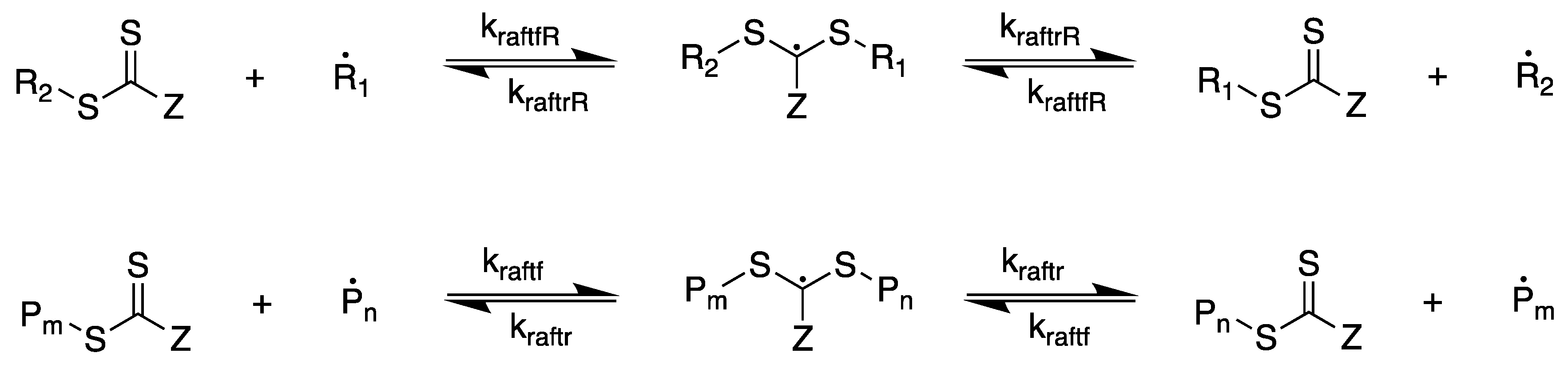
2.1. Modeling RAFT Polymerization—Improving the Model by Zhang and Ray
2.2. Observer Design
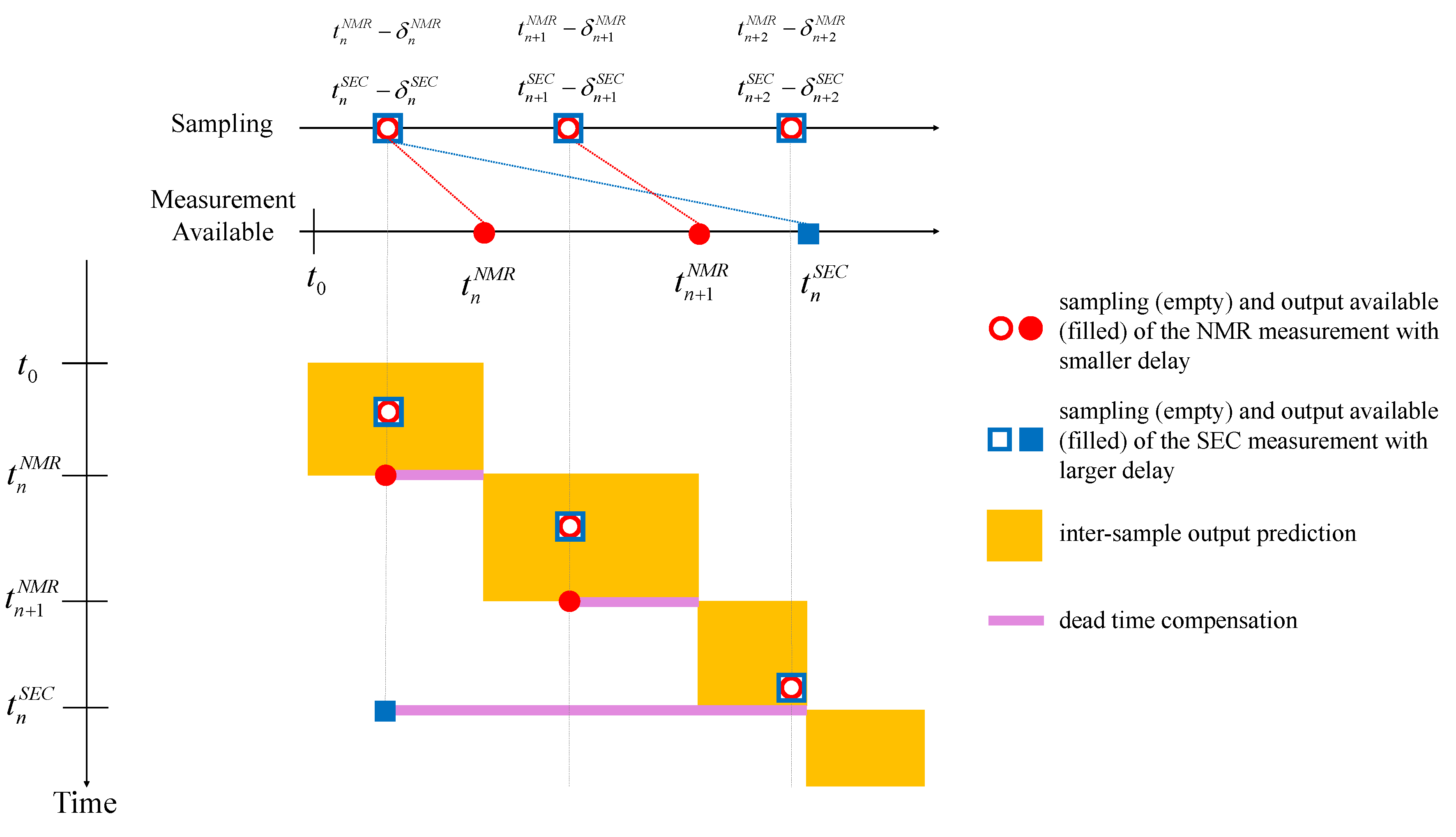
2.2.1. Multi-Rate Multi-Delay Observer—Background
2.2.2. Prediction of RAFT Polymerization
3. Experimental Methods and Results
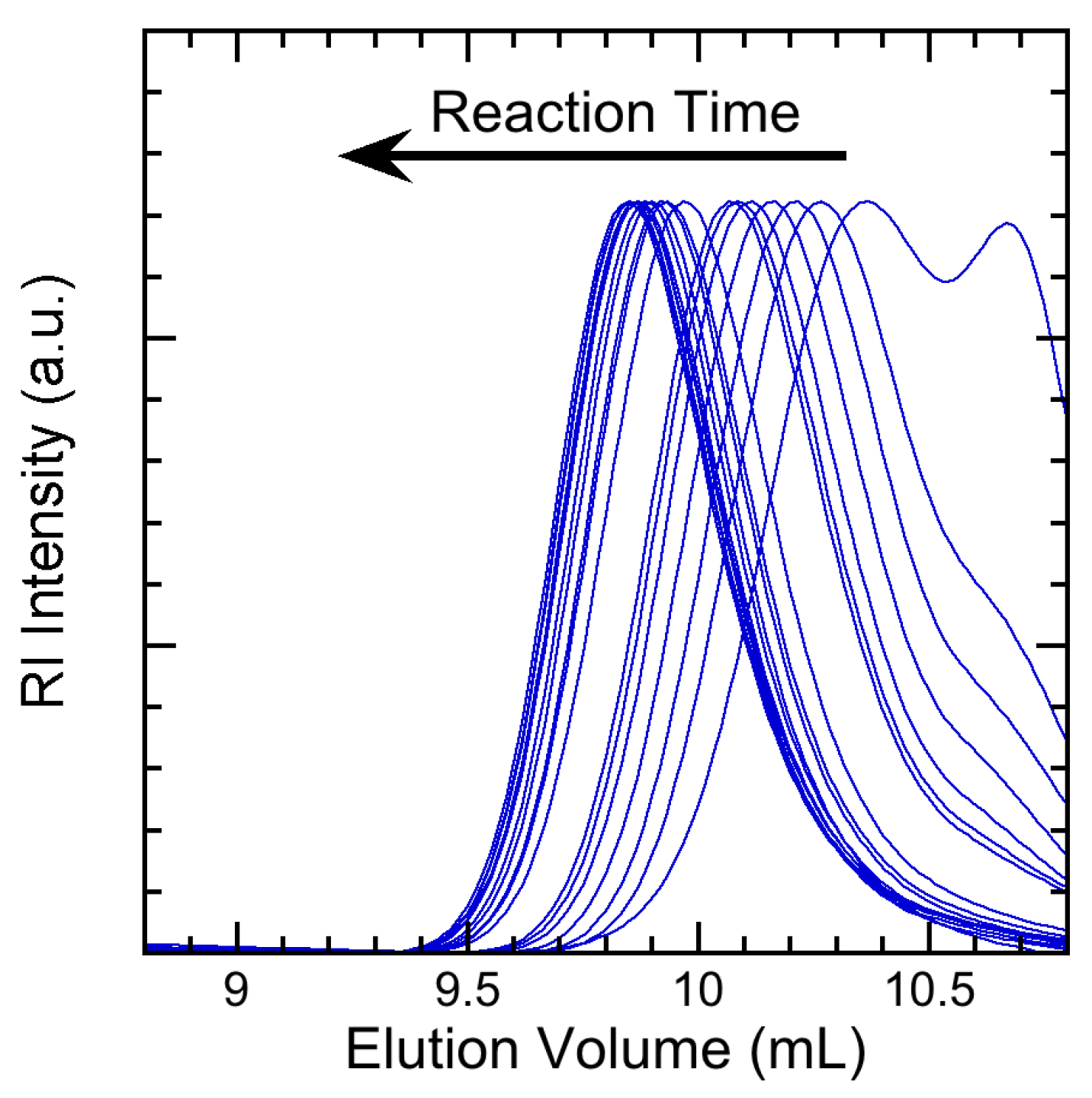
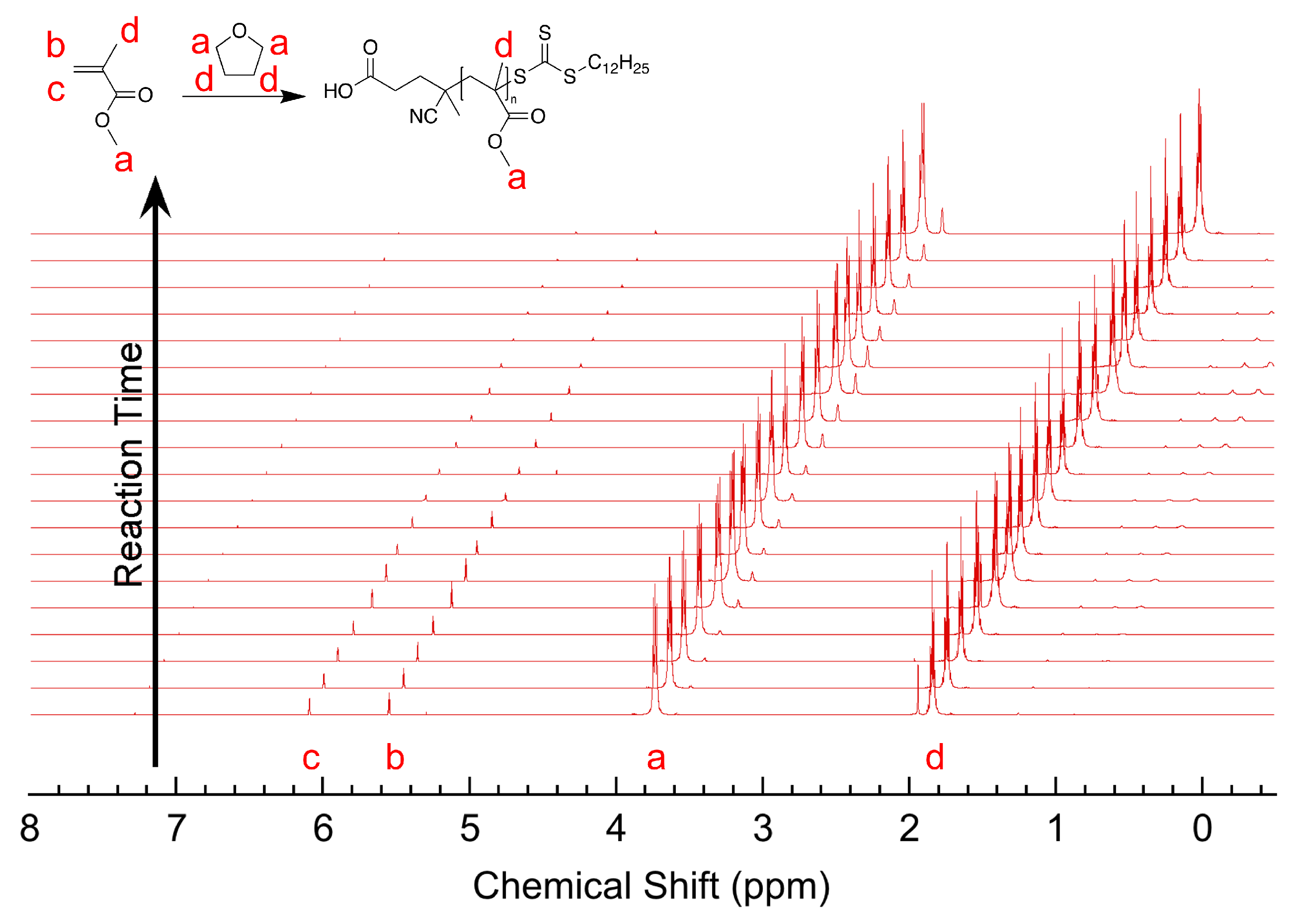
3.1. Materials and Methods
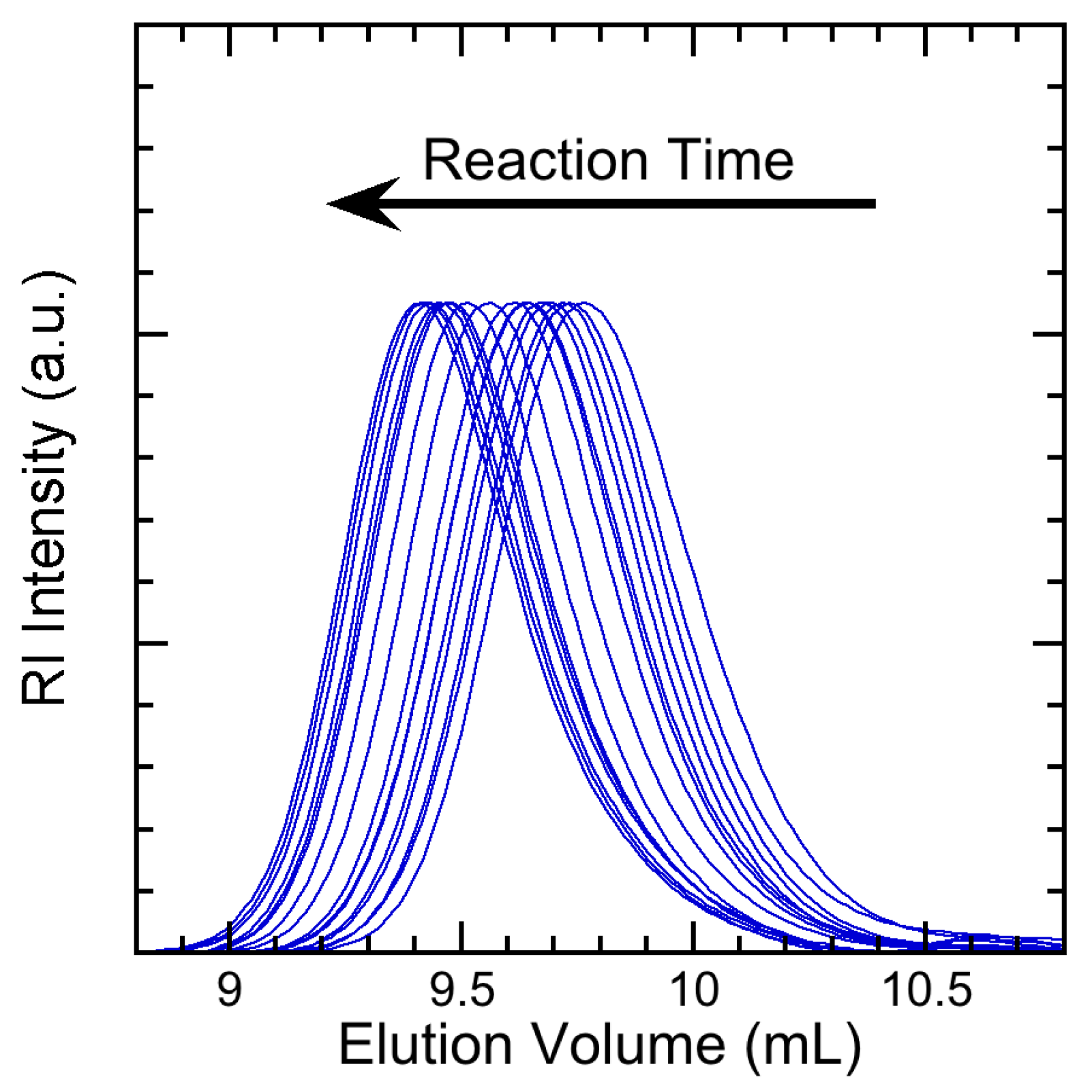
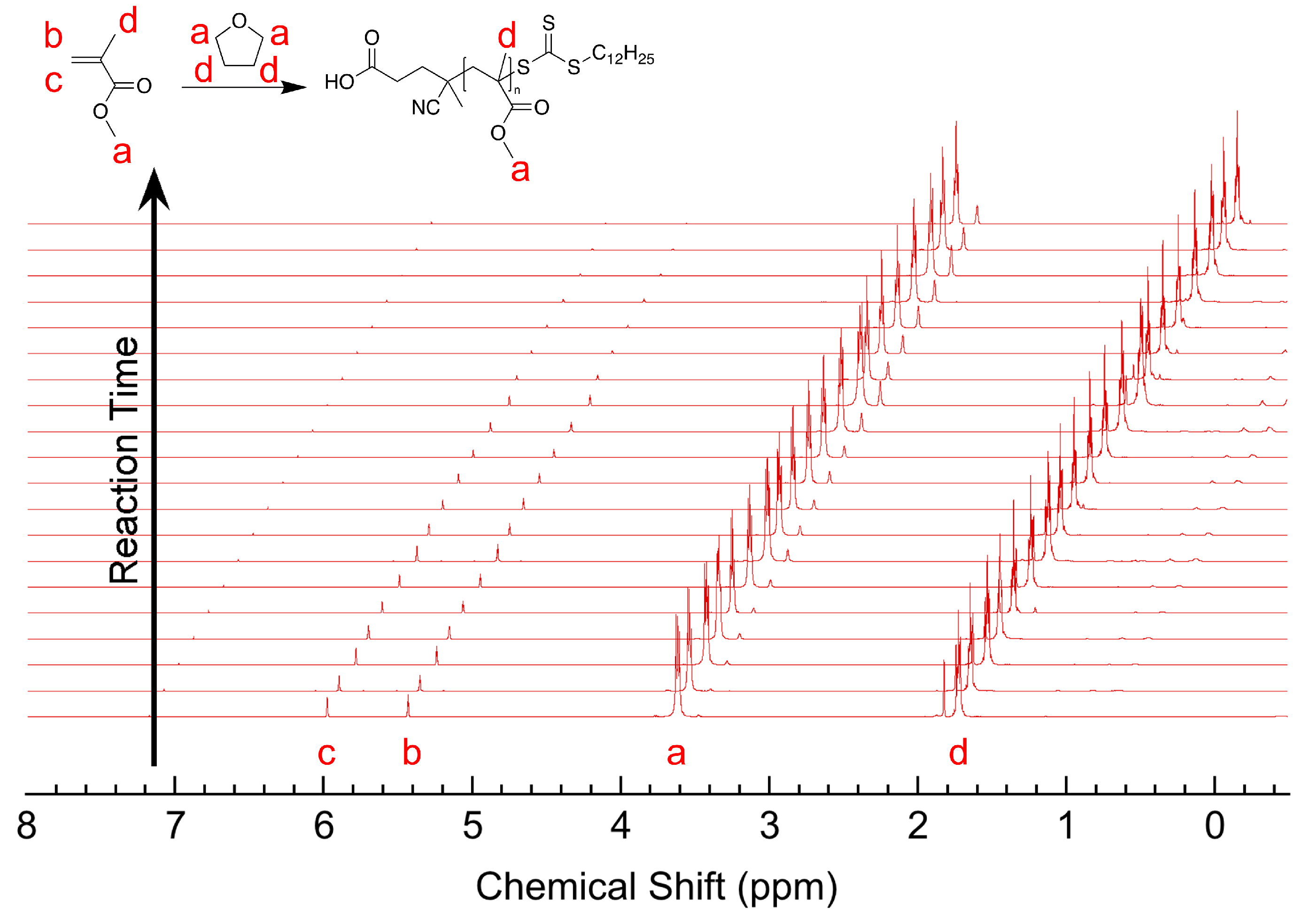
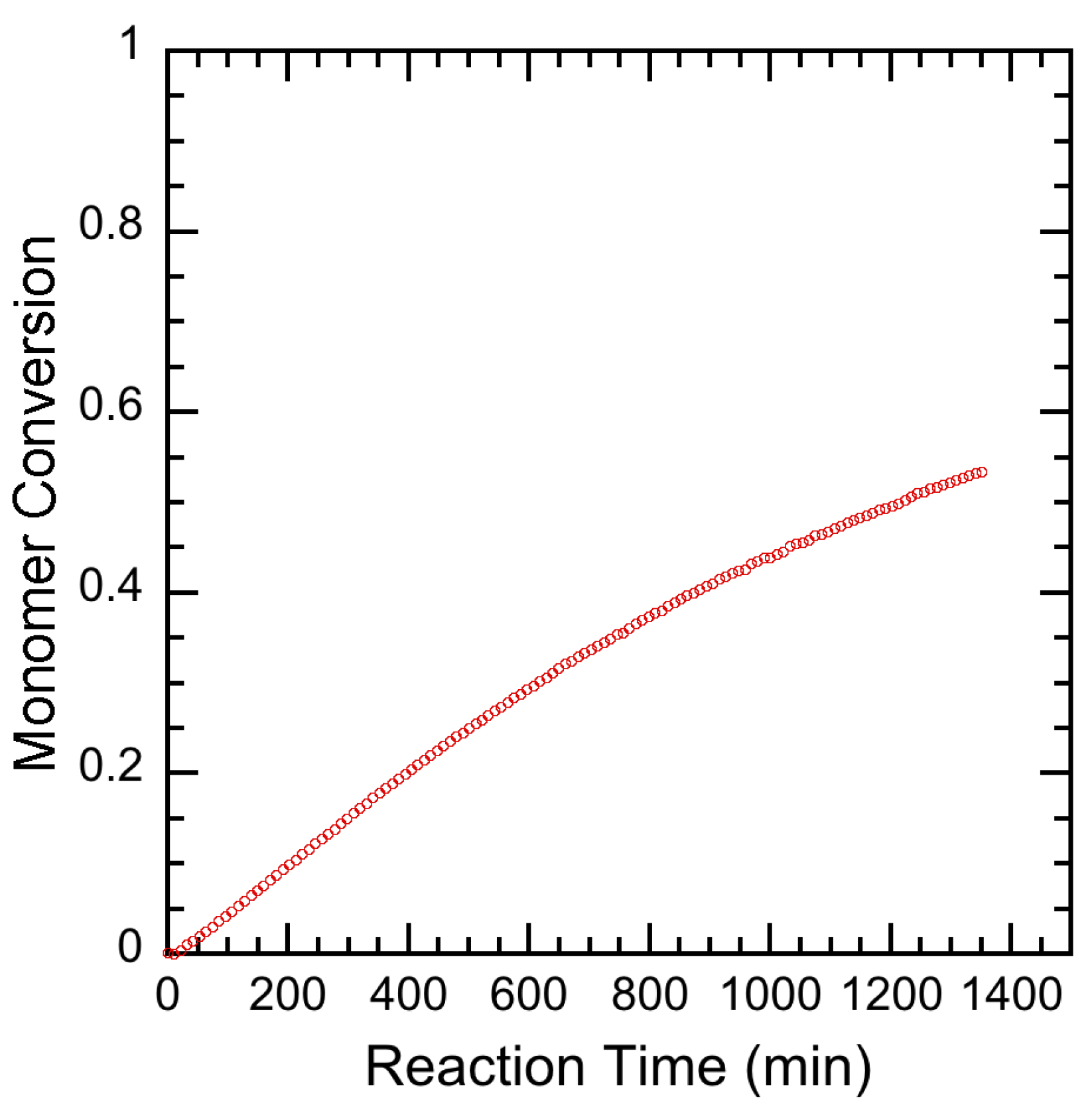
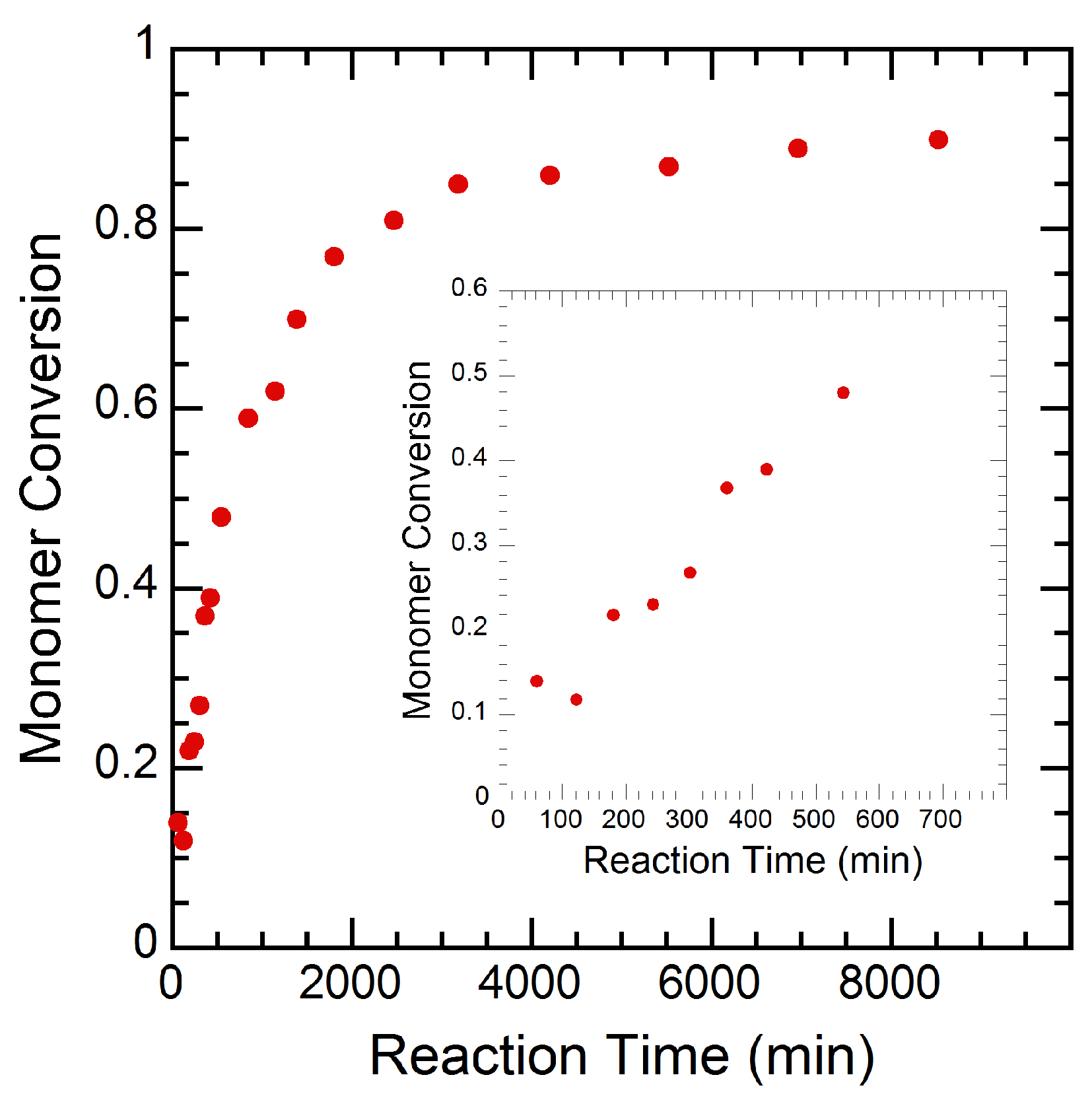
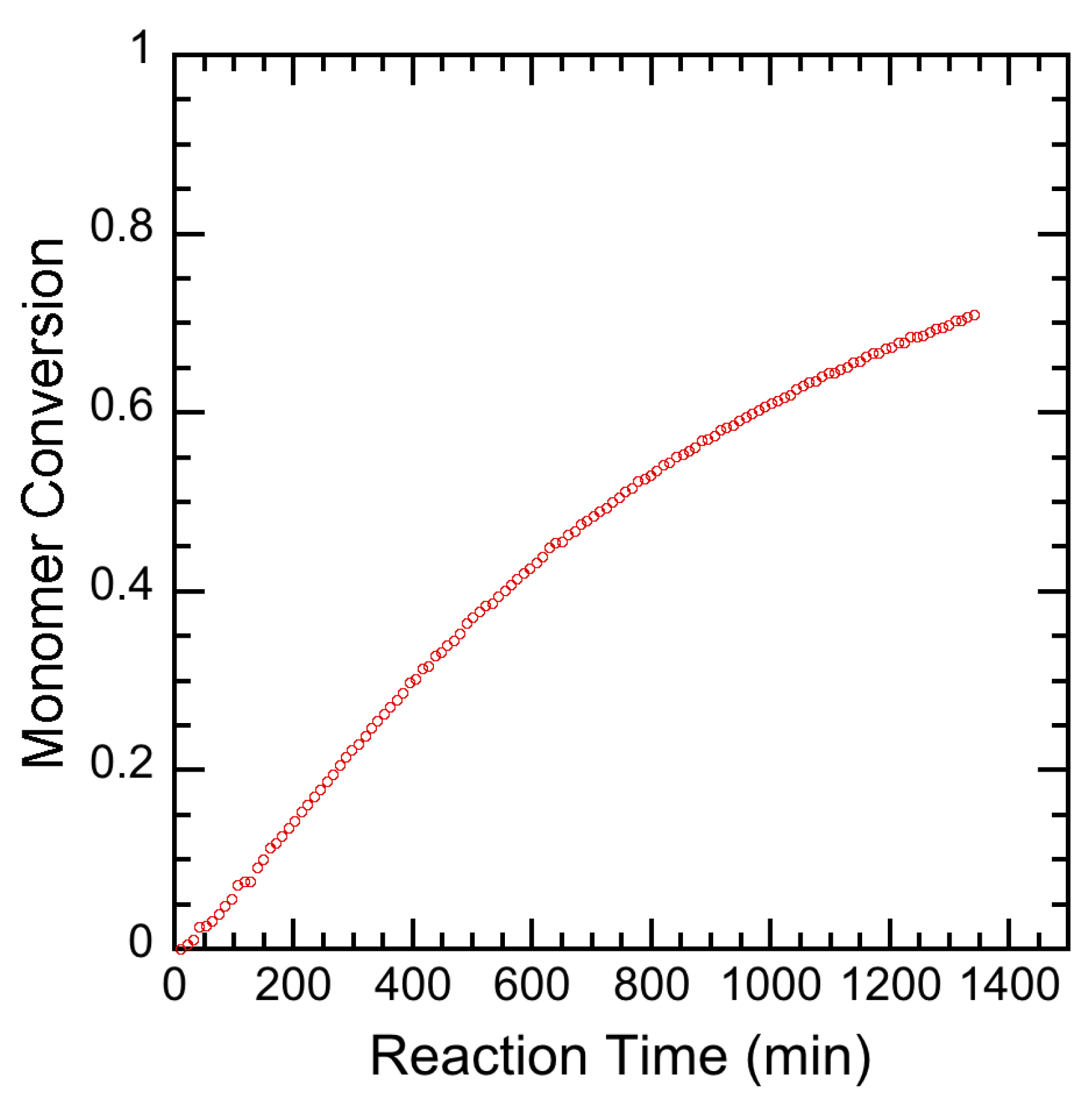
3.2. Experimental Results
4. Results and Discussion
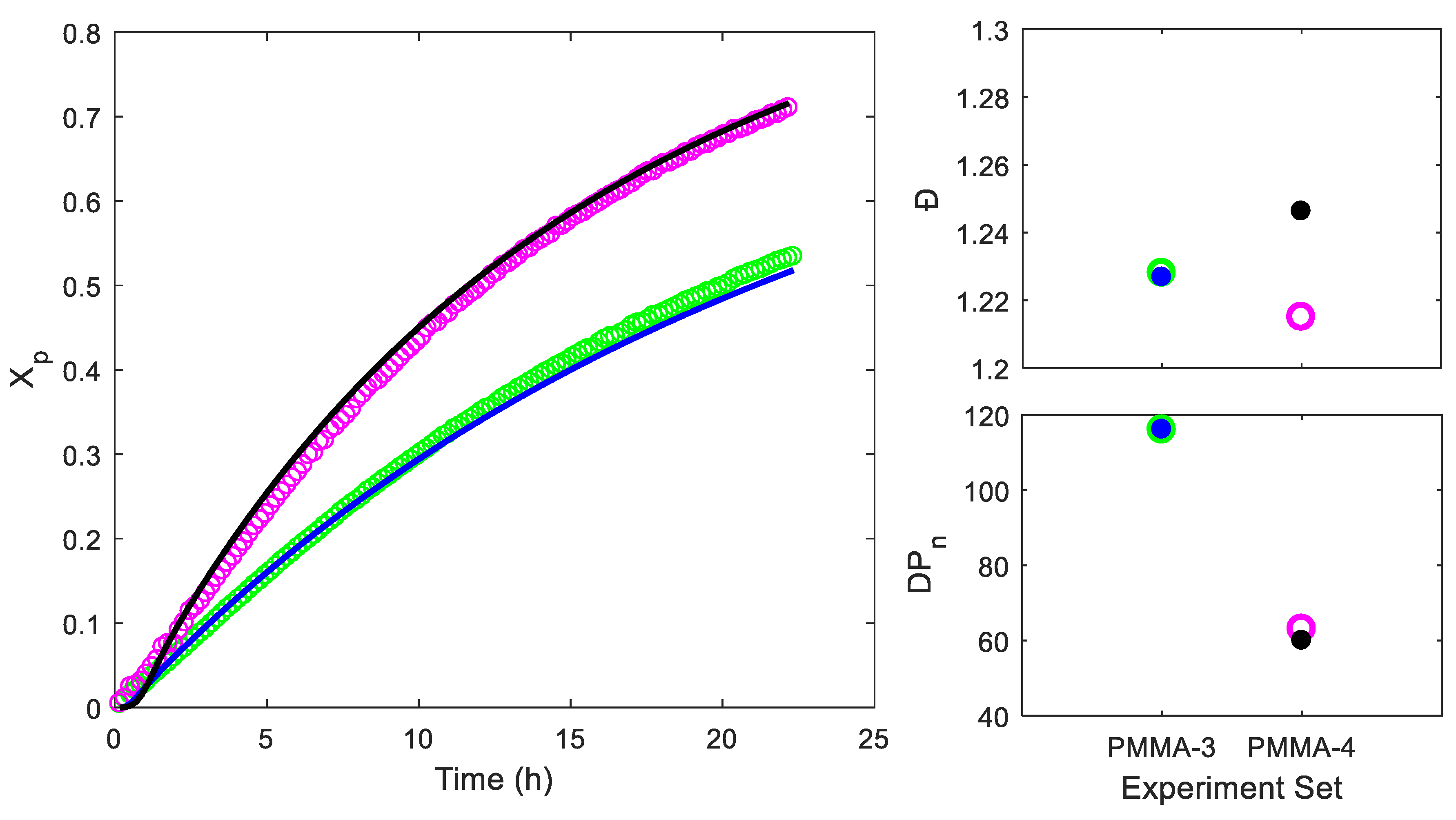
4.1. Parameter Fitting
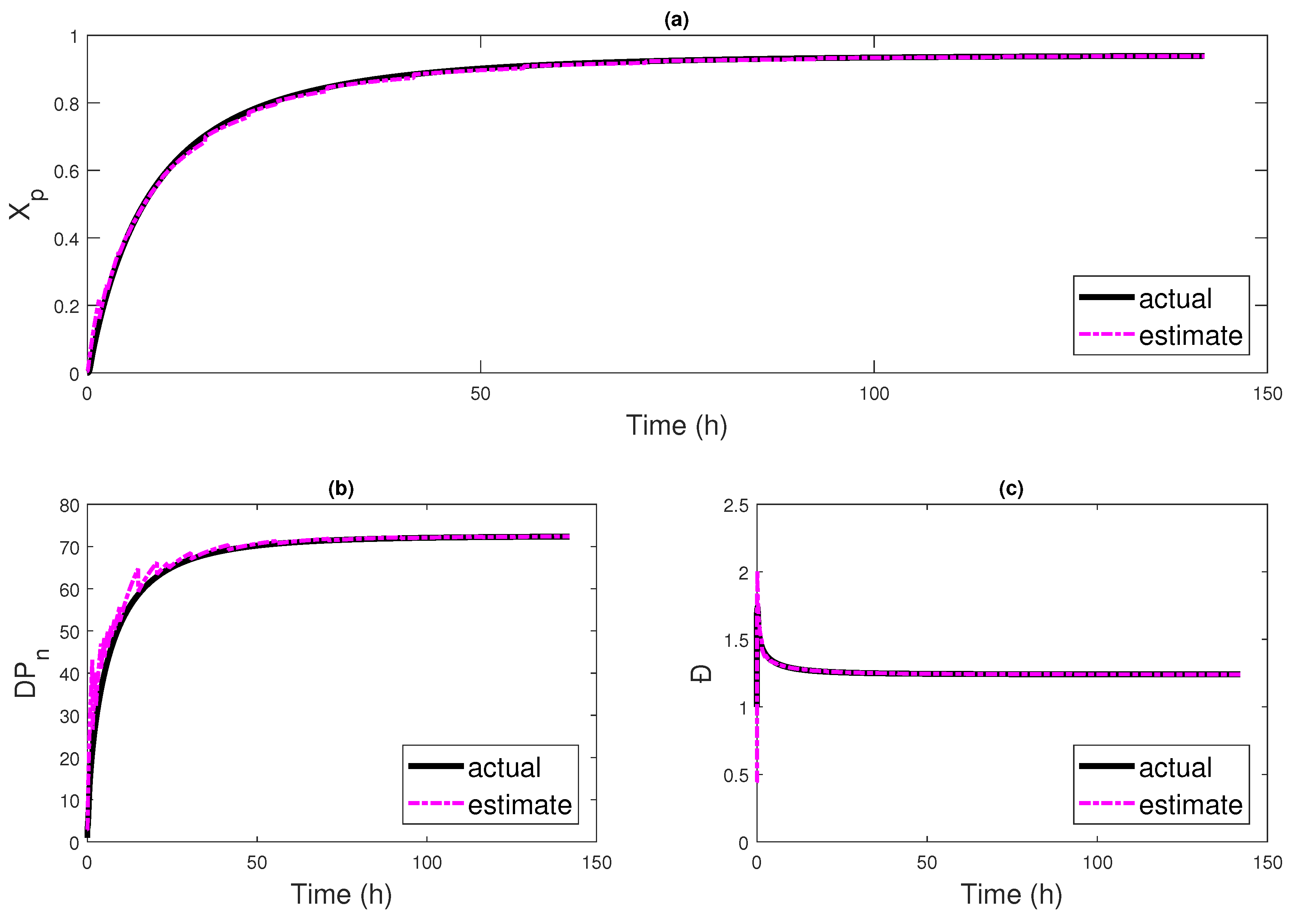
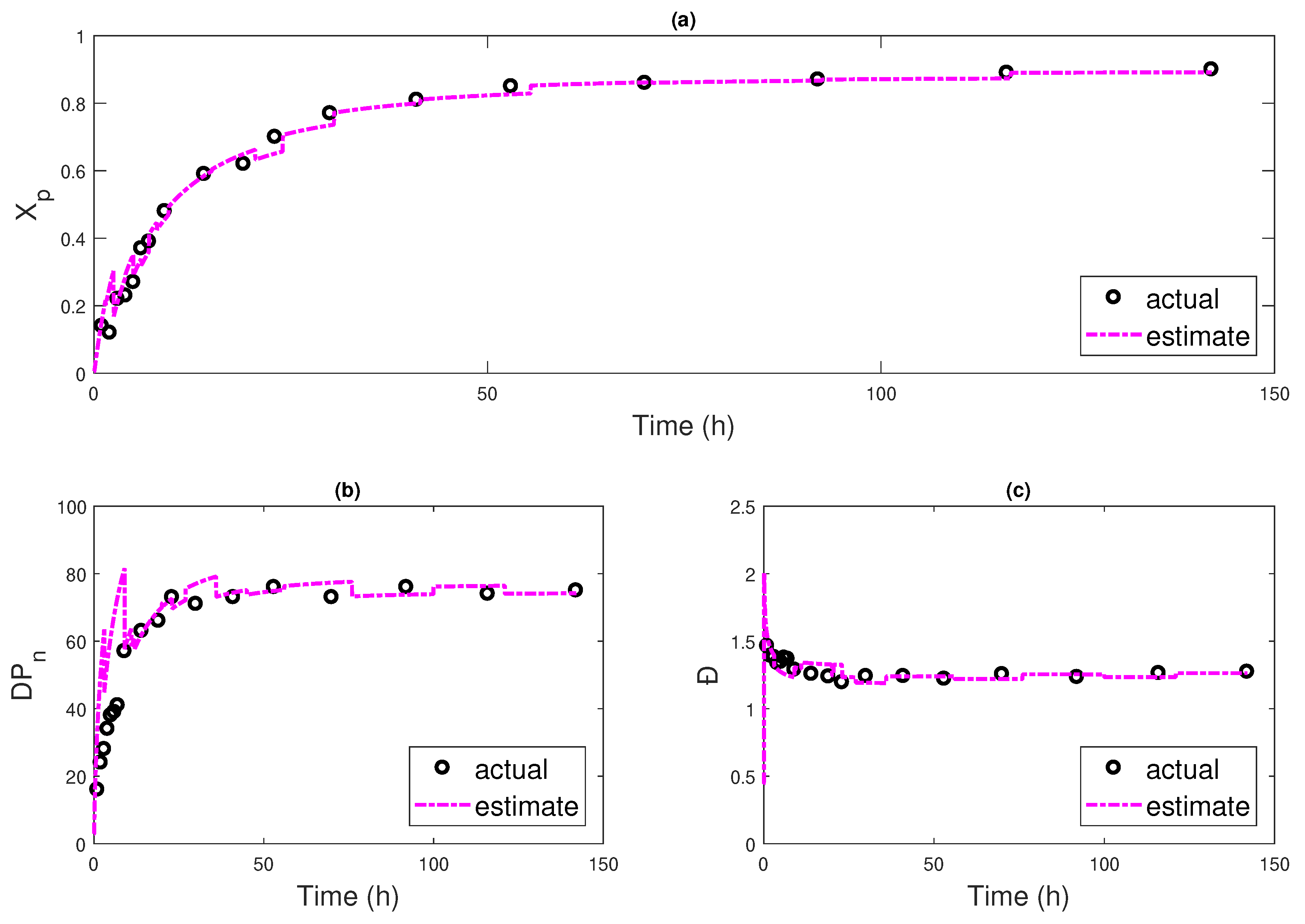
4.2. Multi-Rate Multi-Delay Observer
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| H NMR | Proton Nuclear Magnetic Resonance |
| AIBN | Azobis(isobutyronitrile) |
| CTA | Chain Transfer Agent |
| EKF | Extended Kalman Filter |
| MHE | Moving Horizon Estimation |
| MMA | Methyl Methacrylate |
| PMMA | Poly(methyl methacrylate) |
| RAFT | Reversible Addition–Fragmentation Chain–Transfer |
| SEC | Size Exclusion Chromatography |
| THF | Tetrahydrofuran |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
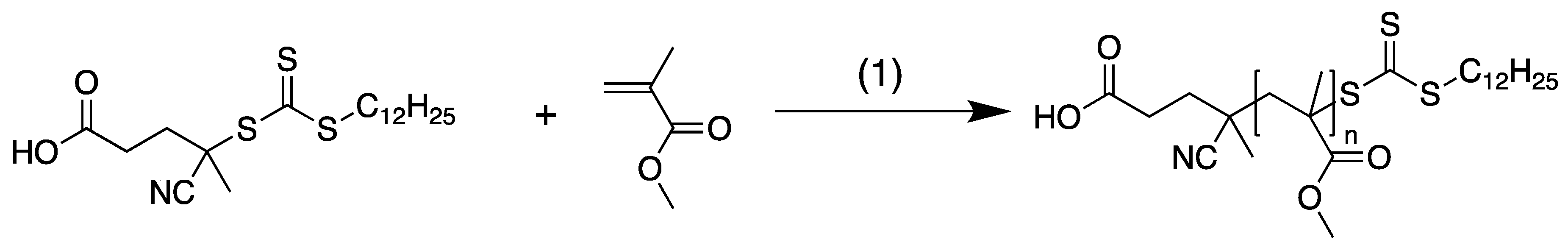{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (h) | Time (min) | NMR Conv. (%) | SEC Conv. (%) | M (g mol) | M (g mol) | Đ | DP |
|---|---|---|---|---|---|---|---|
| 0 | 0 | 0 | - | - | - | - | - |
| 0.5 | 30 | 5 | - | - | - | - | - |
| 1 | 60 | 9 | - | - | - | - | - |
| 1.5 | 90 | 11 | - | - | - | - | - |
| 2 | 120 | 15 | - | - | - | - | - |
| 3 | 180 | 21 | 42.9 | 9637 | 12,409 | 1.288 | 92 |
| 4 | 240 | 26 | 27.5 | 6324 | 9949 | 1.573 | 59 |
| 5 | 300 | 31 | 41.5 | 9334 | 13,227 | 1.417 | 89 |
| 6 | 360 | 33 | 50.4 | 11,261 | 13,954 | 1.239 | 108 |
| 7 | 420 | 38 | 53.5 | 11,927 | 14,858 | 1.246 | 115 |
| 8 | 480 | 41 | 55.6 | 12,383 | 15,382 | 1.242 | 120 |
| 10 | 600 | 47 | 56.3 | 12,523 | 15,800 | 1.262 | 121 |
| 13 | 780 | 59 | 60.1 | 13,346 | 17,018 | 1.275 | 129 |
| 17 | 1020 | 59 | 64.5 | 14,293 | 17,964 | 1.257 | 139 |
| 22 | 1320 | 64 | 68.9 | 15,240 | 19,357 | 1.270 | 148 |
| 33 | 1980 | 75 | 79.7 | 17,560 | 21,400 | 1.219 | 171 |
| 47 | 2820 | 81 | 85.1 | 18,724 | 22,974 | 1.227 | 183 |
| 58 | 3480 | 85 | 86.2 | 18,957 | 23,296 | 1.229 | 185 |
| 66 | 3960 | 87 | 86.6 | 19,052 | 23,744 | 1.246 | 186 |
| 90 | 5400 | 91 | 91.5 | 20,107 | 25,067 | 1.247 | 197 |
| 114 | 6840 | 94 | 94.9 | 20,824 | 25,766 | 1.237 | 204 |
| 143 | 8580 | 95 | 96.2 | 21,121 | 26,347 | 1.247 | 207 |
| Time (h) | Time (min) | NMR Conv. (%) | SEC Conv. (%) | M (g mol) | M (g mol) | Đ | DP |
|---|---|---|---|---|---|---|---|
| 0 | 0 | 0 | - | - | - | - | - |
| 1 | 60 | 14 | 23.4 | 2042 | 2990 | 1.464 | 16 |
| 2 | 120 | 12 | 34.4 | 2814 | 3918 | 1.392 | 24 |
| 3 | 180 | 22 | 40.0 | 3208 | 4433 | 1.382 | 28 |
| 4 | 240 | 23 | 48.1 | 3777 | 5055 | 1.338 | 34 |
| 5 | 300 | 27 | 53.6 | 4162 | 5604 | 1.346 | 38 |
| 6 | 360 | 37 | 55.5 | 4292 | 5910 | 1.377 | 39 |
| 7 | 420 | 39 | 58.1 | 4472 | 6122 | 1.369 | 41 |
| 9 | 540 | 48 | 81.5 | 6112 | 7877 | 1.289 | 57 |
| 14 | 840 | 59 | 90.7 | 6759 | 8496 | 1.257 | 63 |
| 19 | 1140 | 62 | 94.2 | 7003 | 8672 | 1.238 | 66 |
| 23 | 1380 | 70 | 104.1 | 7698 | 9203 | 1.195 | 73 |
| 30 | 1800 | 77 | 101.2 | 7497 | 9304 | 1.241 | 71 |
| 41 | 2460 | 81 | 104.9 | 7754 | 9620 | 1.241 | 73 |
| 53 | 3180 | 85 | 108.9 | 8035 | 9809 | 1.221 | 76 |
| 70 | 4200 | 86 | 104.2 | 7704 | 9671 | 1.255 | 73 |
| 92 | 5520 | 87 | 109.1 | 8051 | 9933 | 1.234 | 76 |
| 116 | 6960 | 89 | 106.0 | 7834 | 9893 | 1.263 | 74 |
| 142 | 8520 | 90 | 106.6 | 7877 | 10,025 | 1.273 | 75 |




References
- Moad, G.; Chiefari, J.; Chong, Y.K.; Krstina, J.; Mayadunne, R.T.A.; Postma, A.; Rizzardo, E.; Thang, S.H. Living free radical polymerization with reversible addition–fragmentation chain transfer (the life of RAFT). Polym. Int. 2000, 49, 993–1001. [Google Scholar] [CrossRef]
- Krstina, J.; Moad, C.L.; Moad, G.; Rizzardo, E.; Berge, C.T.; Fryd, M. A new form of controlled growth free radical polymerization. In Macromolecular Symposia; Hüthig & Wepf Verlag: Basel, Switzerland, 1996; Volume 111, pp. 13–23. [Google Scholar]
- Gardiner, J.; Martinez-Botella, I.; Tsanaktsidis, J.; Moad, G. Dithiocarbamate RAFT agents with broad applicability - the 3,5-dimethyl-1H-pyrazole-1-carbodithioates. Polym. Chem. 2016, 7, 481–492. [Google Scholar] [CrossRef]
- Gardiner, J.; Martinez-Botella, I.; Kohl, T.M.; Krstina, J.; Moad, G.; Tyrell, J.H.; Coote, M.L.; Tsanaktsidis, J. 4-Halogeno-3,5-dimethyl-1H-pyrazole-1-carbodithioates: Versatile reversible addition fragmentation chain transfer agents with broad applicability. Polym. Int. 2017, 66, 1438–1447. [Google Scholar] [CrossRef]
- Moad, G. RAFT Polymerization—Then, and Now. In Controlled Radical Polymerization: Mechanisms; American Chemical Society: Akron, OH, USA, 2015; Volume 1187, pp. 211–246. [Google Scholar]
- Ye, Y.S.; Choi, J.H.; Winey, K.I.; Elabd, Y.A. Polymerized Ionic Liquid Block and Random Copolymers: Effect of Weak Microphase Separation on Ion Transport. Macromolecules 2012, 45, 7027–7035. [Google Scholar] [CrossRef]
- Stace, S.J.; Fellows, C.M.; Moad, G.; Keddie, D.J. Effect of the Z- and Macro-R-Group on the Thermal Desulfurization of Polymers Synthesized with Acid/Base “Switchable” Dithiocarbamate RAFT Agents. Macromol. Rapid Commun. 2018, 39, e1800228. [Google Scholar] [CrossRef]
- Chiefari, J.; Mayadunne, R.T.A.; Moad, C.L.; Moad, G.; Rizzardo, E.; Postma, A.; Skidmore, M.A.; Thang, S.H. Thiocarbonylthio compounds (S=C(Z)S-R) in free radical polymerization with reversible addition–fragmentation chain transfer (RAFT polymerization). Effect of the activating group Z. Macromolecules 2003, 36, 2273–2283. [Google Scholar] [CrossRef]
- Chiefari, J.; Chong, Y.; Ercole, F.; Krstina, J.; Jeffery, J.; Le, T.P.; Mayadunne, R.T.; Meijs, G.F.; Moad, C.L.; Moad, G.; et al. Living free-radical polymerization by reversible addition- fragmentation chain transfer: The RAFT process. Macromolecules 1998, 31, 5559–5562. [Google Scholar] [CrossRef]
- Montgomery, K.S.; Davidson, R.W.M.; Cao, B.; Williams, B.; Simpson, G.W.; Nilsson, S.K.; Chiefari, J.; Fuchter, M.J. Effective macrophage delivery using RAFT copolymer derived nanoparticles. Polym. Chem. 2018, 9, 131–137. [Google Scholar] [CrossRef]
- Nykaza, J.R.; Benjamin, R.; Meek, K.M.; Elabd, Y.A. Polymerized ionic liquid diblock copolymer as an ionomer and anion exchange membrane for alkaline fuel cells. Chem. Eng. Sci. 2016, 154, 119–127. [Google Scholar] [CrossRef]
- Nykaza, J.R.; Savage, A.M.; Pan, Q.W.; Wang, S.J.; Beyer, F.L.; Tang, M.H.; Li, C.Y.; Elabd, Y.A. Polymerized ionic liquid diblock copolymer as solid-state electrolyte and separator in lithium-ion battery. Polymer 2016, 101, 311–318. [Google Scholar] [CrossRef]
- Moehrke, J.; Vana, P. The Kinetics of Surface-Initiated RAFT Polymerization of Butyl acrylate Mediated by Trithiocarbonates. Macromol. Chem. Phys. 2017, 218, 1600506. [Google Scholar] [CrossRef]
- Bates, F.S.; Fredrickson, G.H. Block copolymer thermodynamics: Theory and experiment. Annu. Rev. Phys. Chem. 1990, 41, 525–557. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Ray, W.H. Modeling of “living” free-radical polymerization with RAFT chemistry. Ind. Eng. Chem. Res. 2001, 40, 4336–4352. [Google Scholar] [CrossRef]
- Zetterlund, P.B.; Gody, G.; Perrier, S. Sequence-Controlled Multiblock Copolymers via RAFT Polymerization: Modeling and Simulations. Macromol. Theory Simul. 2014, 23, 331–339. [Google Scholar] [CrossRef]
- Hernandez-Ortiz, J.C.; Jaramillo-Soto, G.; Palacios-Alquisira, J.; Vivaldo-Lima, E. Modeling of Polymerization Kinetics and Molecular Weight Development in the Microwave-Activated RAFT Polymerization of Styrene. Macromol. React. Eng. 2010, 4, 210–221. [Google Scholar] [CrossRef]
- Barner-Kowollik, C.; Quinn, J.F.; Morsley, D.R.; Davis, T.P. Modeling the reversible addition–fragmentation chain transfer process in cumyl dithiobenzoate-mediated styrene homopolymerizations: Assessing rate coefficients for the addition–fragmentation equilibrium. J. Polym. Sci. Part A Polym. Chem. 2001, 39, 1353–1365. [Google Scholar] [CrossRef]
- Wang, A.R.; Zhu, S.P. Effects of diffusion-controlled radical reactions on RAFT polymerization. Macromol. Theory Simul. 2003, 12, 196–208. [Google Scholar] [CrossRef]
- Chaffey-Millar, H.; Busch, M.; Davis, T.P.; Stenzel, M.H.; Barner-Kowollik, C. Advanced computational strategies for modelling the evolution of full molecular weight distributions formed during multiarmed (Star) polymerisations. Macromol. Theory Simul. 2005, 14, 143–157. [Google Scholar] [CrossRef]
- Tobita, H. Modeling Controlled/Living Radical Polymerization Kinetics: Bulk and Miniemulsion. Macromol. React. Eng. 2010, 4, 643–662. [Google Scholar] [CrossRef]
- Lopez-Dominguez, P.; Hernandez-Ortiz, J.C.; Vivaldo-Lima, E. Modeling of RAFT Copolymerization with Crosslinking of Styrene/Divinylbenzene in Supercritical Carbon Dioxide. Macromol. Theory Simul. 2018, 27, 1700064. [Google Scholar] [CrossRef]
- Vana, P.; Davis, T.P.; Barner-Kowollik, C. Kinetic analysis of reversible addition fragmentation chain transfer (RAFT) polymerizations: Conditions for inhibition, retardation, and optimum living polymerization. Macromol. Theory Simul. 2002, 11, 823–835. [Google Scholar] [CrossRef]
- Adebekun, D.K.; Schork, F.J. Continuous solution polymerization reactor control. 2. Estimation and nonlinear reference control during methyl methacrylate polymerization. Ind. Eng. Chem. Res. 1989, 28, 1846–1861. [Google Scholar] [CrossRef]
- Jo, J.H.; Bankoff, S.G. Digital monitoring and estimation of polymerization reactors. AIChE J. 1976, 22, 361–369. [Google Scholar] [CrossRef]
- Kim, K.J.; Choi, K.Y. On-line estimation and control of a continuous stirred tank polymerization reactor. J. Process Control 1991, 1, 96–110. [Google Scholar] [CrossRef]
- Ellis, M.F.; Taylor, T.W.; Jensen, K.F. On-line molecular weight distribution estimation and control in batch polymerization. AIChE J. 1994, 40, 445–462. [Google Scholar] [CrossRef]
- Crowley, T.J.; Choi, K.Y. On-line monitoring and control of a batch polymerization reactor. J. Process Control 1996, 6, 119–127. [Google Scholar] [CrossRef]
- Mutha, R.K.; Cluett, W.R.; Penlidis, A. On-line nonlinear model-based estimation and control of a polymer reactor. AIChE J. 1997, 43, 3042–3058. [Google Scholar] [CrossRef]
- Dimitratos, J.; Georgakis, C.; El-Aasser, M.S.; Klein, A. Dynamic modeling and state estimation for an emulsion copolymerization reactor. Comput. Chem. Eng. 1989, 13, 21–33. [Google Scholar] [CrossRef]
- Kozub, D.J.; MacGregor, J.F. State estimation for semi-batch polymerization reactors. Chem. Eng. Sci. 1992, 47, 1047–1062. [Google Scholar] [CrossRef]
- Astorga, C.M.; Sheibat-Othman, N.; Othman, S.; Hammouri, H.; McKenna, T.F. Nonlinear continuous-discrete observers: Application to emulsion polymerization reactors. Control Eng. Pract. 2002, 10, 3–13. [Google Scholar] [CrossRef]
- Appelhaus, P.; Engell, S. Design and implementation of an extended observer for the polymerization of polyethylenterephthalate. Chem. Eng. Sci. 1996, 51, 1919–1926. [Google Scholar] [CrossRef]
- Van Dootingh, M.; Viel, F.; Rakotopara, D.; Gauthier, J.P.; Hobbes, P. Nonlinear deterministic observer for state estimation: Application to a continuous free radical polymerization reactor. Comput. Chem. Eng. 1992, 16, 777–791. [Google Scholar] [CrossRef]
- Viel, F.; Busvelle, E.; Gauthier, J.P. Stability of polymerization reactors using I/O linearization and a high-gain observer. Automatica 1995, 31, 971–984. [Google Scholar] [CrossRef]
- Tatiraju, S.; Soroush, M. Nonlinear state estimation in a polymerization reactor. Ind. Eng. Chem. Res. 1997, 36, 2679–2690. [Google Scholar] [CrossRef]
- Tatiraju, S.; Soroush, M.; Ogunnaike, B.A. Multirate nonlinear state estimation with application to a polymerization reactor. AIChE J. 1999, 45, 769–780. [Google Scholar] [CrossRef]
- Soroush, M. State and parameter estimations and their applications in process control. Comput. Chem. Eng. 1998, 23, 229–245. [Google Scholar] [CrossRef]
- López-Negrete, R.; Biegler, L.T. A Moving Horizon Estimator for processes with multi-rate measurements: A Nonlinear Programming sensitivity approach. J. Process Control 2012, 22, 677–688. [Google Scholar] [CrossRef]
- Krämer, S.; Gesthuisen, R. Multirate state estimation using moving horizon estimation. In Proceedings of the 16th IFAC World Conference (IFAC 2005), Prague, Czech Republic, 3–8 July 2005; pp. 1–6. [Google Scholar]
- Liu, A.; Zhang, W.; Yu, L.; Chen, J. Moving horizon estimation for multi-rate systems. In Proceedings of the 54th IEEE Conference on Decision and Control, Osaka, Japan, 15–18 December 2015; pp. 6850–6855. [Google Scholar]
- Zambare, N.; Soroush, M.; Grady, M.C. Real-time multirate state estimation in a pilot-scale polymerization reactor. AIChE J. 2002, 48, 1022–1033. [Google Scholar] [CrossRef]
- Ling, C.; Kravaris, C. State observer design for monitoring the degree of polymerization in a series of melt polycondensation reactors. Processes 2016, 4, 4. [Google Scholar] [CrossRef]
- Kahelras, M.; Ahmed-Ali, T.; Giri, F.; Lamnabhi-Lagarrigue, F. Sampled-data chain-observer design for a class of delayed nonlinear systems. Int. J. Control 2018, 91, 1076–1090. [Google Scholar] [CrossRef]
- Ahmed-Ali, T.; Karafyllis, I.; Lamnabhi-Lagarrigue, F. Global exponential sampled-data observers for nonlinear systems with delayed measurements. Syst. Control Lett. 2013, 62, 539–549. [Google Scholar] [CrossRef]
- Nadri, M.; Hammouri, H. Design of a continuous-discrete observer for state affine systems. Appl. Math. Lett. 2003, 16, 967–974. [Google Scholar] [CrossRef]
- Karafyllis, I.; Kravaris, C. From continuous-time design to sampled-data design of observers. IEEE Trans. Autom. Control 2009, 54, 2169–2174. [Google Scholar] [CrossRef]
- Zambare, N.; Soroush, M.; Ogunnaike, B.A. A method of robust multi-rate state estimation. J. Process Control 2003, 13, 337–355. [Google Scholar] [CrossRef]
- Antoniades, C.; Christofides, P.D. Feedback control of nonlinear differential difference equation systems. Chem. Eng. Sci. 1999, 54, 5677–5709. [Google Scholar] [CrossRef]
- Liu, J.; Muñoz de la Peña, D.; Christofides, P.D.; Davis, J.F. Lyapunov-based model predictive control of nonlinear systems subject to time-varying measurement delays. Int. J. Adapt. Control Signal Process. 2009, 23, 788–807. [Google Scholar] [CrossRef]
- Liu, J.; Muñoz de la Peña, D.; Christofides, P.D. Distributed model predictive control of nonlinear systems subject to asynchronous and delayed measurements. Automatica 2010, 46, 52–61. [Google Scholar] [CrossRef]
- Ling, C.; Kravaris, C. Multi-rate observer design using asynchronous inter-sample output predictions. In Proceedings of the 2017 American Control Conference (ACC), Seattle, WA, USA, 24–26 May 2017; pp. 376–381. [Google Scholar]
- Ling, C.; Kravaris, C. Multi-rate observer design for process monitoring using asynchronous inter-sample output predictions. AIChE J. 2017, 63, 3384–3394. [Google Scholar] [CrossRef]
- Ling, C.; Kravaris, C. Multi-rate sampled-data observers based on a continuous-time design. In Proceedings of the 2017 IEEE 56th Annual Conference on Decision and Control (CDC), Melbourne, Australia, 12–15 December 2017; pp. 3664–3669. [Google Scholar]
- Ling, C.; Kravaris, C. Multi-rate Sampled-data Observer Design for Nonlinear Systems with Asynchronous and Delayed Measurements. In Proceedings of the 2019 American Control Conference (ACC), Philadelphia, PA, USA, 10–12 July 2019; pp. 1128–1133. [Google Scholar]
- Ling, C.; Kravaris, C. Multi-Rate Sampled-Data Observer Design Based on a Continuous-Time Design. IEEE Trans. Autom. Control. 2019. [Google Scholar] [CrossRef]
- Ling, C.; Kravaris, C. A dead time compensation approach for multirate observer design with large measurement delays. AIChE J. 2019, 65, 562–570. [Google Scholar] [CrossRef]
- Buback, M.; Kowollik, C. Termination kinetics of methyl methacrylate free-radical polymerization studied by time-resolved pulsed laser experiments. Macromolecules 1998, 31, 3211–3215. [Google Scholar] [CrossRef]
- Beuermann, S.; Buback, M.; Davis, T.P.; Gilbert, R.G.; Hutchinson, R.A.; Olaj, O.F.; Russell, G.T.; Schweer, J.; Van Herk, A.M. Critically evaluated rate coefficients for free-radical polymerization, 2. Propagation rate coefficients for methyl methacrylate. Macromol. Chem. Phys. 1997, 198, 1545–1560. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.; Thang, S.H. Radical addition–fragmentation chemistry in polymer synthesis. Polymer 2008, 49, 1079–1131. [Google Scholar] [CrossRef]
- Perrier, S.; Barner-Kowollik, C.; Quinn, J.F.; Vana, P.; Davis, T.P. Origin of inhibition effects in the reversible addition fragmentation chain transfer (RAFT) polymerization of methyl acrylate. Macromolecules 2002, 35, 8300–8306. [Google Scholar] [CrossRef]
- MARTEN, F.L.; HAMIELEC, A.E. High Conversion Diffusion-Controlled Polymerization. In Polymerization Reactors and Processes; ACS Symposium Series; American Chemical Society: Akron, OH, USA, 1978; Volume 104, pp. 3–43. [Google Scholar]
- Hiorns, R. Polymer Handbook. Polym. Int. 2000, 49, 807. [Google Scholar] [CrossRef]
- Soroush, M. Nonlinear state-observer design with application to reactors. Chem. Eng. Sci. 1997, 52, 387–404. [Google Scholar] [CrossRef]











| Chain Type | Moment Definition |
|---|---|
| Growing Polymer | |
| Dormant Polymer Type 1 | |
| Dormant Polymer Type 2 | |
| Dormant Polymer Type 3 | |
| Dead Polymer |
| Variables | Description |
|---|---|
| Growing polymer chain zeroth moment | |
| Growing polymer chain first moment | |
| Growing polymer chain second moment | |
| Dormant polymer chain type 1 zeroth moment | |
| Dormant polymer chain type 1 first moment | |
| Dormant polymer chain type 1 second moment | |
| Dormant polymer chain type 2 zeroth moment | |
| Dormant polymer chain type 2 first moment | |
| Dormant polymer chain type 2 second moment | |
| Dormant polymer chain type 3 zeroth moment | |
| Dormant polymer chain type 3 first moment | |
| Dormant polymer chain type 3 second moment part one | |
| Dormant polymer chain type 3 second moment part two | |
| Dead polymer chain zeroth moment | |
| Dead polymer chain first moment | |
| Dead polymer chain second moment | |
| Concentration of primary radicals | |
| Concentration of RAFT agent | |
| Concentration of primary intermediate | |
| Concentration of initiator | |
| Concentration of monomer | |
| Concentration of solvent | |
| Concentration of chain transfer agent |
| Reaction Rate Constant | Definition | Equation/Value | Units | Reference |
|---|---|---|---|---|
| Initiator formation | s | [62] | ||
| Live chain propagation | L mol s | [59] | ||
| Radical transfer to monomer | L mol s | [63] | ||
| Radical transfer to solvent | 0, assumed negligible | L mol s | ||
| Radical transfer to CTA | 0, assumed negligible | L mol s | ||
| Termination | L mol s | [58] | ||
| Forward RAFT main equilibrium | Unknown | L mol s | ||
| Reverse RAFT main equilibrium | Unknown | s | ||
| Forward RAFT pre-equilibrium | Unknown | L mol s | ||
| Reverse RAFT pre-equilibrium | Unknown | s | ||
| f | Initiator efficiency | Unknown |
| Reaction | Scale | Recipe | Monomer (g) | CTA (g) | Initiator (g) | Solvent (g) |
|---|---|---|---|---|---|---|
| PMMA-1 | Large | 215:1:0.1 | 250.63 | 4.7 | 0.191 | 626.58 |
| PMMA-2 | Large | 70:1:0.1 | 173.62 | 10.0 | 0.407 | 714.48 |
| PMMA-3 | Small | 215:1:0.1 | 0.5333 | 0.01 | 0.0004 | 1.3331 |
| PMMA-4 | Small | 70:1:0.1 | 0.3472 | 0.02 | 0.0008 | 0.8681 |
| Reaction | Scale | Recipe | Yield (g) | M (g mol) | M (g mol) | Đ |
|---|---|---|---|---|---|---|
| PMMA-1 | Large | 215:1:0.1 | 240 (94%) | 21121 | 26347 | 1.247 |
| PMMA-2 | Large | 70:1:0.1 | 158 (86%) | 7877 | 10025 | 1.273 |
| PMMA-3 | Small | 215:1:0.1 | - | 11971 | 14698 | 1.228 |
| PMMA-4 | Small | 70:1:0.1 | - | 6669 | 8101 | 1.215 |
| Sampling (h) | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 9 | 14 | 19 | 23 | 30 | 41 | 53 | 70 | 92 | 116 | 142 |
| SEC delay (h) | 2 | 3 | 3 | 4 | 4 | 5 | 5 | 6 | 6 | 4 | 4 | 6 | 4 | 3 | 6 | 8 | 5 | 4 |
| NMR delay (h) | 0.5 | 0.5 | 1 | 1 | 1 | 1 | 0.5 | 1 | 1.5 | 1 | 0.5 | 0.5 | 2.5 | 1 | 0.5 | 0.5 | 1.5 | 1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lathrop, P.M.; Duan, Z.; Ling, C.; Elabd, Y.A.; Kravaris, C. Modeling and Observer-Based Monitoring of RAFT Homopolymerization Reactions. Processes 2019, 7, 768. https://doi.org/10.3390/pr7100768
Lathrop PM, Duan Z, Ling C, Elabd YA, Kravaris C. Modeling and Observer-Based Monitoring of RAFT Homopolymerization Reactions. Processes. 2019; 7(10):768. https://doi.org/10.3390/pr7100768
Chicago/Turabian StyleLathrop, Patrick M., Zhaoyang Duan, Chen Ling, Yossef A. Elabd, and Costas Kravaris. 2019. "Modeling and Observer-Based Monitoring of RAFT Homopolymerization Reactions" Processes 7, no. 10: 768. https://doi.org/10.3390/pr7100768
APA StyleLathrop, P. M., Duan, Z., Ling, C., Elabd, Y. A., & Kravaris, C. (2019). Modeling and Observer-Based Monitoring of RAFT Homopolymerization Reactions. Processes, 7(10), 768. https://doi.org/10.3390/pr7100768